

Кинетика и катализ, 2019, T. 60, № 6, стр. 782-787
Кинетика гидрогенолиза глицерина в 1,2-пропиленгликоль на медном катализаторе
В. И. Хаджиев a, Г. С. Дмитриев a, *, И. С. Мельчаков a, Т. Е. Шорина a, Л. Н. Занавескин a, А. Л. Максимов a
a ФГБУН Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
* E-mail: dmitriev.gs@mail.ru
Поступила в редакцию 14.02.2019
После доработки 12.04.2019
Принята к публикации 17.04.2019
Аннотация
Изучена кинетика гидрогенолиза глицерина в 1,2-пропиленгликоль на катализаторе, содержащем 60 мас. % меди на Al2O3. Предложена схема превращений глицерина в условиях каталитического гидрогенолиза. Определен вид кинетических уравнений расходования глицерина по основному направлению в ацетол и 1,2-пропиленгликоль, а также по побочному превращению в этиленгликоль. Показано, что обе реакции гидрогенолиза глицерина имеют нулевой порядок по водороду и первый – по глицерину. Наблюдаемая энергия активации для основного направления расходования глицерина (ацетол и 1,2-пропиленгликоль) и побочного (этиленгликоль) составляют (125 500 ± 200) и (87 500 ± 500) Дж/моль соответственно. Обосновано предположение о том, что лимитирующей стадией процесса получения 1,2-пропиленгликоля является стадия дегидратации глицерина в ацетол.
Водные растворы 1,2-пропиленгликоля (ПГ) – широко распространенные низкозамерзающие теплоносители (антифризы), применяемые в различных отраслях промышленности, в том числе в системах отопления, вентиляции и кондиционирования жилых домов и общественных зданий, в системах охлаждения пищевых производств, а также в другом теплообменном оборудовании. ПГ используется как растворитель природных и синтетических веществ в фармацевтической и косметической промышленности, он применяется в производстве ненасыщенных полиэфирных смол, эластичных полиуретанов, алкидных смол, пластификаторов и т.п.
В промышленности ПГ обычно получают путем гидратации окиси пропилена при температуре от 160 до 200°С и давлении ~1.6 МПа [1]. Выделяют продукт методом вакуумной ректификации. Однако окись пропилена является весьма востребованным и весьма дорогим продуктом. В этой связи разработка новых технологий получения ПГ с использованием других видов доступного сырья является актуальной задачей.
Альтернативный метод получения ПГ основан на каталитическом гидрогенолизе так называемого биоглицерина – отхода производства биодизеля. Этот процесс является типичным процессом “зеленой” химии, поскольку сырьем для получения 1,2-пропиленгликоля служит продукт, получаемый из природного возобновляемого сырья [2, 3].
1,2-Пропиленгликоль из глицерина (ГЛ) получают методом гидрогенолиза при повышенных значениях температуры и давления на металлических катализаторах, которые разделяют на две основные группы. Это катализаторы, содержащие благородные металлы (Pt, Pd, Re и Ru) [4–11], и катализаторы, в состав которых входят металлы переходной валентности (Cu, Co и Ni), нанесенные на пористые носители [4, 12–17]. Катализаторы, содержащие в своем составе благородные металлы, проявляют достаточно высокую активность, однако селективность по ПГ составляет лишь 30–60% из-за протекания побочных реакций с разрывом связей С–С и С–О.
Анализ литературных источников и наши собственные эксперименты показали, что наибольшую селективность (>90%) при гидрогенолизе глицерина показывают медьсодержащие катализаторы на оксиде алюминия, силикагелях или оксиде цинка [18–21]. На основании большого объема экспериментальных данных для процесса получения 1,2-пропиленгликоля гидрогенолизом глицерина был выбран катализатор, содержащий 60 мас. % меди на оксиде алюминия, полученный методом соосаждения. Этот катализатор показал достаточно высокую скорость процесса при селективности по целевому продукту не менее 96%.
Целью настоящей работы являлось изучение кинетики каталитического гидрогенолиза глицерина в 1,2-пропиленгликоль на катализаторе 60% Cu/Al2O3.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализатора 60% Cu/Al2O3
Катализатор готовили методом совместного осаждения гидроксидов меди и алюминия из соответствующих нитратов. Для синтеза использовали водные растворы 1.2 М Cu(NO3)2 и 1.2 М Al(NO3)3 и 20%-ный водный раствор NаОН в стехиометрических количествах. Реагенты смешивали при температуре 40–50°С с последующим нагреванием смеси до 90°С в течение 4 ч. Затем продукт отмывали от нитрата натрия водой с помощью центрифуги с последующим высушиванием при 120°С, прокаливанием при 450°С и восстановлением при 300°С.
Готовый катализатор имел удельную поверхность 133 м2/г, средний объем пор 0.24 см3/г при среднем диаметре пор 66.7 Å.
Схема лабораторной установки
Установка для проведения каталитических экспериментов представляет собой стальной автоклав объемом 300 мл, снабженный мешалкой, рубашкой, термопарой, датчиком давления, штуцерами для подачи газов и сброса давления. Для быстрого нагрева и охлаждения реакционной смеси использовали систему с переключением на один из двух циркуляционных термостатов с горячим и холодным теплоносителями.
Условия проведения каталитических экспериментов
В реактор загружали 150 г 80%-ного водного раствора глицерина и 7.5 г катализатора. Перед началом эксперимента из свободного объема реактора вытесняли воздух азотом в количестве, равном 10-кратному объему свободного пространства реактора. Раствор глицерина с катализатором нагревали при перемешивании до заданной температуры. Началом эксперимента считали момент подачи водорода в реактор. Температура проведения экспериментов составляла 180–220°С, давление меняли в интервале 1.5–4.6 МПа. Скорость вращения мешалки составляла 1000 об/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Экспериментально было показано, что при скорости вращения мешалки более 800 об/мин конверсия глицерина при прочих равных условиях остается постоянной и не зависит от интенсивности перемешивания. Полученные данные свидетельствуют о том, что при указанных условиях внешнедиффузионное торможение отсутствует.
В экспериментах по гидрогенолизу глицерина на катализаторе с размерами частиц <0.1 мм, 0.1–0.16 мм и 0.16–0.25 мм при 200°С и давлении 2.0 МПа при времени контакта 18 ч было показано, что конверсия глицерина на катализаторе с различными размерами частиц практически постоянна и составляет 54.8 ± 0.8%. Полученные данные подтверждают отсутствие внутридиффузионного торможения, что означает, что реакция гидрогенолиза глицерина на выбранном медном катализаторе протекает в кинетической области. Поэтому дальнейшие исследования проводили на катализаторе с размером зерна 0.1–0.16 мм.
Экспериментально установлено, что на катализаторе 60% Cu/Al2O3 основными продуктами реакции гидрогенолиза глицерина являются 1,2-пропиленгликоль, ацетол (АЦ) и этиленгликоль (ЭТГЛ). В следовых количествах в реакционной смеси обнаружен метанол, а в газообразных продуктах среди возможных С1-соединений – только углекислый газ, тогда как метан и оксид углерода отсутствуют.
На схеме 1 представлены превращения глицерина, основанные на экспериментальных данных. Образование 1,2-пропиленгликоля из глицерина через ацетол является одной из наиболее принятых возможных схем взаимных превращений в этом процессе [6, 10, 17–26].
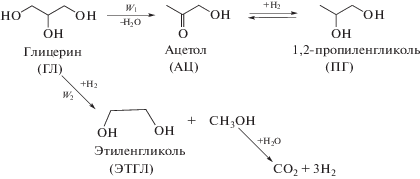
Схема 1. Взаимные превращения при гидрогенолизе глицерина на катализаторе 60% Cu/Al2O3.
Таким образом, глицерин расходуется по двум направлениям, скорости которых обозначены как W1 и W2. Основным направлением является синтез 1,2-пропиленгликоля через образование ацетола, а побочной реакцией – гидрогенолиз С–С-связи с образованием этиленгликоля.
В табл. 1 представлены результаты экспериментов гидрогенолиза глицерина по изучению влияния давления водорода (концентрации водорода) на конверсию глицерина и селективность продуктов реакции при 200°С и времени контакта 18 ч. Видно, что изменение давления водорода не приводит к изменению конверсии глицерина, т.е. скорость превращения глицерина не зависит от концентрации растворенного в реакционной массе водорода.
Таблица 1.
Зависимость конверсии глицерина и селективностей образования продуктов реакции от давления водорода
| Давление общее, МПа | Конверсия ГЛ, % | Селективность образования, % | ||
|---|---|---|---|---|
| ПГ | АЦ | ЭТГЛ | ||
| 1.5 | 55.1 | 95.0 | 2.2 | 2.8 |
| 2.0 | 55.2 | 96.8 | 0.3 | 2.9 |
| 2.7 | 53.9 | 96.7 | 0.4 | 2.9 |
| 3.7 | 55.9 | 97.0 | <0.1 | 3.0 |
| 4.6 | 55.0 | 97.2 | <0.1 | 2.8 |
Уменьшение селективности по ацетолу с одновременным ростом селективности образования 1,2-пропиленгликоля подтверждает, что ацетол является промежуточным соединением. При этом повышение давления водорода приводит к практически полному превращению ацетола, что также согласуется с приведенной схемой 1 .
В соответствии со схемой 1 можно ожидать изменения селективности по этиленгликолю при изменении давления водорода, поскольку водород является реагентом в этой реакции. Однако эксперименты показали, что селективность остается постоянной, а, следовательно, водород не участвует в лимитирующей стадии образования этиленгликоля и в кинетическом уравнении порядок по нему равен нулю. По-видимому, лимитирующей стадией этого превращения является разрыв С–С-связи адсорбированного на катализаторе глицерина.
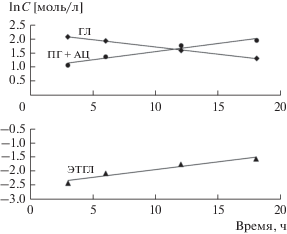
На рис. 1 представлена типичная временная зависимость конверсии глицерина и селективностей продуктов реакции, а на рис. 2 – соответствующие им кинетические кривые расходования глицерина и образования (ПГ + АЦ) и ЭТГЛ в координатах реакции первого порядка lnC–τ. Ацетол является промежуточным соединением, концентрация которого мала, и может быть легко переведен в ПГ путем повышения давления. Сумма концентраций (ПГ + АЦ) определяет скорость расходования глицерина по целевому направлению (W1). Поэтому при изучении кинетики гидрогенолиза глицерина здесь и далее мы рассматривали именно сумму концентраций 1,2-пропиленгликоля и ацетола.
Рис. 1.
Зависимости конверсии глицерина (X) и селективностей образования продуктов реакции (S) от времени проведения процесса. Условия: 150 г 99.5 мас. % водного раствора глицерина, 7.5 г катализатора, температура 200°С, давление 2.0 МПа.
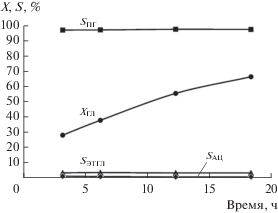
Рис. 2.
Линеаризация кинетических кривых изменения концентрации глицерина и продуктов реакции во времени в координатах реакции первого порядка lnC–τ. Условия реакции те же, что и для рис. 1.
В соответствии с методикой приготовления катализатора перед загрузкой в реактор его восстанавливали и насыщали водородом. Поэтому в начальный период времени наблюдалась повышенная скорость реакции, и нулевую точку в расчет не принимали. Через некоторое время, когда устанавливалось равновесие процессов растворения водорода в реакционной системе и адсорбции–десорбции реагентов на поверхности катализатора, изменение состава реакционной смеси становилось предсказуемым и поддавалось математическому описанию.
Постоянство селективностей образования продуктов реакции во времени (рис. 1) свидетельствует о том, что этиленгликоль образуется из глицерина по независимой реакции, а не в результате деструкции молекулы ПГ.
Данные рис. 2 показывают, что изменения концентраций как глицерина, так и продуктов реакции целевого направления расходования глицерина (ПГ + АЦ) и побочного образования ЭТГЛ подчиняются кинетическому уравнению первого порядка по глицерину. Таким образом, скорости образования продуктов реакции описываются следующими кинетическими уравнениями:
Тогда скорость расходования глицерина описывается как
(3)
${{W}_{{{\text{ГЛ}}}}} = {{W}_{1}} + {{W}_{2}} = {\text{ }}({{k}_{1}} + {{k}_{2}}){{С}_{{{\text{ГЛ}}}}}.$Линеаризация кинетической кривой расходования глицерина позволяет определить сумму констант (k1 + k2). Разделить эти константы в данном случае несложно. Скорости образования продуктов реакции описываются уравнениями:
Поскольку время проведения процесса одинаково, то, поделив уравнения (4) и (5) друг на друга и проинтегрировав, получаем
(6)
$\frac{{{{C}_{{({\text{ПГ + АЦ)}}}}}}}{{{{C}_{{{\text{ЭТГЛ}}}}}}} = \frac{{{{k}_{1}}}}{{{{k}_{2}}}}.$В табл. 2 представлены константы скорости реакции при изменении концентрации глицерина в исходной смеси от 99.5 до 60 мас. % (при прочих равных условиях).
Таблица 2.
Константы скорости гидрогенолиза глицерина при изменении концентрации глицерина в исходной смеси от 60 до 99.5 мас. %*
| Концентрация ГЛ в исходной смеси, мас. % | k1 + k2, ${\text{л}}\,{\text{г}}_{{{\text{кат}}}}^{{ - 1}}\,{{{\text{ч}}}^{{ - 1}}}$ | k1/k2 | k1 | k2 |
|---|---|---|---|---|
| ${\text{л}}\,{\text{г}}_{{{\text{кат}}}}^{{ - 1}}\,{{{\text{ч}}}^{{ - 1}}}$ | ||||
| 99.5 | 1.0 × 10–3 | 32.9 | 9.8 × 10–4 | 3.0 × 10–5 |
| 90 | 1.0 × 10–3 | 32.1 | 9.7 × 10–4 | 3.0 × 10–5 |
| 80 | 1.0 × 10–3 | 30.5 | 9.9 × 10–4 | 3.2 × 10–5 |
| 70 | 9.8 × 10–4 | 33.1 | 9.5 × 10–4 | 2.9 × 10–5 |
| 60 | 9.9 × 10–4 | 31.6 | 9.6 ×10–4 | 3.1 × 10–5 |
| Среднее | (9.7 ± 0.4) ×10–4 | (3.0 ± 0.4) × 10–5 | ||
Данные табл. 2 показывают, что вода выступает только в роли растворителя и не входит в кинетическое уравнение реакций. Достаточно высокая погрешность определения константы k2 является следствием низкой концентрации этиленгликоля в реакционной смеси и, как следствие, погрешностью ее определения. При температуре 200°С скорость целевой реакции в 32 раза превышает скорость побочного превращения глицерина в этиленгликоль.
Для определения энергии активации и предэкспоненциального множителя была проведена серия экспериментов в интервале температур 180–220°С (рис. 3). Полученные кинетические уравнения реакции каталитического гидрогенолиза глицерина на катализаторе 60% Cu/Al2O3 по целевому направлению в 1,2-пропиленгликоль (W1) и побочному в этиленгликоль (W2) имеют следующий вид:
(7)
$\begin{gathered} {{W}_{1}} = 7.16 \times {{10}^{{10}}}{{e}^{{\left( {\frac{{ - (125\,500 \pm 200)}}{{RT}}} \right)}}}{{C}_{{{\text{ГЛ}}}}}, \\ [{\text{моль}}\,\,{\text{г}}_{{{\text{кат}}}}^{{ - 1}}\,\,{{{\text{ч}}}^{{ - 1}}}], \\ \end{gathered} $(8)
$\begin{gathered} {{W}_{2}} = 1.38 \times {{10}^{5}}{{e}^{{\left( {\frac{{ - (87\,500 \pm 500)}}{{RT}}} \right)}}}{{C}_{{{\text{ГЛ}}}}}, \\ [{\text{моль}}\,\,{\text{г}}_{{{\text{кат}}}}^{{ - 1}}\,\,{{{\text{ч}}}^{{ - 1}}}]. \\ \end{gathered} $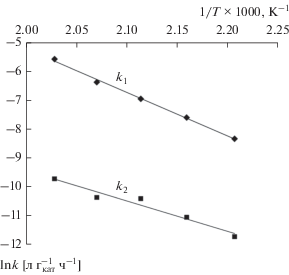
Полученные величины энергий активации подтверждают выдвинутое выше предположение о протекании реакций в кинетической области.
В опубликованных ранее работах по изучению кинетики гидрогенолиза глицерина в присутствии различных медьсодержащих катализаторов энергия активации полученных кинетических уравнений существенно различалась в зависимости от методики исследований и использованных катализаторов. Так, например, на катализаторе 18% Cu/SiO2 [27] энергия активации равнялась 94 кДж/моль, что свидетельствует о протекании реакции в кинетической области. В работе [28] на катализаторе Cu–ZnO–Al2O3 энергия активации дегидратации глицерина в ацетол составила 86.56 кДж/моль, а для гидрирования ацетола в 1,2-пропиленгликоль – 57.8 кДж/моль. Из этого можно сделать вывод, что дегидратация ацетола протекала в кинетической области, а на скорость гидрирования ацетола существенное влияние оказывала уже диффузия водорода. Исследование кинетики в работе [29] на катализаторе 20%Cu–10%ZrO2–MgO, по-видимому, было проведено в условиях внешней диффузии, о чем свидетельствует крайне низкая величина энергии активации 22.8 кДж/моль. Однако при этом скорости вращения мешалки были небольшими (500 об/мин), и убедительных доводов об отсутствии диффузионных торможений не было приведено.
В приведенных литературных данных кинетические уравнения также существенно отличаются. Так, например, в работе [29] получено кинетическое уравнение W = $kC_{{{\text{ГЛ}}}}^{{0.7}}C_{{\text{Н}}}^{{0.6}},$ но не показано, каким образом была определена концентрация растворенного водорода в реакционной смеси. Кроме того, зависимость скорости реакции от давления и близкие порядки по глицерину и водороду в кинетическом уравнении свидетельствуют о том, что скорости образования ацетола из глицерина и его гидрирования близки.
Исследование кинетики в работе [27] было проведено в растворах глицерина в бутаноле-1 при низких концентрациях глицерина в исходной смеси 10–40 мас. % и было показано, что, с одной стороны, с ростом концентрации бутанола растворимость водорода в реакционной смеси увеличивается в разы (что приводит к росту скорости), но, с другой стороны, концентрация глицерина уменьшается. Следовательно, скорость должна тоже уменьшаться, что, по нашему мнению, находит отражение в порядках по реагентам. Порядок по глицерину составил 0.17, а по водороду 1.06.
В работе [28] на катализаторе Cu–ZnO–Al2O3 значения энергии активации и порядков реакции по реагентам наиболее близки к полученным нами. Скорость дегидратации глицерина в ацетол пропорциональна концентрации глицерина по первому порядку, а скорость гидрирования ацетола в 1,2-пропиленгликоль – концентрации ацетола по первому порядку и давлению водорода по первому порядку. При этом гидрирование ацетола протекает на порядок быстрее, чем дегидратация ацетола, что также соответствует полученным нами данным.
В соответствии с кинетическими уравнениями (7) и (8) энергия активации реакции образования 1,2-пропиленгликоля выше, чем образования побочного продукта этиленгликоля. Это значит, что рост температуры способствует повышению селективности образования целевого продукта. Так, например, при увеличении температуры проведения процесса с 200 до 220°С селективность образования этиленгликоля уменьшается с 2.9 до 1.5%.
Полученное кинетическое уравнение реакции образования 1,2-пропиленгликоля (а именно первый порядок по глицерину и нулевой по водороду), и низкая концентрация промежуточного соединения ацетола свидетельствуют о том, что лимитирующей стадией процесса является дегидратация глицерина в ацетол. Реакция гидрирования ацетола протекает быстро и без образования каких-либо побочных продуктов. Следовательно, модифицирование катализатора 60% Cu/Al2O3 с целью повышения скорости процесса должно быть направлено на увеличение скорости дегидратации глицерина в ацетол.
ЗАКЛЮЧЕНИЕ
Изучение кинетики гидрогенолиза глицерина позволило установить, что расходование глицерина идет, преимущественно, по двум направлениям. Основным направлением является дегидратация глицерина с образованием ацетола с последующим гидрированием ацетола в 1,2-пропиленгликоль. На катализаторе 60% Cu/Al2O3 лимитирующей стадией процесса является дегидратация глицерина с образованием ацетола. Гидрирование ацетола в 1,2-пропиленгликоль является быстрой реакцией, что обеспечивает низкую концентрацию ацетола в реакционной смеси. В качестве побочного продукта по параллельной реакции с разрывом С–С-связи образуется этиленгликоль. Кинетика гидрогенолиза глицерина по целевому и побочному направлениям описывается кинетическими уравнениями первых порядков по глицерину. Величины энергий активации на изученном катализаторе для основного направления (ацетол и 1,2-пропиленгликоль) и побочного (этиленгликоль) составляют (125 500 ± 200) и (87 500 ± 500) Дж/моль соответственно. Рост температуры способствует повышению селективности образования целевого 1,2-пропиленгликоля.
Список литературы
Евразийский химический рынок. Международный деловой журн. 2018. № 2 (161). С. 30.
Len C., Luque R. // Sustainable Chemical Processes. 2014. V. 2. P. 1.
Konstantinovic S.S., Danilovic B.R., Ciric J.T., Ilic S.B., Savic D.S., Veljkovic V.B. // Chem. Ind. Chem. Eng. Q. 2016. V. 22. P. 461.
Dasari A.M., Kiatsimkul P.-P., Sutterlin W.R., Suppes G.J. // Appl. Catal. A. General. 2005. V. 281. P. 225.
Shmidt S.R., Tanielyan S.K., Marin N., Alvez G., Augustine R.L. // Top. Catal. 2010. V. 53. P. 1214.
Chaminand J., Djakovitch L., Gallezot P., Marion P., Pinel C., Rosier C. // Green Chem. 2004. V. 6. P. 359.
Maris E.P., Davis R.J. // J. Catal. 2007. V. 249. P. 328.
Kusunaki Y., Miayazawa T., Kunimori K., Tomishige K. // Catal. Commun. 2005. V. 6. P. 645.
Miayazawa T., Kusunaki Y., Kunimori K., Tomishige K. // J. Catal. 2009. V. 240. P. 213.
Wang S., Yin K., Zhang Y., Liu H. // ACS Catal. 2013. V. 3. P. 2112.
Ryneveld E., Mahomed A.S., Heerden P.S., Friendrich H.B. // Catal. Lett. 2011. V. 141. P. 958.
Huang L.H., Zhu Y.-L., Zheng H.-Y., Li Y.-W., Zeng Z.-Y. // J. Chem. Techol. Biot. 2008. V. 83. P. 1670.
Bienholz A., Hofmann H., Claus P. // Appl. Catal. A. General. 2011. V. 391. P. 153.
Guo L., Zhou J., Mao J., Guo X., Zhang S. // Appl. Catal. A. General. 2009. V. 367. P. 93.
Vasiliadou E.S., Lemonidou A.A. // Appl. Catal. A. General. 2011. V. 396. P. 177.
Bienholz A., Schwab F., Claus P. // Green Chem. 2010. V. 12. P. 290.
Balaraju M., Rekha V., Prasad P.S.S. // Catal. Lett. 2008. V. 126. P. 119.
Huang Z., Cui F., Kang H., Chen J., Xia C. // Appl. Catal. A. General. 2009. V. 366. P. 288.
Akiyama M., Sato S., Takahashi R., Inui K., Yokota M. // Appl. Catal. A. General. 2009. V. 371. P. 60.
Huang Z., Cui F., Kang H., Chen J., Zhang X., Xia C. // Chem. Mater. 2008. V. 20. P. 5090.
Yuan Z., Wang L., Wang J., Xia S., Chen P., Hou Z., Zheng X. // Appl. Catal. B. Environ. 2011. V. 101. P. 431.
Ryneveld E., Mahomed A.S., Heerden P.S., Green M.G., Friendrich H.B. // Green Chem. 2011. V. 13. P. 1819.
Wang S., Liu H. // Catal. Lett. 2007. V. 117. № 1–2. P. 62.
Xia S., Yuan Z., Wang L., Chen P., Hou Z. // Appl. Catal. A. General. 2011. V. 403. P. 173.
Alhanash A., Koshevnikova E.F., Kozhevnikov I.V. // Catal. Lett. 2008. V. 120. P. 307.
Feng Y., Yin H., Wang A., Shen L., Yu L., Jiang T. // Chem. Eng. J. 2011. V. 168. P. 403.
Vasiliadou E.S., Lemonidou A.A. // Chem. Eng. J. 2013. V. 231. P. 103.
Zhou Z., Li X., Zeng T., Hong W., Cheng Z. // Chinese J. Chem. Eng. 2010. V. 18. P. 384.
Rekha V., Raju N., Sumana C., Douglas S.P., Lingaiah N. // Catal Lett. 2016. V. 146. P. 1487.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ