

Кинетика и катализ, 2019, T. 60, № 6, стр. 725-740
Удаление закиси азота в производстве азотной кислоты
Л. А. Исупова a, *, Ю. А. Иванова a
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: isupova@catalysis.ru
Поступила в редакцию 01.04.2019
После доработки 11.04.2019
Принята к публикации 11.04.2019
Аннотация
Производство азотной кислоты является одним из основных промышленных источников поступления в атмосферу закиси азота, озонразрушающего газа с потенциалом в 310 раз более высоким, чем СО2. В обзоре рассмотрены каталитические системы и однореакторные способы (высокотемпературный и низкотемпературный) для снижения выбросов закиси азота в производстве азотной кислоты. Показано, что существующие катализаторы обеспечивают 80–90% разложение закиси азота. Дальнейшее увеличение степени разложения связано с разработкой более активных и стабильных катализаторов.
ВВЕДЕНИЕ
С активизацией индустриальной деятельности человека в атмосферу стали выбрасываться значительные объемы парниковых газов (CO2, CH4, N2O). Статистика глобальных антропогенных выбросов парниковых газов, собранная C2ES (независимый центр климатических и энергетических решений, некоммерческая организация), свидетельствует, что за последние 20 лет их количество увеличилось приблизительно в четыре раза. Ожидается, что к концу столетия это приведет к повышению температуры планеты на 1.5–2.0°C [1, 2].
В конце XX в. было выработано международное соглашение о сокращении выбросов парниковых газов в атмосферу для сдерживания глобального потепления, подписанное в 1997 г. в Киото (Япония), и намечены основные пути для этого, такие как:
– изменение структуры топливного баланса стран мира путем перехода к менее “грязным” технологиям (переход от сжигания угля к сжиганию газа, использование АЭС и ГЭС, ветровой энергии и др.);
– широкое внедрение энергосберегающих технологий и очистных сооружений.
Основной объем выбросов парниковых газов приходится на промышленность и сельское хозяйство и составляет около 76% CO2, 16% CH4 и 6% N2O. Парниковые газы различаются “силой” влияния на атмосферу; для сравнения парникового воздействия их эффект рассчитывается относительно наиболее распространенного – углекислого газа СО2. Закись азота N2O обладает самым высоким потенциалом глобального потепления, в 310 раз превышающим таковой для СО2 [2, 3]. В стратосфере N2O подвергается фотолизу и реагирует с атомами кислорода с образованием оксида азота (NO), который входит в известный цикл разрушения озонового (O3) слоя [4].
Сокращение выбросов N2O в сельскохозяйственном секторе до сих пор не имеет значимого прогресса, поэтому единственной возможностью является снижение выбросов в химических производствах, составляющих примерно 30% от общей эмиссии закиси азота [5]. К выбросам N2O приводят два основных производственных процесса – получение адипиновой и азотной кислот. На долю адипиновой кислоты приходится почти три четверти (70%) промышленных выбросов, а на долю азотной – оставшаяся часть (30%). Общемировые выбросы закиси азота от обоих производств составляют более 900 кт/год, от синтеза азотной кислоты – около 300 кт/год [6, 7].
В РФ существует три основные схемы получения азотной кислоты, отличающиеcя по мощности и рабочему давлению [8].
Общая технологическая линия производства слабой азотной кислоты состоит из трех стадий:
– каталитическое окисление аммиака до оксида азота;
– последующее окисление оксида азота до диоксида азота;
– адсорбция диоксида азота водой с образование азотной кислоты.
Закись азота при синтезе азотной кислоты формируется как побочный продукт на стадии каталитического окисления аммиака, которое протекает в интервале температур 800–1000°С на сетках, состоящих из Pt–Rh(–Pd)-сплавов, в соответствии с реакцией:
Помимо этого образование закиси азота возможно во вторичных реакциях в результате взаимодействия продукта реакции NO и непрореагировавшего (проскочившего) аммиака:
Мониторинг содержания N2O по газовому тракту в производстве неконцентрированной азотной кислоты (рис. 1) показал, что после катализаторного пакета концентрация N2O варьируется от 500 до 3000 ppm в зависимости от качества и количества платиноидных сеток в пакете и газодинамических условий работы реактора окисления аммиака [9, 10]. В работе [11] было показано, что за время пребывания нитрозных газов в горячей зоне реактора окисления аммиака около 10% N2O термически разлагается под действием высокой температуры, однако для более полного термического разложения требуется значительное увеличение размера реактора, что нецелесообразно.
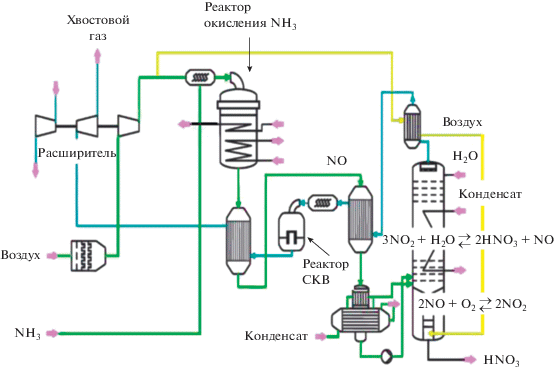
Далее нитрозные газы проходят пароперегреватель, котел-утилизатор, охлаждаясь при этом от 900 до 250–270°С, затем окислитель, подогреватель отходящих газов, холодильники-конденсаторы, где освобождаются от основной массы реакционной влаги. Окисленный и охлажденный газ при 50–60°С поступает в абсорбционную колонну, в которую также подают кислый конденсат на орошение и воздух с оксидами азота после отдувочной колонны, где NO2 связывается водой. При прохождении этого пути наблюдается снижение концентрации N2O до 500–1500 ppm [10]. Дальнейшее содержание N2O в выхлопных газах зависит от условий их обязательной очистки от NOх. Поскольку ПДК оксидов азота NO и NO2 в атмосфере составляет всего 0.04–0.086 мг/м3 [12], в технологические линии производств азотной кислоты включены схемы De-NOx, при этом, как показали результаты обследования (табл. 1), особенности реализации процессов De-NOx могут приводить как к уменьшению, так и к увеличению содержания N2O в отходящих газах.
Таблица 1.
Уровень эмиссии закиси азота в типовых схемах производства азотной кислоты до и после реактора каталитической очистки [9]
| Схема производства |
Способ очистки от NOх | Концентрация N2O, ppm | |
|---|---|---|---|
| на входе | на выходе | ||
| Комбинированная 1/3.5 |
Селективное восстановление NH3 (~280–300°С) |
500–800 | 900–1200 |
| УКЛ-7.3 | Высокотемпературное восстановление СН4 (~700°С) |
870–1390 | 20–50 |
| УКЛ-7.3 | Селективное восстановление NH3 (~280–300°С) |
~560 | ~780 |
| УКЛ-7.3 | Селективное восстановление NH3 (~280–300°С) |
900–1200 | 900–1000 |
| АК-72 | Высокотемпературное восстановление СН4 (~700°С) |
1000–1200 | 3–50 |
При высокотемпературном (неселективном) каталитическом восстановлении NOx – производится методом высокотемпературного восстановления NOx природным газом (CH4) на Pd-содержащих катализаторах при 720–770°C – одновременно с NOх удаляется и N2O (табл. 1) [9, 10, 13].
Селективное каталитическое восстановление NOх аммиаком, протекающее при средних и низких температурах (250–520°С), в зависимости от используемого катализатора (в российских реакторах применяются WO3–V2O5, нанесенные на Al2O3 или TiO2 [14], в зарубежных – Fe-цеолитные катализаторы в технологии Uhde EnviNOх® [10, 15–18]), в лучшем случае не влияет на содержание закиси азота в отходящих газах. Из-за колебания содержания оксидов азота в отходящих газах при недостаточно точной регулировке дозирования аммиака возможна подача избыточного количества NH3 для восстановления NOх, что приводит к продуцированию в реакторе дополнительного количества закиси азота; при точном дозировании аммиака в реактор селективной очистки образования закиси азота практически не происходит.
Таким образом, в технологических схемах производств азотной кислоты, включающих неселективное удаление оксидов азота, дополнительной очистки от закиси азота не требуется; при селективном удалении NOx снижения содержания закиси азота, помимо оптимизации стадии окисления аммиака на платиноидных сетках (за счет подбора пакета сеток, улучшения газораспределения в пакете и др.), можно достичь путем введения дополнительной стадии очистки газов от N2O [10].
Исходя из технологической схемы производств азотной кислоты, в литературе рассматриваются различные возможности для осуществления процесса удаления закиси азота со значительно различающимися по температуре и составу газов условиями. Так, описаны варианты высокотемпературного и низкотемпературного удаления закиси азота путем организации двухступенчатых каталитических систем в реакторах окисления аммиака или СКВ, а также с помощью установки дополнительного реактора для очистки отходящих газов в конце газового тракта.
Высокотемпературное разложение N2O протекает в условиях процесса окисления аммиака при температурах 800–1000°С и давлении 1–13 бар, GHSV = 60 000–600 000 ч−1. Высокотемпературный способ удаления N2O предполагает, что в реакторе окисления аммиака после (по ходу газа) платиноидных сеток устанавливают вторую каталитическую ступень для разложения закиси азота. Удаление N2O непосредственно в реакторе после платиновых сеток технологически удобный способ. Он не требует больших дополнительных вложений и изменений в технологической линии. Катализаторы разложения N2O в условиях процесса окисления аммиака, кроме высокой активности, должны удовлетворять требованиям термостабильности, устойчивости в агрессивной среде (окислы азота, вода, непрореагировавший аммиак), инертностью по отношению к NO (не разлагать NO), а также характеризоваться низким газодинамическим сопротивлением слоя катализатора. Использование блочных катализаторов в таких условиях предпочтительнее.
Низкотемпературная очистка (при 200–520°C) может быть организована в реакторе СКВ NOх аммиаком либо в дополнительном реакторе очистки хвостовых газов. Технологии комплексной очистки под торговой маркой Uhde EnviNOх® [18, 19], ориентированные на однореакторные схемы, включают: 1) разложение N2O в 1-м слое Fe-цеолитного катализатора и селективное каталитическое восстановление NOх аммиаком во 2-м слое; 2) восстановление оксидов азота аммиаком на катализаторе в 1-м слое и восстановление закиси углеводородами (метаном или пропаном) во 2-м слое. Технологическим трендом на сегодняшний день является возможность осуществления комплексной низкотемпературной каталитической очистки хвостовых газов от NOх и N2O в реакторе СКВ, когда в 1-м слое происходит каталитическое восстановление NOх аммиаком, а во 2-м слое – каталитическое разложения N2O [10, 20]. Для такого подхода катализаторы 1 и 2 слоя должны работать в близком температурном интервале. В технологии Uhde Envinox, применяющей цеолитные катализаторы для удаления оксидов азота, процессы протекают при температурах около 400°C. Для российских схем, использующих низкотемпературное СКВ NOх аммиаком при давлении 2.8–0.56 МПа, температурах 200–300°С и GHSV = = 7200 ч–1, организация однореакторной схемы комплексной очистки потребует разработки специальных низкотемпературных катализаторов разложения, поскольку цеолитные катализаторы функционируют при температуре выше 350°С [20, 21]. Типичный состав отходящих газов в производствах азотной и адипиновой кислот представлен в табл. 2. Важно также то, что в реакторе СКВ есть свободное пространство, которое позволяет загрузить до 1 м катализатора второй ступени по высоте. Таким образом, гранулированный катализатор второго слоя реактора СКВ должен быть активным в области температур 250−300°С, устойчивым в реакционной среде и обеспечивать снижение закиси азота до 50 ppm при высоте слоя не более 1 м.
Таблица 2.
Типичные составы отходящих хвостовых газов в производствах азотной и адипиновой кислот [6]
| Процесс производства | Температура, °С | Концентрации компонентов, % | |||||
|---|---|---|---|---|---|---|---|
| N2O | NOx | O2 | H2O | CO | NH3 | ||
| Адипиновая кислота | 200–300 | 30–50 | 0.7 | 4 | 2–3 | 0.03 | 0 |
| Азотная кислота | 180–300 | 0.03–0.3 | 0.03–0.3 | 2–4 | 2–3 | 0 | 0–0.12* |
* Состав газов после каталитической очистки от NOx [20].
МЕХАНИЗМ РАЗЛОЖЕНИЯ ЗАКИСИ АЗОТА
Разложение N2O (ΔH298 = −163 кДж/моль) – экзотермическая необратимая реакция. Этот процесс может протекать по двум механизмам: по механизму Лэнгмюра–Хиншельвуда (Л–Х), характеризующемуся низкой энергией активации стадии диффузии поверхностного кислорода и его рекомбинации (стадия II), или по механизму Элей–Ридела (Э–Р), отличающемуся высокой энергией активации поверхностной диффузии, что требует больших затрат энергии для диффузии кислорода (стадия III) [22–24]:
(I)
${{{\text{N}}}_{2}}{\text{O}} + {\text{S}} \to {{{\text{N}}}_{2}} \uparrow + {\text{ S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}},~$(II)
$2{\text{S}} \ldots {{{\text{O}}}_{{{\text{surf\;}}}}}\,\,\begin{array}{*{20}{c}} \leftarrow \\ \to \end{array}\,\,2{\text{S}} + {{{\text{O}}}_{{2~}}}({\text{Л}}{\kern 1pt} --{\kern 1pt} {\text{Х}}),$(III)
${\text{S}} \ldots {{{\text{O}}}_{{{\text{surf\;}}}}} + {{{\text{N}}}_{2}}{\text{O}} \to {\text{S}} + {{{\text{O}}}_{2}}{\kern 1pt} \uparrow + {\text{ }}{{{\text{N}}}_{2}}{\kern 1pt} \uparrow ({\text{Э}}{\kern 1pt} --{\kern 1pt} {\text{Р}}),$Установлено, что для различных оксидных систем в области температур 400–600°C стадии (II) и (III) относительно медленные по сравнению со стадией (I) [25–29]. Другими словами, десорбцию O2 можно считать стадией, определяющей скорость каталитического разложения N2O независимо от механизма образования O2, что было показано еще в ранних работах [29, 30], где установили линейную зависимость между скоростью обмена кислорода c поверхностью и скоростью разложения N2O для ряда оксидов и перовскитов в интервале температур 300–600°С.
Литературные данные свидетельствуют о том, что скорость отдельных стадий и, следовательно, их вклад в процесс разложения, зависят от природы активных центров (кислородные вакансии или катионы металла) и температуры процесса [31–33]. Kiwi-Minsker и соав. [33, 34] продемонстрировали с помощью математической модели, основанной на экспериментальных данных, полученных для Fe–ZSM-5, что скоростьопределяющая стадия разложения N2O может изменяться в зависимости от температуры реакции. В работе [6], кроме того, было показано, что стадия десорбции кислорода может также лимитироваться переносом кислорода по поверхности при образовании O2surf.
(IV)
${\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}} + {\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}}\,~\begin{array}{*{20}{c}} \leftarrow \\ \to \end{array}~\,{{{\text{O}}}_{{2{\text{surf}}}}} + 2{\text{S}}~~\begin{array}{*{20}{c}} \leftarrow \\ \to \end{array}~~{{{\text{O}}}_{2}}{\kern 1pt} \uparrow + {\text{ }}2{\text{S}}.$Это позволяет объяснить особенности некоторых оксидных систем, например, таких как CeO2 [35]. Оксидная система CeO2 обладает высокой кислородной емкостью, обусловленной легкостью окислительно-восстановительного процесса Се3+/Се4+ с подвижным реакционноспособным кислородом на границах раздела фаз активного компонента (центра) и носителя – CeO2 [36]. Активный центр ответственен за диссоциацию молекул N2O и образование интермедиатов кислорода (I), тогда как на границе раздела фаз носитель/активный центр за счет диффузии кислорода может протекать кислородный перенос (стадии (V) и (VI)). Стадии (VI) и (VII) приводят к рекомбинации кислорода и закрытию каталитического цикла [37–40]:
(V)
${\text{S}} + {\text{Ce}}{\kern 1pt} --{\kern 1pt} {\text{O}}\,~\begin{array}{*{20}{c}} \leftarrow \\ \to \end{array}~\,\,{\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}} + {\text{Ce*}},$(VI)
${\text{S}} \ldots {{{\text{O}}}_{{{\text{surf\;}}}}} + {\text{Ce}}--{\text{O}} \to {\text{S}} + {\text{Ce*}} + {{{\text{O}}}_{2}},$(VII)
${\text{Ce*}} + {{{\text{N}}}_{2}}{\text{O}}~\, \to \,~{\text{Ce}}{\kern 1pt} --{\kern 1pt} {\text{O}} + {{{\text{N}}}_{2}},$Механизм поверхностного переноса кислорода часто рассматривается и для систем с нанесенными металлами платиновой группы [41–44].
Влияние поверхностного переноса на скорость реакции было отмечено также в работах [31, 45, 46] при исследовании высокотемпературного (700–900°С) разложения закиси азота на оксидах со структурой перовскита. Так, в работе [31] с помощью метода изотопных релаксаций SSITKA (Steady-State Isotopic Transient Kinetic Analysis) была установлена корреляция между скоростью разложения N2O и коэффициентом диффузии кислорода в решетке перовскитов La1– xSrxMnO3 ± δ:
В то же время для перовскитов La1– x SrxFeO3 ± δ корреляции между скоростью разложения N2O и коэффициентом диффузии кислорода не отмечалось [45]. Чтобы объяснить наблюдаемую корреляцию с точки зрения механизма реакции авторы [46] выполнили динамические эксперименты для изучения переноса кислорода от N2O к O2 в процессе каталитического разложения N2O на LaMnO3 + δ и показали наличие взаимосвязи между коэффициентом диффузии кислорода в объеме (D) и коэффициентом гетерообмена (R) на поверхности, зависящими от наличия кислородных вакансий. Таким образом, в случае LaMnO3 + δ скоростьопределяющей стадией десорбции кислорода является поверхностный перенос, в то время как для La1 – x SrxFeO3 ± δ – десорбция кислорода.
Исследования каталитических систем в разложении N2O ориентированы на их практическое применение, когда помимо каталитической активности имеет значение влияние на их стабильность других газов, которые обычно встречаются в отходящих газах и оказывают ингибирующее действие. Наибольшей способностью дезактивировать многие оксидные системы обладает вода, затем O2 и наименьший эффект оказывают NOx [26, 47, 48]. Авторы [22, 39, 49] объясняют это конкурентной адсорбцией ингибиторов на активных центрах активации N2O и их блокировкой. Некоторые системы обратимо дезактивируются после воздействия ингибиторов, например, Ru/Al2O3 при 480°С [50], а CuO/CeO2 при 400°С [22, 39].
Введение добавок щелочных металлов и CeO2 увеличивает устойчивость оксидных систем к воздействию ингибиторов. Исходя из механизма процесса разложения N2O этот эффект объясняется тем, что низкая энергия ионизации атомов щелочных металлов ослабляет связь активного центра с адсорбированным кислородом, облегчая регенерацию центра [36, 49, 51]. Оксид церия, обладая высокой подвижностью решеточного кислорода, способен быстро подводить кислород к активному центру, обеспечивая образование молекулы кислорода [39, 52–55].
Таким образом, увеличение окислительно-восстановительных способностей каталитических систем облегчает как десорбцию кислорода, так и регенерацию активных центров.
КАТАЛИЗАТОРЫ НИЗКОТЕМПЕРАТУРНОГО РАЗЛОЖЕНИЯ N2O
Каталитические системы, которые исследователи описывают как активные в реакции разложения N2O, можно разбить на несколько групп: цеолиты с нанесенным активным компонентом; чистые и смешанные оксидные системы; оксидные системы с нанесенными металлами, в том числе благородными (табл. 3). Большинство работ посвящено изучению активности катализаторов в области низких температур с целью определения возможности их применения для очистки отходящих газов в производствах азотной и адипиновой кислот, а также в однореакторных схемах De-NOx и De-N2O.
Таблица 3.
Катализаторы и условия каталитического разложения N2O
| Система | Состав реакционной смеси | Т50%,°С | Ссылка |
|---|---|---|---|
| Оксидные системы, содержащие металлы платиновой группы | |||
| Rh/ZnO, Rh/CeO, Rh/ZSM-5 (0.1–2% Rh) | N2O – 950 ppm (5% O2, 1% H2O), 0.1 г, 80 мл/мин | 220 | [60] |
| Ru/MCM-41 | 10% N2O, GHSV = 4000 ч−1 | 315 | [128] |
| Rh/Al2O3, Pd/Al2O3, Pt/Al2O3 (5% Me) | N2O – 60 ppm (21% O2), 0.050 г, 50 мл/мин | 270 | [56] |
| Rh/HAP-10.5, Rh/Al2O3, Rh/TiO2,Rh/SiO2, Rh/Zn/Al2O3 (1% Rh, 0.5–20% Zn) | 0.5% N2O, GHSV = 7000 ч−1 | 250 | [57–59] |
| Rh + Na2O/Al2O3 (0–0.099 мол. % Na2O) | 1% N2O, GHSV= 4000 ч−1 | 300 | [129, 130] |
| 0.5 мас. % M/MeOx (M = Rh, Pt, Pd;Me = CeO2, CeO2(La), CeO2(Pr), Al2O3;10 мас. % La или Pr) | N2O – 1000 ppm, GHSV = 10 000 ч−1 | 250 | [41, 42] |
| 1–4% Ru/Al2O3 | Механизм отравления катализатора реагентами: O2, CO, CO2, SO2, H2O | 340 | 50 |
| 2% Ru/γ-Al2O3 | N2O – 500 ppm (5% O2, 50 ppm SO2, 10% H2O), GHSV = 56 000 ч–1 | 350–500 | 131 |
| Rux/Co3O4 (x = 0–0.3) | N2O – 500–5000 ppm (2.1% H2O, 1.5% O2), GHSV = 54 000 ч−1 | 280 | [61] |
| Rh/CeO2, Rh/La2O3–CeO2, Rh/CeO2–Al2O3, Rh/Al2O3 (1% Rh) | N2O – 500 ppm (6% O2, 6% H2O), GHSV = 60 000 ч−1 | 220 | [132, 133] |
| Ag/Al2O3, (1–8 мас. % Ag), Rh/Al2O3, Rh–Ag/Al2O3 (0.05–0.5% Rh) | N2O – 0.01 атм (0.01 атм. CO), 150 мл/мин | 325–410 | [134] |
| Rh2O3/CePO4 | 0.5% N2O (92% CO2, 5% O2, 2% H2O), 0.25 г, 60 мл/мин | 290 | [135] |
| 0.5% Ir/Al2O3, Ir/CeO2/Al2O3, Ir/CeO2 | N2O – 1000 ppm (2% O2), GHSV = 40 000 ч−1 | 460 | [136] |
| Ir/TiO2, Ir/Al2O3, Ir/P25 (0.1–5 мас. % Ir) | 30% N2O, GHSV = 30 000 ч−1 | 270 | [44] |
| Ag/FexAl2 – xO3 (0.0 ≤ x ≤ 2.0) | N2O – 1000–3000 ppm, 50 г, 200 мл/мин | 400 | 137 |
| 5% Ag/SiO2 | N2O – 5000 ppm, GHSV = 7200 ч−1 | 490 | 138 |
| Au/Al2O3, Au/CeO2, Au/Fe2O3, Au/TiO2, Au/ZnO (1 мас. % Au/MeOx) | N2O – 1000 ppm (2% O2), GHSV = 10 000–40 000 ч−1 | 520 | 139 |
| RE/NiO, RE/CuO (RE = Gd, La, Sm, Nd, Pr, Tb, Y) | N2O – 500 ppm, 0.5 г, 200 мл/мин | 320–440 | [66–69] |
| Pr1 − xBaxMnO3 (x = 0–0.4) | N2O – 5000 ppm (200 ppm NO, 5% O2) | 450 | [140] |
| Оксидные системы на основе переходных металлов | |||
| AB2O4 (A = Mg, Ca, Mn, Co, Ni, Cu, Cr, Fe, Zn; B = Cr, Fe, Co) | N2O – 5000 ppm (5% O2), GHSV = 80 000 ч–1 | 440–780 | [72] |
| MCO3–Co3O4 (M = Ca, Sr, Ba; M/Co = 0.1, 0.15, 0.2, 0.3, 0.4) | Влияние природы оксидной добавки и степени замещения | 370 | [73] |
| MgxCo1 – xCo2O4 (х = 0–1); K/Co = 0.05–0.2 | N2O – 500 ppm, 0.5 г, 200 мл/мин | 280 | [74] |
| NixCo1 – xCo2O4 (x = 0–1) | N2O – 500 ppm, 0.5 г, 200 мл/мин | 250 | [75] |
| K/CuxCo1 –xCo2O4 (x = 0–1) | N2O – 1000 ppm, 0.5 г, GHSV = 54 000 ч−1 | 300 | [47] |
| CuxCo1 – xCo2O4 (x = 0–1) | N2O – 1500 ppm, 0.5 г, 200 мл/мин | 280 | [76] |
| ZnxCo1− xCo2O4 (x = 0.25–1) | N2O – 500 ppm, 0.5 г, 200 мл/мин | 300 | [77] |
| Me/NiO (Me = Li, Na, Cs, K; Me/Ni = 0–0.2) | N2O – 500 ppm, 0.5 г, 200 мл/мин | 355 | [119] |
| MgСo3 – xO4 (x = 0.2–1), MgMnyCo2 – yO4 (y = 0.2–1) | 2% N2O (4% O2, 8.8% H2O), 1 г, 140 мл/мин | 380 | [78] |
| Me/CuxCo3 − xO4 (Me = Ba, K, Na, Sr; x = 0–1) | N2O – 1000 ppm (200 ppm NO, 2% O2, 0.5% H2O), GHSV = 54 000 ч−1 | ~250 | [79] |
| Co3O4 | 0.1% N2O, 60 л г−1 ч−1 | 375 | [80] |
| K/Co3O4–TiO2, K/Co3O4–SSWM | 0.1 мол. % N2O, GHSV = 10 909 ч−1 | 390 | [81] |
| K/Co–Mn–Al (SBA-15, MMT, SiO2, TiO2, Al2O3) | 0.1 мол. % N2O (0.1 мол. % N2O, 0.005 мол. % NO, 0.9 мол. % H2O, 0.5 мол. % O2 ), GHSV = 40 380 ч–1 | 350 | [82–84] |
| CuO–CeO2Cu/Ce = 0.3–0.7 | N2O – 2000 ppm (2% O2), GHSV = 70 000 ч−1 | 375 | [63] |
| M/Cu–Fe–Ox (M = Ce, La, Nd; Cu/Fe = 0.25–4) | N2O – 2000 ppm, GHSV = 2400 ч−1 | 320 | [64] |
| 5 мол. % CuO/CeO2 | 5% N2O/He (1% H2O), 350 мг, 30 мл/мин | 430 | [65] |
| CuO/CeO2 (25–40 мол. % Cu) | N2O – 2500 ppm (H2O или NO = 1.5%), GHSV = 30 000–45 000 ч−1 | 440 | [39, 54] |
| CoCexO (x = 0–1) | N2O – 1000 ppm (10% O2, 3% H2O), GHSV =15 000–60 000 ч−1 | 210 | [52, 53] |
| Co3O4, Co-CeO2, K/Co3O4, K/Co-CeO2 | N2O – 500 ppm, GHSV = 45 000 ч−1 | 220 | [85] |
| Ce–Co–O | N2O – 1000 ppm, GHSV = 80 000 ч−1 | 270 | [38] |
| MxCo1 − xCo2O4 (M = Ni, Mg; x = 0–0.99) | N2O – 1000 ppm (10% O2, 5% H2O), GHSV = 15 000 ч−1 | 150 | [87] |
| K/Co3O4 (K/Co = 0–0.1) | N2O – 5000 ppm (2% O2), W/F = 0.3 г см л–1 | 150 | [49] |
| Mn3O4, Fe3O4, Co3O4 ( Li, Na, K, Cs) | 5% N2O, GHSV = 7000 ч−1 | 184 | [88–91] |
| Co3 − xFexO4 | 5% N2O (1% H2O), GHSV = 7000 ч−1 | 397 | [92] |
| MgCo2O4, MgCoAlO4, ЭCoAl2O4, ЭMg0.5Co0.5, Al2O4, ЭMgAl2O4 | 5% N2O, GHSV = 7000 ч−1 | 350 | [93] |
| Co-Al2O3 (1.4–6.4% Co) | 5% N2O, GHSV = 7000 ч−1 | 513 | [94] |
| Co3O4/α-Al2O3 (15% Co3O4) | 5% N2O, GHSV = 7000 ч−1 | 410 | [95] |
| Mx–Co3O4 (M = Bi, Ce, K) | N2O – 2000 ppm (5% O2, 2% H2O, 10% CO2), GHSV = 20 000 ч−1 | 210 | [96] |
| CoOx/ZrO2, CuOx/ZrO2, FeOx/ZrO2, MnOx/ZrO2 (K+) | N2O – 4000 ppm (4000 ppm O2, 4000 ppm NO, 2% H2O), GHSV = 24 000 ч−1 | 400 | [97] |
| K–Co2.6Zn0.4O4/α-Al2O3, K–Zn0.4Co2.6O4/α-Al2O3, K–Co3O4/α-Al2O3 | 1.5–5% N2O, GHSV = 7000 ч−1 | 250 | [98–100] |
| Co3O4, Me0.5Co2.5O4 (Me = Mg, Ca, Sr, Ba) | 0.68% N2O (3% O2), 0.2 г, 80 мл/мин | 380 | [101] |
| MexCo (Me = Pb, Ba, Ag, Ca; x = 0–8.91) | N2O – 2000 ppm (500 ppm NOx, 2% H2O, 5% O2, 10% CO2, 50 ppm SO2), GHSV = 20 000 ч−1 | 320 | [102–106] |
| SnO2–Co3O4 | 0.1% N2O (2.3% H2O), GHSV = 10 000 ч−1 | 260 | [107] |
| Co3O4/CeO2 (0–20% Co3O4) | N2O – 1000 ppm, GHSV = 30 000 ч−1 | 400 | [37] |
| CeyBaxNi9 (y = 0–2, x = 0–1.5) | N2O – 2000 ppm (5% O2), GHSV = 20 000 ч−1 | 230 | [121, 122] |
| Zn1Co2ZrxOδ (x = 0–0.15) | N2O – 500 ppm, 100 мг, 200 мл/мин | 420 | [123] |
| Al2O3–Fe2O3 | He : N2O = 2 : 1, 0.4 г, 180 мл/мин | 675** | [124] |
| FexCe1 – хO2 (x = 0–1) | 0.15% N2O, GHSV = 23 800 ч−1 | 470 | [125] |
| BaFe1 – xSnxO3 – δ (x = 0–1) | 30% N2O, GHSV= 30 000 ч−1 | 550** | [126] |
| ${\text{La}}{{{\text{B}}}_{{1 - x}}}{\text{B}}_{x}^{'}{{{\text{O}}}_{3}},$ CaB1 – xCuxO3, (B = Mn, Fe и B' = Cu, Ni) | 20% N2O, GHSV = 22 100 ч−1 | 460–480 | [127] |
| Каталитические системы, нанесенные на цеолиты | |||
| Fe/MFI (Fe/Al = 0.75) | N2O – 1000 ppm (4% O2, 0.5% H2O), GHSV = 15 000 ч−1 | 475 | [136] |
| Fe-ZSM-5 (Fe/Al = 0.18) | N2O – 1500 ppm (200 ppm NOx), GHSV = 13 000 ч−1 | 350 | [137] |
| Fe-ZSM-5 (0.58–5 мас. % Fe) | 0.15% N2O, GHSV = 36 000 ч−1 Реальные составы (O2, CO2, H2O, SO2 ) | 370–430 | [7] |
| Fe/HZSM-5 | 2% N2O (2% O2, 10% NO, ~3% H2O) | 250** | [138, 139] |
| Fe-ZSM-5, Fe-ZSM-12, Fe-BEA (2.6–3.6 мас. % Fe) | N2O – 5000 ppm, GHSV = 20 000 ч−1 | 400** | [140] |
| Fe-ZSM-5 (0.3 мас. % Fe) | 2% N2O, 20 мл/мин Исследована природа центров Fe | 250** | [141] |
| Fe-ZSM-5 (0.1–3.2 мас. % Fe) | N2O – 1000 ppm (4% O2), GHSV = 42 000 ч−1 | 450 | [142] |
| Fe/ZSM-5 (Fe/Al = 0.61) | 5% N2O, 24 000 мл г−1 ч−1 | 415 | [143] |
| Fe-ZSM-5 (0.07–0.33 мас. % Fe) | N2O – 5000 ppm (5% O2), GHSV = 30 000 ч−1 | 475–525 | [144] |
| Fe-FER (0.9 мас. % Fe) | N2O – 1000 ppm (0.5% NO, 2% O2, 10% H2O), GHSV = 350 000 ч−1 | 400 | [145, 146] |
| Fe-ZSM-5, Fe-RTH, Fe-SSZ-3, Fe-LTA, Fe-FER, Fe-PST-7 (0.7–1% Fe) | N2O – 1000 ppm (4 об. % O2), GHSV = 42 000 ч−1 | 450–520 | [147] |
| Cu-ZSM-5 (Cu/Al > 1) | Исследован механизм и кинетика разложения N2O | 400** | [148] |
| Cu-ZSM-5 (1.6–9.4 мас. % Cu) | 0.5% N2O, 60 мл/мин | 350 | [149, 150] |
| Cu-APSO-34, Cu-ZSM-5 | N2O – 600 ppm | 420 | [151] |
| Cu-ZSM-5 (1.48 мас. % Cu) | 0.5% N2O, 60 мл/мин | 410 | [152] |
| Cu-ZSM-5 | Теоретическое исследование механизма и кинетики разложения N2O | 700** | [153, 154] |
Оксидные системы, содержащие металлы платиновой группы
Имеется довольно большое количество работ, в которых показана способность каталитических систем, содержащих металлы платиновой подгруппы (Ru, Rh, Pd, Ir, Pt), разлагать N2O. Некоторые из них демонстрируют высокую активность при низкой (до 300°С) температуре [41, 42, 56–59]. На каталитическую активность таких нанесенных катализаторов значительное влияние оказывает оксидный носитель, от природы которого зависит дисперсное распределение металлических активных центров [60, 61] и их состояние (кислотно-оснóвные и окислительно-восстановительные свойства) [41, 42, 62].
Как и многие каталитические системы, катализаторы, содержащие благородные металлы и элементы редкоземельной подгруппы, дезактивируются в присутствии ингибиторов (H2O, NOx, O2 и др.) и имеют к тому же высокую стоимость, что сдерживает их использование в процессах очистки хвостовых выбросов в реальных условиях производств азотной или адипиновой кислот [70].
Оксидные системы на основе переходных металлов
Оксидные каталитические системы на основе 3d-металлов, как правило, обладают хорошей каталитической активностью в реакции разложения закиси, при этом имеют низкую стоимость относительно металлов платиновой группы [71]. Такие катализаторы могут применяться в широкой температурной области в зависимости от их свойств. Особый интерес представляют оксиды со структурой шпинели, проявляющие высокую активность благодаря способности легко образовывать поверхностные кислородные вакансии, которые играют роль активных центров в каталитическом разложении закиси азота [52, 72]. Большинство исследований было направлено на изучение оксидных систем на основе Co3O4 для низкотемпературного разложения закиси азота [22, 37, 38, 49, 52, 53, 72–109 ]. Специфическое химическое состояние поверхности объясняет замечательные характеристики Co3O4 в реакции разложения закиси азота [23, 24]. Как было показано, дополнительное влияние на активность систем на основе Co3O4 оказывают метод приготовления [80, 108, 109], природа и количество легирующих примесей (таких как Mg, Cu, Ni, Fe, Zn, Ba, Bi, Ca, Sr, Sn, Pb, Ag, Ce) [52, 53, 64, 74–79, 85–87, 92, 102–107], а также наличие различных промоторов (в качестве которых выступают катионы щелочных металлов) [49, 74, 79, 81–85, 88–91, 98–100]. Было обнаружено, что кобальтсодержащие шпинели превосходят шпинели других переходных металлов не только по активности в разложении N2O, но и по устойчивости к воздействию O2 [72, 73, 89]. Добавки CeO2 [51–53], К [36, 49] или Cs [108] дополнительно увеличивают резистентность кобальтовой шпинели к действию ингибиторов (воды и кислорода). Интерес к элементам редкоземельной подгруппы был вызван результатами работ многих авторов, отмечающих положительный эффект добавки CeO2 [37, 39, 52, 54, 63–65]. Abu-Zied и соавт. был продемонстрирован также позитивный эффект введения других редкоземельных элементов и найдена корреляция между электрической проводимостью и способностью допированных оксидных систем восстанавливать активные центры разложения N2O [66–69].
Каталитические системы, нанесенные на цеолиты
Среди катализаторов разложения закиси азота особое место занимают цеолиты, содержащие Cu, Сo и особенно Fe [10, 112–120]. Цеолитные катализаторы, в состав которых входит Cu и Fe, способны выступать как бифункциональные системы для De-NOx и De-N2O [18, 19, 116].
Преимущество каталитических систем на основе цеолитов – высокоразвитые поверхности, что позволяет получать системы с высокодисперсным активным компонентом, а также их относительно низкая стоимость. Однако такие системы дезактивируются в присутствии воды [115] и разлагают закись азота при температурах выше 350°С [117, 118]. Учитывая требование регламента низкотемпературной селективной каталитической очистки от NOx в производстве азотной кислоты в России [9], системы на основе цеолитов могут быть использованы в качестве катализаторов разложения закиси азота только после дополнительного подогрева газов поcле стадии СКВ, что нежелательно [20].
Таким образом, литературные данные позволяют заключить, что системы на основе оксида кобальта наиболее перспективны для разработки однореакторной схемы низкотемпературного (250–300°С) удаления закиси азота в технологических схемах получения азотной кислоты в России.
КАТАЛИЗАТОРЫ ВЫСОКОТЕМПЕРАТУРНОГО РАЗЛОЖЕНИЯ N2O
Условия функционирования высокотемпературного катализатора разложения закиси азота в составе двухступенчатой системы в реакторе окисления аммиака выдвигают к ним жесткие требования. Катализаторы должны характеризоваться не только высокой активностью, термостабильностью и устойчивостью в реакционной среде, но также низким перепадом давления в слое, соответствующим гидродинамическим требованиям процесса Освальда. Как правило, катализатор должен быть в виде колец Рашига либо блоков сотовой структуры. Использование блочных катализаторов предпочтительнее, поскольку при меньшем перепаде давления блоки обеспечивают бóльшую однородность потока реакционной газовой смеси, что способствует также улучшению работы катализаторного пакета.
Оксиды со структурой перовскита ${{{\text{A}}}_{{1 - x}}}{\text{A}}_{x}^{'}{{{\text{B}}}_{{1 - y}}}{\text{B}}_{y}^{'}{{{\text{O}}}_{3}}$ (A = La или Ca, B = Mn, Co или Fe, A' = Sr, B' = Cu или Ni), шпинели AB2O4 (A = Mg, Ca, Mn, Co, Ni, Cu, Cr, Fe, Zn; B = Cr, Fe, Co), корунда, флюорита, гексаалюминатов наиболее часто рассматриваются в качестве перспективных каталитических систем для высокотемпературного разложения закиси азота (табл. 4). Хорошие показатели демонстрировали нанесенные катализаторы, содержащие Rh и Pd [155, 156], однако применение вышеуказанных катализаторов экономически невыгодно.
Таблица 4.
Катализаторы и условия для высокотемпературного разложения N2O в реакторе окисления аммиака
| Система | Состав реакционной смеси | Рабочий интервал Т, °С | Ссылка |
|---|---|---|---|
| Rh(Pd)/MeOx (Me = Ti, Al, Zr) |
Условия процесса Освальда* | 850–900 | [155, 156] |
| Pd–Rh/MeOx (Me = Se, Al, Zr) |
Условия процесса Освальда* | 750–1000 | [157] |
| La1 – xSrxFeO3 ± δ, La1 – xSrxMnO3 ± δ | 0.15% N2O, 1 атм, 4000 с/м | 800–1000 | [31, 45, 46] |
| LaCo1 – xFexO3 (x = 0–0.2) |
0.1% N2O (5% NO, 6% O2, 5% H2O), GHSV = 30000 ч–1 |
700–900 | [188, 189] |
| La2O3–MnO–CoO | N2O – 2100 ppm (1.5% O2, 0.8% NO), GHSV = 12500 ч–1 |
700–900 | [190] |
| La0.8Ce0.2CoO3 | Условия процесса Освальда* | 750–1000 | [176] |
| AB2O4 (A = Mg, Ca, Mn, Co, Ni, Cu, Cr, Fe, Zn; B = Cr, Fe, Co) |
N2O – 1200–5000 ppm (4–5% O2, 10% NOx), GHSV = 80 000–280 000 ч–1 | 440–900 | [158–160] |
| Y2O3–ZrO2 | N2O – 200 ppm (1400 ppm NO, 1% O2, 15% H2O), GHSV = 36 000 ч–1 | 750 | [191] |
| CexZr1 – xO2 | N2O – 2000 ppm (1.4% NO, 1% O2, 15% H2O), GHSV = 36 000 ч–1 | 650–900 | [192] |
| BaRu0.2FeAl10.8O19 | 30% N2O, GHSV= 30 000 ч–1 | 500–1200 | [181] |
| MeOx/Al2O3, MeOx/CeO2 (Me = Fe, Co, Ni) |
Условия процесса Освальда* | 800–900 | [183–186] |
| Fe2O3–MeOx (Me = Al, Ce, La, Cu, Cr) |
N2O – 1500 ppm (1500 ppm NO), GHSV = 10 000 ч–1 |
500–900 | [187, 193] |
| CuO–Al2O3 | Условия процесса Освальда* | ~900 | [182] |
| Co2AlO4/CeO2 | Условия процесса Освальда* | ~900 | [158–160] |
| Na/CaO | 0.2% N2O (10% O2, 1% NO), GHSV = 37 500 ч–1 | 400–750 | [194] |
| ${{{\text{M}}}^{1}}{\text{M}}_{{1 - x}}^{2}{\text{M}}_{x}^{3}{{{\text{O}}}_{3}}$ (М1 = La, Се, Nd, Pr, Sm; М2 = Fe, Ni; М3 = Cu, Со, Mn; х = 0.05–0.9) |
Условия процесса Освальда* | ~800–900 | [173] |
| ${\text{LaM}}_{{1 - x}}^{1}{\text{M}}_{x}^{2}{{{\text{O}}}_{3}}$ (М1 = Fe, Mn, Со; М2 = Со, Cu, Ni, Zn, Mg; х = 0–0.4) |
Условия процесса Освальда* | 800–920 | [183] |
Так, “Norsk Hydro” запатентовал серию катализаторов со структурой шпинели Co3 – xMxO4 (где M = Fe или Al, а x = 0–2), нанесенных на CeO2. Присутствие небольшого количества ZrO2 повышало стабильность их работы [157–160]. Катализаторы, как заявляли авторы, не разлагали NO, были активны, стабильны в широком интервале температур и составов газа, а также в присутствии кислорода и воды. Катализаторы NH-1 и NH-2 были успешно коммерциализированы “Norsk Hydro”.
Катализатор на основе Cu–Zn–Al был разработан, запатентован и коммерциализирован “BASF” в 1999 [161, 162]. В состав лучшего катализатора в виде экструдатов (О3-80, О3-85, О3-86) входит 8% CuO, 30% ZnO и 62% Al2O3.
Катализатор на основе оксидов церия и кобальта коммерциализирован “Yara International” под маркой Yara 58-Y1 (рис. 2). Катализатор содержит >80% оксида церия, <1% оксида кобальта и <1% алюмокобальтового оксида [163]. Сообщалось, что он обеспечивает 90% степень разложения закиси азота. Гранулированный катализатор на основе кобальтита лантана (“Amoxis”) также разработан и применяется для высокотемпературного разложения закиси азота.
Рис. 2.
Внешний вид гранулированных катализаторов “Yara International” и “Jonson Matthey” для разложения закиси азота.

Созданные в Институте керамики [164, 165] катализаторы, нанесенные на пористый керамический носитель, для разложения закиси азота и содержащие оксиды Cr, Fe, Mn, Co, Ni, Cu, La в пилотной установке при температуре выше 800оС обеспечивали 100% степень разложения.
В патентной литературе можно найти и другие примеры высокотемпературных катализаторов разложения закиси азота [166–173].
Следует отметить работу [173], в которой были предложены блочные (массивные и нанесенные на носитель) перовскитсодержащие катализаторы состава ${{{\text{M}}}^{1}}{\text{M}}_{{1 - x}}^{2}{\text{M}}_{x}^{3}$ (M1 = La, Nd, Sm, Pr и их смеси, М2 = Fe, Ni и их смеси, М3 = Сu, Co, Mn и их смеси) для высокотемпературного разложения N2O. Катализатор в условиях агрегата среднего давления, установленный после катализаторного пакета, обеспечивает степень разложения более 90% и демонстрирует высокую стабильность каталитических свойств в процессе его работы (рис. 3).
Рис. 3.
Внешний вид блочного высокотемпературного перовскитсодержащего катализатора разложения закиси азота (“Umicore”) и реактор окисления аммиака с загруженным катализатором [174].

Основной проблемой высокотемпературного каталитического удаления закиси азота является снижение активности катализатора под влиянием жестких условий реакционной среды. Большинство катализаторов имеют достаточную начальную активность в реакции разложения N2О, но только незначительная их часть обладает необходимой стабильностью, как, например, катализатор Co2AlO4/CeO2 (“Yara International”) [159]. Применение его в качестве второго слоя после катализаторного пакета в реакторе окисления аммиака при производительности 90 000 тон HNO3/год позволяло достигать 80% конверсии закиси азота. Установка блочного массивного катализатора фирмы “Umicore” (рис. 3.) обеспечивала разложение закиси азота до 90% [173, 174].
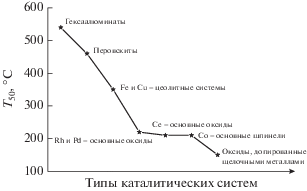
J. Perez-Ramirez в работе [175] отмечал конкурентную способность гексаалюминатов на основе LaAl11O18 и BaAl11O18, содержащих Fe и Mn, наряду с коммерческими катализаторами Co2AlO4/CeO2 [159, 160] и La0.8Ce0.2CoO3 [176]. Указанные образцы обладали высокой стабильностью (до 1100°С), превосходя оксидные системы со структурой шпинели и перовскита, благодаря особенностям их кристаллической структуры. Структура гексаалюминатов ABxAl12– xO19 – α обеспечивает устойчивость оксидов к спеканию и термическим перепадам, а замещение ионов Al на ионы переходных металлов (A = щелочные, щелочено-земельные и редкоземельные металлы; B = Mn, Fe, Ni, Cr, Co, Pt, Pd, Ru) позволяет получать катализаторы с различными свойствами для окислительно-восстановительных процессов [177–179], в том числе для разложения N2O [167, 175, 180]. Zhang и соавт. [181] сообщали о способности структуры гексаалюмината прочно фиксировать активные центры Ru, например, в BaRu0.2FeAl10.8O19, который может работать при температурах до 1200°С без испарения RuO2. Кроме того, гексаалюминатные системы просты в приготовлении и относительно дешевы, однако в целом их активность ниже активности перовскитов (табл. 4, рис. 4).
Рис. 4.
Сравнительные характеристики каталитических систем для разложения N2O (условия приведены в табл. 3 и 4).
Еще одной проблемой при эксплуатации катализатора является унос (химический или механический) активного компонента [10]. Поэтому использование любых медьсодержащих систем проблематично, поскольку медь при попадании в конечный продукт – нитрат аммония – может катализировать его разложение [10], что небезопасно. Это напрямую касается системы CuO–Al2O3, предлагаемой фирмой “BASF” [182]. В работе [183] содержание меди в катализаторе ограничивается 0.3 мас. %.
Среди нанесенных катализаторов особый интерес вызывают железосодержащие оксидные системы, которые обладают хорошими показателями не только в реакции разложения N2O, но и в окислении остаточного аммиака, проскочившего после сеток. Системы Fe2О3/Al2O3, Fe2О3/CeО2/Al2O3, Fe2О3/CeO2, [183–187] являются бифункциональными катализаторами, поскольку обеспечивают не только снижение содержания закиси азота, но также повышение выхода целевого продукта – NO. В длительных испытаниях (катализатор нанесенного типа Fe2О3/Al2O3 был размещен в составе 2-й каталитической ступени в реакторе УКЛ-7) после пробега в течение ~4000 ч катализатор продемонстрировал хорошую стабильность [183]. В работе [186] было показано существенное влияние условий синтеза на свойства катализатора. Так, замена при приготовлении нитратной соли железа на оксалатную, хотя и приводит к уменьшению содержания активного компонента в катализаторе, но не снижает его активность. Поэтому можно предполагать, что оптимизация химического состава катализатора и процедуры его синтеза позволят дополнительно улучшить селективность и термостойкость нанесенного блочного железосодержащего катализатора FeОх/Al2O3.
Таким образом, из литературных данных следует, что в области высоких температур наиболее перспективными катализаторами могут быть гексаалюминаты, перовскиты и оксид железа, а в области низких температур – Co-содержащие шпинели, допированные катионами щелочных металлов (рис. 4).
ЗАКЛЮЧЕНИЕ
Производство азотной кислоты является одним из основных промышленных источников поступления закиси азота – озонразрушающего газа – в атмосферу. Среди рассматриваемых в литературе способов каталитического удаления закиси азота в технологии получения азотной кислоты наиболее привлекательными являются однореакторные (высокотемпературная или низкотемпературная) схемы, не требующие установки дополнительного реактора. Они предусматривают организацию высокотемпературного разложения закиси азота путем установки высокотемпературного катализатора второго слоя после платиноидного катализаторного пакета в реакторе окисления аммиака (способ 1) или организацию низкотемпературного разложения путем установки низкотемпературного катализатора после катализатора СКВ в реакторе СКВ NO аммиаком (способ 2).
Реализация высокотемпературного удаления N2O предъявляет дополнительные требования к катализаторам, такие как термостабильность и инертность по отношению к NO (катализатор не разлагает NO), а каталитический слой не должен оказывать значительное газодинамическое сопротивление. Поэтому предпочтительно применение блочных катализаторов – массивных или нанесенных – для высокотемпературного разложения закиси азота. Наличие у такого катализатора также активности в реакции окисления аммиака представляет особый интерес, поскольку может позволить снизить содержание платиноидов в составе основного катализаторного пакета.
Однореакторная схема низкотемпературного удаления NO и N2O реализована на практике фирмой “Uhde” с использованием Fe-цеолитного катализатора при 450°С и обеспечивает разложение на уровне 90%. Однако для реализации однореакторной схемы комплексной очистки в реакторе СКВ на Российских предприятиях требуется разработка низкотемпературного (250–300°С) катализатора каталитического разложения закиси азота. Катализаторы на основе Co3O4 наиболее перспективны для указанных условий. Такой низкотемпературный катализатор разложения закиси азота может быть использован и для очистки хвостовых газов в производстве адипиновой кислоты.
Приведенные примеры демонстрируют, что каталитические технологии могут быть весьма успешно применены для решения экологических проблем, связанных с эмиссией опасных азотсодержащих газовых выбросов, в существующих схемах промышленного производства азотной кислоты. Обе схемы обеспечивают разложение 80–90% закиси азота. Для более глубокой очистки необходимо разрабатывать катализаторы с повышенной активностью и стабильностью.
Список литературы
Tian H., Chen G., Lu C., Xu X., Ren W., Zhang B., Banger K., Tao B., Pan S., Liu M., Zhang C., Bruhwiler L., Wofsy S. // Ecosystem Health and Sustainability. 2014. V. 1. № 4. C. 1.
Tuckett R. Greenhouse Gases / Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. Elsevier, 2018. https://doi.org/10.1016/B978-0-12-409547-2.14031-4
Ravishankara A.R., Daniel J.S., Portmann R.W. // Science. 2009. V. 326. P. 123.
Linak W.P., Kramlich J.C. // Stud. Env. Sci. 1998. V. 72. P. 265.
A report produced for DGXI / Options to Reduce Nitrous Oxide Emissions – Final Report: November. 1998. V. 3.
Kapteijn F., Rodriguez-Mirasol J., Moulijn J.A. // Appl. Catal. B: Env. 1996. V. 9. P. 25.
Pérez-Ramırez J., Kapteijn F., Mulb G., Xub X., Moulijn J.A. // Catal. Today. 2002. V. 76. P. 55.
Справочник азотчика. Mосква: Химия, 1987. Т. 2.
Бруштейн Е.А., Ванчурин В.И., Ященко А.В. // Катализ в промышленности. 2012. Т. 4. С. 7.
Perez-Ramırez J., Kapteijn F., Schoffel K., Moulijn J.A. // Appl. Catal. B: Env. 2003. V. 44. P. 117.
Schwefer M., Maurer R., Groves M. // Nitrogen. Vienna. 12–14 March, 2000.
Гигиенические нормативы ГН 2.1.6.695-98. Москва: Минздрав России. 1998.
Li Y., Armor J.N. // Appl. Catal. B: Env. 1993. V. 3. P. 55.
Shutilov R.A., Shutilov A.A., Zenkovets G.A. // Mater. Today: Proceedings. 2017. V. 4. P. 11490.
Sankar S., Putluru R., Schill L., Godiksen A., Poreddy R., Mossin S., Jensen A.D, Fehrmann R. // Appl. Catal. B: Env. 2016. V. 183. P. 282.
Huang X., Zhang S., Chen H., Zhong Q., Huang X. // J. Mol. Structure. 2015. V. 1098. P. 289.
Grzybek T., Papp H. // Appl. Catal. B: Env. 1992. V. 1. P. 271.
Groves M.C.E., Sasonow A. // J. Integrative Env. Sci. 2010. V. 7. № S1. P. 211.
Perbandt C., Bacher V., Groves M., Schwefer M., Siefert R., Turek T. // Chem. Ing. Technik. 2013. V. 585. P. 705.
Овчинникова Е.В., Чумаченко В.А., Пирютко Л.В., Харитонов А.С., Носков А.С. // Катализ в промышленности. 2009. Т. 1. С. 47.
Liu N., Zhang R., Chen B., Li Y.// J. Catal. 2012. V. 294. P. 99.
Yu H., Wang X. // Catal. Commun. 2018. V. 106. P. 40.
Zasada F., Piskorz W., Janas J., Grybos J., Indyka P., Sojka Z. // ACS Catalysis. 2015. V. 5. P. 6879.
Kaczmarczyk J., Zasada F., Janas J., Indyka P.A., Piskorz W., Kotarba A., Sojka Z. // ACS Catalysis. 2016. V. 6. P. 1235.
Dandl H., Emig G. // Appl. Catal. A: Gen. 1998. V. 168. P. 261.
Piskorz W. Zasada F., Stelmachowski P., Kotarba A., Sojka Z. // Catal. Today. 2008. V. 137. P. 418.
Yamashita T., Vannice A. // J. Catal. 1996. V. 161. P. 254.
Raj S.L., Srinivasan V. //J. Catal. 1980. V. 65. № 1. P. 121.
Winter E.R.S. // J. Chem. Soc. A. 1968. V. 0. P. 2889.
Сазонов Л.А., Москвина З.В., Артамонов Е.В. // Кинетика и катализ. 1974. Т. 15. № 1. С. 120.
Ivanov D.V., Sadovskaya E.M., Pinaeva L.G., Isupova L.A. // J. Catal. 2009. V. 267. P. 5.
Li G.N., Pidko E.A., Filot I.A.W, van Santen R.A., Li C., Hensen E.J.M. // J. Catal. 2013. V. 308. P. 386.
Guesmi H., Berthomieu D., Kiwi-Minsker L. // J. Phys. Chem. C. 2008. V. 112. № 51. P. 20319.
Guesmi H., Berthomieu D., Bromley B., Coqa B., Kiwi-Minsker L. // Phys. Chem. Chem. Phys. 2010. V. 12. P. 2873.
Catalysis by Ceria and Related Materials. Catalytic Science Series. Eds. Trovarelli A., Fornasiero P. London: Imperial College Press, 2013. V. 12.
Pasha N., Lingaiaha N., Seshu Babua N., Siva Sankar Reddya P., Sai Prasad P.S. // Catal. Commun. 2008. V. 10. P. 132.
Grzybek G., Stelmachowski P., Gudyka S., Indyka P., Sojka Z., Guillén-Hurtado N., Rico-Pérez V., Bueno-López A., Kotarba A. // Appl. Catal. B: Env. 2016. V. 180. P. 622.
You Y., Chang H., Ma L., Guo L., Qin X., Li J., Li J. // Chem. Eng. J. 2018. V. 347. P. 184.
Zabilskiy M., Djinovi P., Tchernychova E., Pintar A. // Appl. Catal. B: Env. 2016. V. 197. P. 146.
Iwanek E., Krawczyk K., Petryk J., Sobczak J.W., Kaszkur Z. // Appl. Catal. B: Env. 2011. V. 106. P. 416.
Parres-Esclapez S., Illán-Gómez M.J., Salinas-Martínez de Lecea C., Bueno-López A. // Appl. Catal. B: Env. 2010. V. 96. P. 370.
Parres-Esclapez S., Such-Basanez I., Illan-Gomez M.J., Salinas-Martínez de Lecea C., Bueno-Lopez A. // J. Catal. 2010. V. 276. P. 390.
Nobukawa T., Tanaka S., Ito S., Tomishige K., Kameoka S., Kunimori K. // Catal. Lett. 2002. V. 83. № 1–2. P. 5.
Liu S., Tang N., Shang Q., Wu C., Xu G., Cong Y. // Chin. J. Catal. 2018. V. 39. P. 1189.
Ivanov D.V., Pinaeva L.G., Isupova L.A., Sadovskaya E.M., Prosvirin I.P., Gerasimov E.Yu., Yakovleva I.S. // Appl. Catal. A: Gen. 2013. V. 457. P. 42.
Ivanov D.V., Pinaeva L.G., Sadovskaya E.M., Isupova L.A. // J. Mol. Catal. A: Chem. 2016. V. 412. P. 34.
Franken T., Palkovits R. // Appl. Catal. B: Env. 2015. V. 176–177. P. 298.
Amrousse R., Tsutsumi A., Bachar A., Lahcene D. // Appl. Catal. A: Gen. 2013. V. 450. P. 253.
Asano K., Ohnishi C., Iwamoto S., Shioya Y., Inoue M. // Appl. Catal. B: Env. 2008. V. 78. P. 242.
Marnellos G.E., Efthimiadis E.A., Vasalos I.A. // Appl. Catal. B: Env. 2003. V. 46. P. 523.
Imamura S., Shono M., Okamoto N., Hamada R., Ishida S. // Appl. Catal. A: Gen. 1996. V. 142. P. 279.
Xue L., Zhang C., He H., Teraoka Y. // Appl. Catal. B: Env. 2007. V. 75. P. 167.
Xue L., He H., Liu C., Zhang C., Zhang B. // Env. Sci. Technol. 2009. V. 43. № 3. P. 890.
Zabilskiy M., Erjavec B., Djinovic P., Pintar A. // Chem. Eng. J. 2014. V. 254. P. 153.
Liu Z., He C., Chen B., Liu H. // Catal. Today. 2017. V. 297. P. 78.
Doi K., Wu Y. Y., Takeda R., Matsunami A., Arai N., Tagawa T., Goto S. // Appl. Catal. B: Env. 2001. V. 35. P. 43.
Huang C., Ma Z., Xie P., Yue Y., Hua W., Gao Z. // J. Mol. Catal. A: Chem. 2015. V. 400. P. 90.
Huang C., Ma Z., Miao C., Yue Y., Hua W., Gao Z. // RSC Adv. 2017. V. 7. P. 4243.
Huang C., Jiang Y., Ma Z., Xie P., Lin Y., Meng T., Miao C., Yue Y., Hua W., Gao Z. // J. Mol. Catal. A: Chem. 2016. V. 420. P. 73.
Oi J., Obuchi A., Bamwenda G.R., Ogata A., Yagita H., Kushiyama S., Mizuno K. // Appl. Catal. B: Env. 1997. V. 12. P. 277.
Sui C., Zhang T., Dong Y., Yuan F., Niu X., Zhu Y. // Mol. Catal. 2017. V. 435. P. 174.
Wood B.R., Reimer J.A., Bell A.T. // J. Catal. 2002. V. 209. P. 151.
Liu Z., He C., Chen B., Liu H. // Catal. Today. 2017. V. 297. P. 78.
Xue Z., Shen Y., Shen S., Li C., Zhu S. // J. Indust. Eng. Chem. 2015. V. 30. P. 98.
Adamski A., Zaj W., Zasada F., Sojka Z. // Catalysis Today. 2012. V. 191. P. 129.
Abu-Zied B.M., Bawaked S.M., Kosa S.A., Ali T.T., Schwieger W. // Catalysts. 2016. V. 6. P. 70.
Abu-Zied B.M., Bawaked S.M., Kosa S.A., Ali T.T., Schwieger W., Aqlan F.M. // Appl. Surf. Sci. 2017. V. 419. P. 399.
Abu-Zied B.M., Bawaked S.M., Kosa S.A., Ali T.T., Schwieger W. // Int. J. Electrochem. Sci. Int. J. Electrochem. Sci. 2016. V. 11. P. 1568.
Abu-Zied B.M., Bawaked S.M., Kosa S.A., Ali T.T., Schwieger W. // Int. J. Electrochem. Sci. 2016. V. 11. P. 2230.
Xu X., Xu H., Kapteijn F., Moulijn J.A. // Appl. Catal. B: Env. 2004. V. 53. P. 265.
Li Y., Armor J.N. // Appl. Catal. B: Env. 1992. V. l. P. 21.
Russo N., Fino D., Saracco G., Specchia V. // Catal. Today. 2007. V. 119. P. 228.
Abu-Zied B.M., Soliman S.A. // Catal. Lett. 2009. V. 132. P. 299.
Abu-Zied B.M. // Chin. J. Catal. 2011. V. 32. P. 264.
Abu-Zied B.M., Soliman S.A., Abdellah S.E. // Chin. J. Catal. 2014. V. 35. P. 1105.
Abu-Zied B.M., Soliman S.A., Abdellah S.E. // J. Indust. Eng. Chem. 2015. V. 21. P. 814.
Abu-Zied B.M., Soliman S.A., Abdellah S.E. // Modern Res. Catal. 2017. V. 6. 47.
Li Z., Cang-cang W., Xiu-feng X. // J. Fuel Chem. Technol. 2016. V. 44. № 12. 1494.
Franken T., Palkovits R. // Appl. Catal. B: Env. 2015. V. 176–177. P. 298.
Chromcakova Z., Obalova L., Kovanda F., Legut D., Titov A., Ritz M., Fridrichova D., Michalik S., Kustrowski P., Jiratova K. // Catal. Today. 2015. V. 257. P. 18.
Klyushina A., Pacultova K., Krejcova S., Slowik G., Jiratova K., Kovanda F., Ryczkowski J., Obalova L. // Catal. Today. 2015. V. 257. P. 2.
Obalova L., Karaskova K., Jiratova K., Kovanda F. // Appl. Catal. B: Env. 2009. V. 90. P. 132.
Obalova L., Jiratova K., Karaskova K., Kovanda F. // Chin. J. Catal. 2011. V. 32. P. 816.
Pacultova K., Karaskova K., Strakosova J., Jiratova K., Obalova L. // C. R. Chim. 2015. V. 18. P. 1114.
Kim M.-J., Lee S.-J., Ryu I.-S., Jeon M.-W., Moon S.-H., Roh H.-S., Jeon S.G. // Mol. Catal. 2017. V. 442. P. 202.
Yan L., Ren T., Wang X., Gao Q., Ji D., Suo J. // Catal. Commun. 2003. V. 4. P. 505.
Yan L., Ren T., Wang X., Ji D., Suo J. // Appl. Catal. B: Env. 2003. V. 45. P. 85.
Kotarba A., Kruk I., Sojka Z. // J. Catal. 2002. V. 211. P. 265.
Stelmachowski P., Maniak G., Kotarba A., Sojka Z. // Catal. Commun. 2009. V. 10. P. 1062.
Maniak G., Stelmachowski P., Zasada F., Piskorz W., Kotarba A., Sojka Z. // Catal. Today. 2011. V. 176. P. 369.
Maniak G., Stelmachowski P., Kotarba A., Sojka Z., Rico-Pérez V., Bueno-López A. // Appl. Catal. B: Env. 2013. V. 136–137. P. 302.
Maniak G., Stelmachowski P., Stanek J.J., Kotarba A., Sojka Z. // Catal. Commun. 2011. V. 15. P. 127.
Stelmachowski P., Maniak G., Kaczmarczyk J., Zasada F., Piskorz W., Kotarba A., Sojka Z. // Appl. Catal. B: Env. 2014. V. 146. P. 105.
Grzybek G., Wojcik S., Ciura K., Grybos J., Indyka P., Oszajca M., Stelmachowski P., Witkowski S., Inger M., Wilk M., Kotarba A., Sojka Z. // Appl. Catal. B: Env. 2017. V. 210. P. 34.
Gudyka S., Grzybek G., Grybos J., Indyka P., Leszczynski B., Kotarba A., Sojka Z. // Appl. Catal. B: Env. 2017. V. 201. P. 339.
Tursun M., Wang X., Zhang F., Yu H. // Catal. Commun. 2015. V. 65. P. 1.
Pietrogiacomi D., Campa M.C., Carbone L.R., Tuti S., Occhiuzzi M. // Appl. Catal. B: Env. 2016. V. 187. P. 218.
Grzybek G., Wójcik S., Legutko P., Grybos J., Indyka P., Leszczynski B., Kotarba A., Sojka Z. // Appl. Catal. B: Env. 2017. V. 205. P. 597.
Wojcik S., Grzybek G., Grybos J., Kotarba A., Sojka Z. // Catal. Commun. 2018. V. 110. P. 64.
Wojcik S., Ercolino G., Gajewska M., Moncada Quintero C.W., Specchia S., Kotarba A. // Chem. Eng. J. 2019. https://doi.org/10.1016/j.cej.2018.10.025
Si-xuan L., Xia L., Li J., Liu X., Sun J., Wang H., Chi Y., Li C., Song Y. // J. Fuel Chem. Technol. 2018. V. 46. № 11. P. 1377.
Yu H., Tursun M., Wang X., Wu X. // Appl. Catal. B: Env. 2016. V. 185. P. 110.
Yu H., Wang X., Wu X. // Mol. Catal. 2018. V. 460. P. 69.
Yu H., Wang X., Wu X., Chen Y. // Chem. Eng. J. 2018. V. 334. P. 800.
Yu H., Wang X., Li Y. // Catal. Today. 2019. https://doi.org/10.1016/j.cattod.2018.10.036
Yu H., Wang X. // Catal. Commun. 2018. V. 106. P. 40.
Wang Y., Hu X., Zheng K., Wei X., Zhao Y. // Catal. Commun. 2018. V. 111. P. 70.
Иванова Ю.А., Сутормина Е.Ф., Исупова Л.А., Вовк Е.И. //Кинетика и катализ. 2017. Т. 58. № 6. С. 773.
Иванова Ю.А., Сутормина Е.Ф., Исупова Л.А., Рогов В.А. //Кинетика и катализ. 2018. Т. 59. № 3. С. 365.
Shen Q., Li L., Li J., Tian H., Hao Z., Hazard J. // J. Hazardous Mater. 2009. V. 163. P. 1332.
Papadatos K., Shelstad K.A. // J. Catal. 1973. V. 28. P. 116.
Sun K., Xia H., Hensen E., van Santen R., Li C. // J. Catal. 2006. V. 238. P. 186.
Sobolev V.I., Panov G.I., Kharitonov A.S., Romannikov V.N., Volodin A.M., Ione K.G. // J. Catal. 1993. V. 139. P. 435.
Dubkov K.A., Ovanesyan N.S., Shteinmaan A.A., Starokon E.V., Panov G.I. // J. Catal. 2002. V. 207. P. 341.
Roy P.K., Prins R., Pirngruber G.D. // Appl. Catal. B. 2008. V. 80. P. 226.
Ahrens M., Marie O., Bazin P., Daturi M. // J. Catal. 2010. V. 271. P.1.
Xie P., Luo Y., Ma Z., Huang C., Miao C., Yue Y., Hua W., Gao Z. // J. Catal. 2015. V. 330. P. 311.
Zhang B., Liu F., He H., Xue L. // Chin. J. Catal. 2014. V. 35. P. 1972.
Abu-Zied B.M., Asiri A.M. // Chin. J. Catal. 2015. V. 36. P. 1837.
Rutkowska M., Piwowarska Z., Micek E., Chmielarz L. // Micropor. Mesopor. Mater. 2015. V. 209. P. 54.
Zhang F., Wang X., Zhang X., Turxun M., Yu H., Zhao J. // Chem. Eng. J. 2014. V. 256. P. 365.
Zhang F., Wang X., Zhang X., Tursun M., Yu H. // Chin. J. Catal. 2015. V. 36. P. 344.
Basahel S.N., Abd El-Maksod I.H., Abu-Zied B.M., Mokhtar M. // J. Alloys and Compounds. 2010. V. 493. P. 630.
Ladavos A., Bakas T. // React. Kinet. Catal. Lett. 2001. V. 73. № 2. P. 229.
Perez-Alonso F.J., Melián-Cabrera I., Lopez Granados M., Kapteijn F., Fierro J.L.G. // J. Catal. 2006. V. 239. P. 340.
Huanga C., Zhuc Y., Wang X., Liu X., Wang J., Zhang T. // J. Catal. 2017. V. 347. P. 9.
Alini S., Basile F., Blasioli S., Rinaldi C., Vaccari A. // Appl. Catal. B: Env. 2007. V. 70. P. 323.
Kawi S., Liu S.Y., Shen S.-C. // Catal. Today. 2001. V. 68. P. 237.
Haber J., Machej T., Janas J., Nattich M. // Catal. Today. 2004. V. 90. P. 15.
Haber J., Nattich M., Machej T. // Appl. Catal. B: Env. 2008. V. 77. P. 278.
Komvokis V.G., Marnellos G.E., Vasalos I.A., Triantafyllidis K.S. // Appl. Catal. B: Env. 2009. V. 89. P. 627.
Kim S.S., Lee S.J., Hong S.C. // J. Indust. Eng. Chem. 2012. V.18. P. 1263.
Kim S.S., Lee S.J., Hong S.C. // Chem. Eng. J. 2011. V. 169. P. 173.
Angelidis T.N., Tzitzios V. // Appl. Catal. B: Env. 2003. V. 41. P. 357.
Liu H., Ma Z. // Chin. J. Chem. Eng. 2018. V. 26. P. 109.
Kogel M., Abu-Zied B.M., Schwefer M., Turek T. // Catal. Commun. 2001. V. 2. P. 273.
van den Brink R.W., Booneveld S., Pels J.R., Bakker D.F., Verhaak M.J.F.M. // Appl. Catal. B: Env. 2001. V. 32. P. 73.
Bulushev D.A., Kiwi-Minsker L., Renken A. // J. Catal. 2004. V. 222. P. 389.
Kiwi-Minsker L., Bulushev D.A., Renken A. // J. Catal. 2003. V. 219. P. 273.
Pirngruber G.D. // J. Catal. 2003. V. 219. P. 456.
Berrier E., Ovsitser O., Kondratenko E.V., Schwidder M., Grünert W., Brückner A. // J. Catal. 2007. V. 249. P. 67.
Park J.-H., Choung J.-H., Nam I.-S., Ham S.-W. // Appl. Catal. B: Env. 2008. V. 78. P. 342.
Xia H., Sun K., Liu Z., Feng Z., Ying P., Li C. // J. Catal. 2010. V. 270. P. 103.
Shen Q., Wu M., Wang H., Sun N., He C., Wei W. // Appl. Surf. Sci. 2018. V. 441. P. 474.
Sadovska G., Bernauer M., Bernauer B., Tabor E., Vondrova A., Sobalik Z. // Catal. Commun. 2018. V. 112. P. 58.
Tabora E., Sadovska G., Bernauera M., Sazama P., Novakova J., Filab V., Kmjecc T., Kohout J., Zaveta K., Sobalik Z. // Appl. Catal. B: Env. 2019. V. 240. P. 358.
Lim J.B., Cha S.H., Hong S.B. // Appl. Catal. B: Env. 2019. V. 243. P. 750.
Fanson P.T., Stradt M.W., Lauterbach J., Delgass W.N. // Appl. Catal. B: Env. 2002. V. 38. P. 331.
Meng T., Ren N., Ma Z. // J. Mol. Catal. A: Chem. 2015. V. 404–405. P. 233.
Meng T., Ren N., Ma Z. // Chin. J. Chem. Eng. 2018. V. 26. P. 1051.
Palella B.I., Cadoni M., Frache A., Pastore H.O., Pirone R., Russo G., Coluccia S., Marchese L. // J. Catal. 2003. V. 217. P. 100.
Zou W., Xie P., Hua W., Wang Y., Kong D., Yue Y., Ma Z., Yang W., Gao Z. // J. Mol. Catal. A: Chem. 2014. V. 394. P. 83.
Liu X., Yang Z., Zhang R., Li Q., Li Y. // J. Phys. Chem. C. 2012. V. 116. P. 20262.
Liu X., Yang Z., Li Y., Zhang F. // J. Mol. Catal. A: Chem. 2015. V. 396. P. 181.
Gorywoda M., Jantsch U., Kraus M., Lund J. // Patent DE 102004024026 (A1), 2004.
Jantsch U., Lund J., Gorywoda M., Kraus M. // Patent US 20050202966 (A1), 2005.
Янч У., Лунд Д., Горивода М., Краус М. // Патент RU 2304465 (C2), 2005.
Nirisen O., Schoffel K., Waller D., Ovrebo D. // Patent WO 0202230, 2002.
Nirisen O., Schoffel K., Waller D., Ovrebo D. // Patent WO 02/02230 (A1), 2002.
Nirisen O., Schoffel K., Waller D., Ovrebo D. // Patent US 2004/0023796 (A1), 2004.
Schumacher V., Bűrger G., Fetzer T., Baier M., Hesse M. // Patent WO 99/55621, 1999.
Schumacher V., Bűrger G., Fetzer T., Baier M., Hesse M. // Patent US 6743404 (B1), 2004.
Åbø K.M. / Investigation of the Yara 58-Y1 nitrous oxide decomposition catalyst. Master thesis. Norwegian University of Science and Technology. Department of Chemical Engineering. Trondheim, June. 2014. https://ntnuopen.ntnu.no/ntnu-xmlui/bitstream/ handle/11250/2351725/10844_FULLTEXT.pdf?sequence=1&isAllowed=y
Burckhardt W., Froehlich F., Seifert F. // Key Eng. Mater. 1997. V. 132–136. P. 775.
Burckhardt W., Seifert F., Winterstein G. // Patent WO 00/13789. 2000.
Neveu G. // Patent WO 99/64139, 1999.
Sermon P.A. // Patent US 4088604, 1978.
Vernooy P.D. // Patent US 2002/0123424 (A1), 2002.
Vernooy P.D. // Patent US 6429168 (B1), 2002.
Hamon C., Duclos D. // Patent WO 2004/052512 (A1), 2004.
Schwefer M., Maurer R., Turek T. // Patent WO 01/51415 (A1), 2001.
Jantsch U., Lund J., Gorywoda M., Kraus M. // Patent US 2009/0130010 (A1), 2009.
Neumann J., Isupova L., Pinaeva L., Kulikovskaya N., Zolotarskii I. // Patent WO 2007/104403 (A1), 2007.
Jantzen S., Neumann J., Novel A. / Multifunctional Product Family of Umicore’s MKS Concept: Beyond a Mere Solution for N2O Abatement. Uhde Fertiliser Symposium, Dortmund, Germany, May 17–19. 2006.
Perez-Ramırez J., Santiago M. // Chem. Commun. 2007. V. 6. P. 619.
Axon S., Coupland D., Foy J., Ridland J., Wishart I. // Patent WO 2004/096703 A2, 2004.
Machida M., Eguchi K., Arai H. // J. Catal. 1990. V. 123. P. 477.
Groppi G., Cristiani C., Forzatti P. // J. Catal. 1997. V. 168. P. 95.
Kikuchi R., Iwasa Y., Takeguchi T., Eguchi K. // Appl. Catal. A: Gen. 2005. V. 281. P. 61.
Konsolakis M. // ACS Cataysis. 2015. V. 5. № 11. P. 6397.
Zhang Y., Wang X., Zhu Y., Zhang T. // Appl. Catal. B: Env. 2013. V. 129. P. 382.
Baier M., Fetzer T., Hofstadt O., Hesse M., Buerger G., Harth K., Schumacher V., Wistuba H., Otto B. // Patent WO 0023176, 2000.
Пинаева Л.Г., Исупова Л.А., Куликовская Н.А., Марчук А.А. // Патент РФ № 28. 2430781, 2011.
Sadykov V.A., Isupova L.A., Zolotarskii I.A., Bobrova L.N., Noskov A.S., Parmon V.N., Brushtein E.A., Telyatnikova T.V., Chernyshev V.I., Lunin V.V. // Appl. Catal. A: Gen. 2000. V. 204. P. 59.
Pinaeva L.G., Prosvirin I.P., Dovlitova L.S., Danilova I.G., Sadovskay E.M., Isupova L.A. // Catal. Sci. Technol. 2016. V. 6. P. 2150.
Пинаева Л.Г., Довлитова Л.С., Исупова Л.А. // Кинетика и катализ. 2017. Т. 58. № 2. С. 183.
Giecko G., Borowiecki T., Gac W., Kruk J. // Catal. Today. 2008. V. 137. P. 403.
Wu Y., Ni X., Beaurain A., Dujardin C., Granger P. // Appl. Catal. B: Env. 2012. V. 125. P. 149.
Wu Y., Cordier C., Berrier E., Nuns N., Dujardin C., Granger P. // Appl. Catal. B: Env. 2013. V. 140–141. P. 151.
Catalysist for the decomposition of nitrogen protoxid. Patent EP 2616165 (B1), 2011.
Granger P., Esteves P., Kieger S., Navascues L., Leclercq G. // Appl. Catal. B: Env. 2006. V. 62. P. 236.
Esteves P., Wu Y., Dujardin C., Dongare M.K., Granger P. // Catal. Today. 2011. V. 176. P. 453.
Kruk J., Stolecki K., Michalska K., Konkol M., Kowalik P. // Catal. Today. 2012. V. 191. P. 125.
Kondratenko E.V., Gçlden V., Sokolov S. // ChemCatChem. 2010. V. 2. P. 633.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ