

Кинетика и катализ, 2019, T. 60, № 5, стр. 634-640
ТЕОРЕТИЧЕСКОЕ ИССЛЕДОВАНИЕ МЕХАНИЗМА ДЕОКСИГЕНАЦИИ ПРОПАНОВОЙ КИСЛОТЫ НА ПОВЕРХНОСТИ ПАЛЛАДИЯ
Р. С. Шамсиев a, *, И. Е. Соколов a, Ф. О. Данилов a, В. Р. Флид a
a ФГБОУ ВО “МИРЭА – Российский технологический университет”, Институт тонких химических технологий
им. М.В. Ломоносова
119571 Москва, пр-т Вернадского, 86, Россия
* E-mail: Shamsiev.R@gmail.com
Поступила в редакцию 11.01.2019
После доработки 12.04.2019
Принята к публикации 06.05.2019
Аннотация
Методом DFT-PBE с использованием скалярно-релятивистского приближения выполнено моделирование механизмов двух основных направлений деоксигенации пропановой кислоты на шероховатой и плоской (111) поверхностях палладия. Согласно расчетам, в механизме декарбоксилирования на шероховатой и плоской поверхностях предпочтительно образование следующих интермедиатов: C2H5COO, C2H4COO и C2H4. Для второго направления деоксигенации – реакции декарбонилирования – механизмы на разных поверхностях палладия отличаются. Так, на шероховатой поверхности наиболее вероятно протекание стадий C2H5COOH → C2H5CO → C2H5 → C2H4, а на поверхности Pd(111) – стадий C2H5COOH → C2H4COOH → C2H4CO → C2H4. Координационная ненасыщенность атомов палладия способствует понижению активационных барьеров реакции на 8–13 ккал/моль. Таким образом, плоская поверхность палладиевых частиц менее активна в деоксигенации карбоновых кислот. На селективность направлений деоксигенации тип поверхности палладия влияет незначительно. На шероховатой поверхности скорость декарбонилирования немного превышает скорость декарбоксилирования, а на поверхности Pd(111) – наоборот. Разница в активационных барьерах этих направлений деоксигенации невелика (0.7 ккал/моль).
Неослабевающий интерес к возобновляемым видам топлива связан с экологическими и экономическими соображениями. За последние 15–20 лет были разработаны различные способы производства биотоплива [см. обзоры 1, 2], основанные на крекинге биомассы. Среди многих недостатков получаемого такими способами биодизеля отмечают низкую теплоту сгорания из-за высокого содержания кислорода в нем по сравнению с нефтяным топливом. В связи с этим особый интерес представляют процессы гидродеоксигенации и деоксигенации [3], в которых кислород удаляется в виде СО (декарбонилирование), СО2 (декарбоксилирование) и Н2О (декарбонилирование, гидродеоксигенация) с образованием углеводородов. Полученное топливо, называемое “зеленым” или “возобновляемым” дизелем, превосходит биодизель и по теплоте сгорания и по экологичности. По сравнению с гидродеоксигенацией деоксигенация триглицеридов жирных кислот через реакции декарбоксилирования (ДКК) и декарбонилирования (ДКН) позволяет использовать более простые катализаторы и требует меньше водорода. Одними из наиболее активных и часто применяемых в деоксигенации жирных кислот являются палладиевые катализаторы, нанесенные на различные носители [4–6].
В последнее время достигнуты определенные успехи в понимании механизма деоксигенации, без которых трудно представить разработку высокоэффективных катализаторов. Ранее методом теории функционала плотности (DFT) были исследованы механизмы реакций ДКК и ДКН пропановой кислоты (в качестве модели жирных кислот) на неплоских поверхностях кластеров Pd4 [7] и Pd15 [2, 8] и было показано, что для разрыва С–С-связи необходим предварительный отрыв атома H от β-атома С молекулы пропановой кислоты. В допущении, что в реакциях ДКК и ДКН образуется общий интермедиат (И) с адсорбированной частицей COOH, была установлена предпочтительность реакции ДКН. Разрыв по связи C–СOOH имеет наибольший активационный барьер (Δ≠G623 = 32.8 ккал/моль).
Согласно микрокинетическому анализу деоксигенации пропановой кислоты на поверхностях Pd(111) и Pd(211) [9] на основе периодических расчетов DFT-PW91, стадиям диссоциации связей OС–OH (в реакции ДКН) и C–СOO (в реакции ДКК) предшествует глубокое дегидрирование молекулы пропановой кислоты до частиц CHCHCOOH или CH3CCOO соответственно. По нашему мнению, это довольно спорный механизм, который может осуществляться только при достаточно прочной адсорбции молекулы пропановой кислоты. Кроме того, несмотря на игнорирование энтропийного фактора в работе получены ничтожно малые значения частоты оборотов катализатора (TOF, ~10–7 с–1).
Целью настоящей работы являлось моделирование механизмов реакций декарбоксилирования и декарбонилирования пропановой кислоты на различных моделях поверхности Pd и оценка их относительной каталитической активности.
МЕТОДИЧЕСКАЯ ЧАСТЬ
Квантово-химические расчеты выполняли в программе PRIRODA [10, 11] в рамках полноэлектронного скалярно-релятивистского приближения метода теории функционала плотности. Использовали обменно-корреляционный функционал PBE [12] и базисный набор L11 [13] со следующими схемами группирования орбиталей: Pd (26s23p16d5f)/[7s6p4d1f], C, O (10s7p3d)/[4s3p1d] и H (6s2p)/[2s1p]. Адекватность данной методики была проверена на палладий-гидридных системах [14].
В качестве моделей палладиевой поверхности использовали икосаэдрический кластер Pd13 (модель шероховатой поверхности) и двухслойный кластер Pd30. Все поверхностные атомы кластера Pd13 являются эквивалентными и имеют пониженное координационное число (КЧ), равное 6, по сравнению с атомами поверхности Pd(111) (КЧ = 9). Основное электронное состояние кластеров Pd13 и Pd30 имеет спиновую мультиплетность 9 и 17 соответственно. Выбор этих моделей обусловлен тем, что их структура почти не деформируется при адсорбции ненасыщенных молекул (например, фенилацетилена [15]), что позволяет не рассматривать в расчетах энергию искажения кластера.
Соответствие оптимизированных структур минимумам или переходным состояниям (ПС) подтверждали частотным анализом. Координаты атомов палладия в расчетах не фиксировали.
Частоту оборотов катализатора (TOF) каждого маршрута рассчитывали с использованием модели energetic span [16] в программе AUTOF [17]. Энергии Гиббса интермедиатов (И) и ПС рассчитаны для температуры 623.15 К, соответствующей большинству экспериментальных исследований деоксигенации жирных кислот, и отсчитаны относительно суммы энергий молекулы C2H5COOH и палладиевого кластера. Результирующее изменение энергии Гиббса в реакциях ДКК (ΔrG623 = –37.5 ккал/моль) и ДКН (ΔrG623 = –28.8 ккал/моль) пропановой кислоты рассчитано в наиболее строгом методе – CCSD/L11, поскольку для такой оценки нет необходимости в расчете энергии палладиевых частиц.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Моделирование взаимодействия молекулы пропановой кислоты с кластером Pd13 и поверхностью Pd(111) показало, что в обоих случаях возможно образование всего 3 адсорбционных комплексов (АК). Оптимизированные структуры обнаруженных адсорбционных комплексов представлены на рис. 1, а их энергии – в табл. 1.
Рис. 1.
Структуры адсорбционных комплексов (АК) пропановой кислоты с кластером Pd13 и поверхностью Pd(111). Межатомные расстояния указаны в Å.
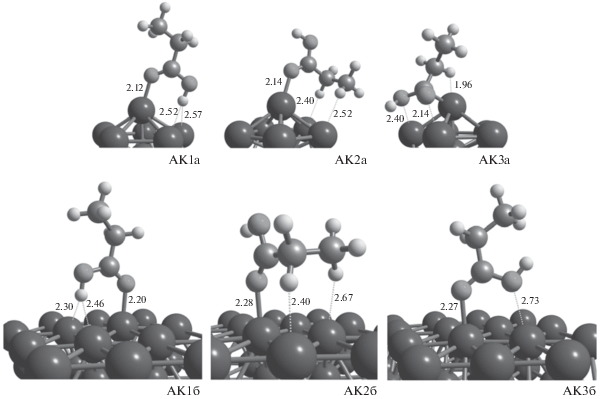
Таблица 1.
Энергии адсорбции (ккал/моль) пропановой кислоты на палладиевых поверхностях
| Поверх- ность | АК1 | АК2 | АК3 | |||
|---|---|---|---|---|---|---|
| ΔадсE0 | ΔадсG623 | ΔадсE0 | ΔадсG623 | ΔадсE0 | ΔадсG623 | |
| Pd13 (а) | –25.8 | –0.6 | –18.4 | 7.3 | –19.8 | 10.8 |
| Pd(111) (б) | –14.7 | 8.5 | –6.7 | 16.8 | –3.0 | 20.3 |
Как следует из табл. 1, самые прочные комплексы получаются за счет взаимодействия атомов O и H карбоксильной группы с атомами Pd при перпендикулярном расположении молекулы кислоты (АК1а и AК1б, рис. 1). При этом только на вершинах кластера Pd13 значение ΔадсG623 является отрицательным (–0.6 ккал/моль для АК1а, табл. 1), в остальных случаях, особенно для плоской поверхности (АК1б–АК3б, табл. 1), энергии адсорбции ΔадсE0 не компенсируют потери энтропии при образовании адсорбционного комплекса. Следовательно, при таких значениях ΔадсG623 сложно представить и смоделировать механизмы, инициируемые стадиями отщепления атомов H от α- и β-атомов C молекулы пропановой кислоты [9].
Механизмы декарбоксилирования и декарбонилирования на Pd13
Поскольку в реальных условиях деоксигенации в системе всегда имеется водород, а также для ограничения миграции адсорбатов, была смоделирована предварительная диссоциативная адсорбция молекулы H2 на поверхности Pd13. В присутствии атомов Н энергия адсорбции молекулы пропановой кислоты на Pd13 незначительно увеличивается (на 0.3 ккал/моль).
На рис. 2 представлена схема превращения пропановой кислоты в реакции ДКК. На первой стадии с низким активационным барьером (8.8 ккал/моль) отщепляется атом H и на поверхности Pd13 формируется частица C2H5COO* (И1а, рис. 2), μ2-координация которой на атомах Pd приводит к заметному понижению энергии.
Рис. 2.
Механизм декарбоксилирования пропановой кислоты на Pd13. Рядом с обозначениями структур приведены значения ΔG623, а рядом со стрелками – Δ≠G623 (в ккал/моль).
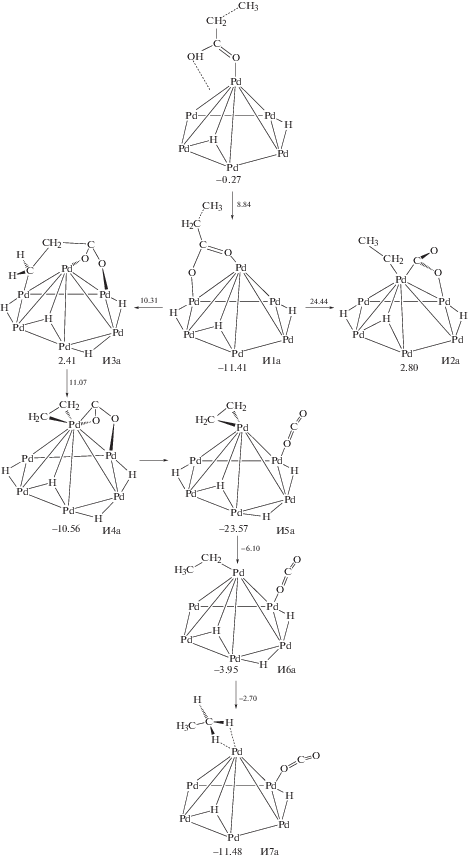
Стадия разрыва связи С–С в интермедиате И1а протекает с высоким активационным барьером (35.9 ккал/моль для И1а → И2а, рис. 2). Существенно меньший барьер для разрыва С–С-связи потребуется, если предварительно отрывается атом H от β-атома С частицы C2H5COO* (И1а → И3а, рис. 2). Вероятно, отрыв атома H от β-атома C позволяет частице C2H4COO* прочнее связаться с металлом и тем самым активировать С–С-связь. В этом случае разрыв связи углерод–углерод в частице C2H4COO* (И3а) связан с преодолением невысокого барьера в 13.5 ккал/моль и заметным экзотермическим эффектом (И4а) за счет π-координации этилена. Далее возможно практически безбарьерное элиминирование молекулы СО2, что приводит к дополнительному понижению энергии системы до -23.6 ккал/моль (И5а, рис. 2). Последующие стадии превращения интермедиата И5а связаны с последовательным гидрированием адсорбированной молекулы этилена до этана (И5а → И6а → И7а, рис. 2).
На рис. 3 представлен наиболее вероятный механизм превращения молекулы C2H5COOH в реакции ДКН. Первая стадия этого механизма связана с изменением координации карбоксильной группы на палладии (И1б). С энергетической точки зрения эта стадия невыгодна, поскольку энергия системы возрастает до 14.2 ккал/моль, однако она позволяет создать структурные предпосылки к последующим стадиям разрыва С–С-связи (И1б → И2б, рис. 3) или отрыва OH-группы (И1б → И3б, рис. 3). С точки зрения кинетики разрыв С–С-связи в И2б (24.1 ккал/моль) менее вероятен по сравнению с разрывом связи C–OH (21.5 ккал/моль). В последнем случае формируется частица C2H5CO* с небольшим понижением энергии (И3б, рис. 3).
Рис. 3.
Механизм декарбонилирования пропановой кислоты на Pd13. Рядом с обозначениями структур приведены значения ΔG623, а рядом со стрелками – Δ≠G623 (в ккал/моль).
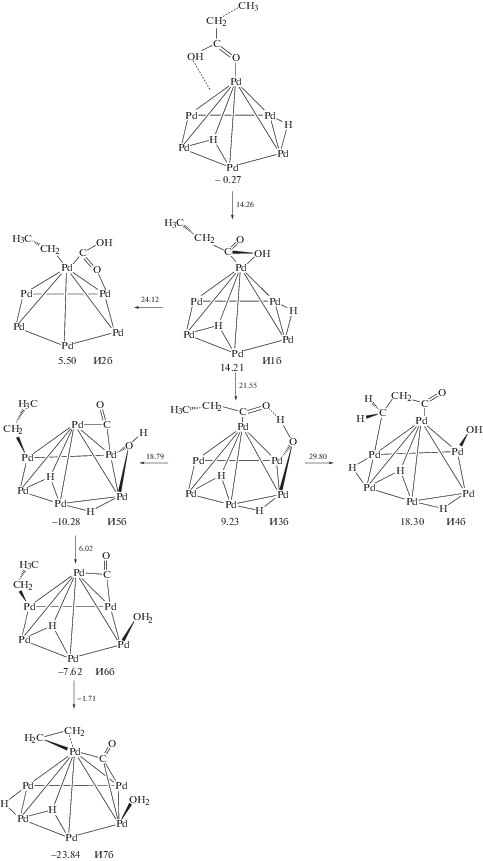
Отрыв атома H от β-атома С частицы C2H5CO* протекает с более высоким активационным барьером (20.6 ккал/моль для И3б → И4б, рис. 3) по сравнению с барьером стадии разрыва С–С-связи (9.6 ккал/моль для И3б → И5б). Кроме того, стадия И3б → И5б сопровождается заметным понижением энергии системы в отличие от стадии И3б → И4б. Заключительные стадии образования молекулы воды (И5б → И6б, рис. 3) и дегидрирования этила до этилена (И6б → И7б, рис. 3) не требуют преодоления значительных активационных барьеров. Таким образом, максимальные энергетические барьеры реакций ДКК и ДКН на поверхности Pd13 составляют 22.5 и 21.8 ккал/моль соответственно.
Механизмы реакций ДКК и ДКН на поверхности Pd(111)
На рис. 4 и 5 представлены энергетические профили и оптимизированные структуры интермедиатов механизмов реакций ДКК и ДКН на поверхности Pd(111). Как показали расчеты, механизм реакции ДКК на плоской поверхности (рис. 4), в целом, соответствует механизму ДКК на шероховатой поверхности (рис. 2). Основное отличие заключается в резком увеличении активационных барьеров каждой стадии (на 8–12 ккал/моль), начиная с адсорбции молекулы пропановой кислоты. Очевидно, главной причиной этих изменений является низкая координационная доступность атомов плоской поверхности палладия.
Рис. 4.
Энергетический профиль реакции декарбоксилирования пропановой кислоты на поверхности Pd(111).
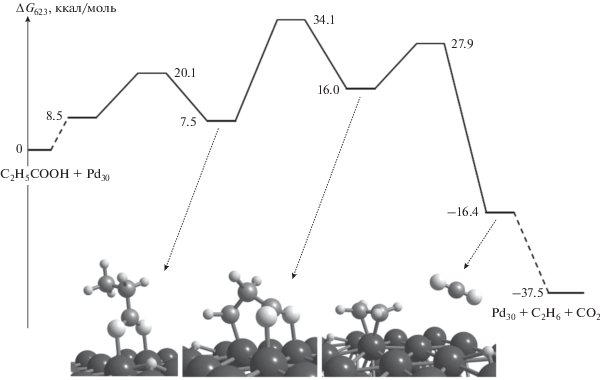
Рис. 5.
Энергетические профили реакции декарбонилирования пропановой кислоты на поверхности Pd(111).
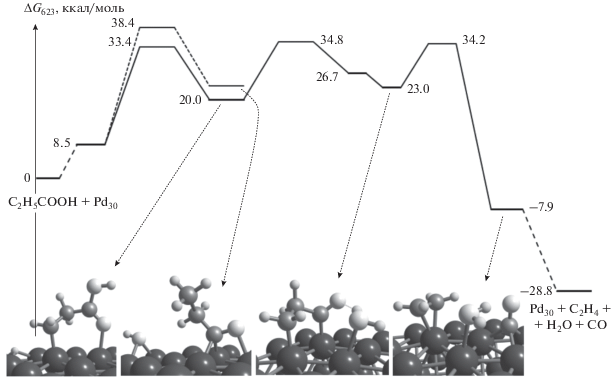
Что касается механизма реакции ДКН, то на плоской поверхности не удалось обнаружить интермедиат И1б с активированной связью С–ОН (рис. 3). В связи с этим разрыву С–OH-связи должна предшествовать стадия отрыва атома H от β-атома C молекулы пропановой кислоты с образованием частицы C2H4COOH (рис. 5). В противном случае, энергия ПС стадии разрыва С–OH-связи в молекуле C2H5COOH достигает величины в 38.4 ккал/моль (рис. 5).
На рис. 4 и 5 видно, что итоговые активационные барьеры реакций ДКК и ДКН достаточно близки (34.1 и 34.8 ккал/моль), а соответствующие рассчитанные значения TOF (14.1 и 4.2 c–1) на 7–8 порядков превосходят аналогичные величины, приведенные в работе [9].
ЗАКЛЮЧЕНИЕ
Квантово-химическое моделирование (метод DFT-PBE/L11) деоксигенации пропановой кислоты на поверхности палладия позволило выявить наиболее вероятные (низкоэнергетические) механизмы основных направлений процесса – декарбоксилирования и декарбонилирования. Механизм декарбоксилирования включает стадии адсорбции пропановой кислоты, последовательный отрыв атомов H от карбоксильной группы и от β-атома углерода, разрыв С–C-связи с элиминированием молекулы СО2 и, наконец, стадии гидрирования этилена до этана. Наибольшие активационные барьеры имеют стадии β-элиминирования атома H (в случае Pd(111)) и разрыва С–С-связи (в случае Pd13).
Механизм реакции ДКН на шероховатой поверхности включает последовательность следующих интермедиатов: C2H5COOH → C2H5CO → → C2H5 → C2H4. На поверхности Pd(111) последовательность интермедиатов несколько иная, а именно: C2H5COOH → CH2CH2COOH → C2H4CO → C2H4. Максимальным активационным барьером обладает стадия отрыва OH-группы.
Согласно расчетам, координационная доступность поверхностных атомов палладия кластера Pd13 способствует понижению активационных барьеров на 8–13 ккал/моль. Таким образом, тип структуры поверхности палладия оказывает значительное влияние на скорость процесса, и плоские поверхности палладиевых частиц менее активны в деоксигенации карбоновых кислот. В то же время величины TOF реакций декарбоксилирования и декарбонилирования на Pd(111) достаточно велики и равны 14.1 и 4.2 c–1 соответственно.
Несмотря на различную природу лимитирующих стадий направлений деоксигенации, их активационные барьеры очень близки в пределах одного типа поверхности, что объясняет экспериментально наблюдаемый набор продуктов, характерный для обоих направлений. Если для шероховатой поверхности скорость реакции декарбонилирования незначительно больше скорости декарбоксилирования, то для поверхности Pd(111) – наоборот. В обоих случаях разница в величине активационных барьеров этих направлений деоксигенации невелика (0.7 ккал/моль) и находится в пределах ошибки метода теории функционала плотности.
Список литературы
Santillan-Jimenez E., Crocker M. // J. Chem. Technol. Biotechnol. 2012. V. 87. P. 1041.
Беренблюм А.С., Данюшевский В.Я., Кузнецов П.С., Кацман Е.А., Шамсиев Р.С. // Нефтехимия. 2016. Т. 56. № 5. С. 433.
Беренблюм А.С., Подоплелова Т.А., Шамсиев Р.С., Кацман Е.А., Данюшевский В.Я., Флид В.Р. // Катализ в пром-сти. 2012. Т. 4. № 3. С. 84.
Snåre M., Kubicková I., Mäki-Arvela P., Eränen K., Murzin D.Yu. // Ind. Eng. Chem. Res. 2006. V. 45. P. 5708.
Беренблюм А.С., Подоплелова Т.А., Шамсиев Р.С., Кацман Е.А., Данюшевский В.Я. // Нефтехимия. 2011. Т. 51. № 5. С. 342.
Ford J.P., Immer J.G., Lamb H.H. // Top. Catal. 2012. V. 55. P. 175.
Беренблюм А.С., Шамсиев Р.С., Подоплелова Т.А., Данюшевский В.Я. // Журн. физ. хим. 2012. № 8. С. 1340.
Беренблюм А.С., Подоплелова Т.А., Кацман Е.А., Шамсиев Р.С., Данюшевский В.Я. // Кинетика и катализ. 2012. Т. 53. № 5. С. 634.
Behtash S., Lu J., Williams C.T., Monnier J.R., Heyden A. // J. Phys. Chem. C. 2015. V. 119. № 4. P. 1928.
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. P. 151.
Лайков Д.Н., Устынюк Ю.А. // Изв. АН. Сер. хим. 2005. № 3. С. 804.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Laikov D.N. // Chem. Phys. Lett. 2005. V. 416. P. 116.
Шамсиев Р.С., Данилов Ф.О. // Изв. АН. Сер. хим. 2017. № 3. С. 395.
Шамсиев Р.С., Данилов Ф.О. // Кинетика и катализ. 2018. Т. 59. № 3. С. 340.
Uhe A., Kozuch S., Shaik S. // J. Comput. Chem. 2011. V. 32. P. 978.
Kozuch S., Martin J.M.L. // ACS Catal. 2011. V. 1. P. 246.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ