

Кинетика и катализ, 2020, T. 61, № 2, стр. 195-224
Полимер-иммобилизованные кластеры и наночастицы металлов в катализе
Г. И. Джардималиева a, *, А. К. Жармагамбетова b, С. Е. Кудайбергенов c, d, И. Е. Уфлянд e
a ФГБУН Институт проблем химической физики РАН
142432 Черноголовка, просп. Акад. Н.Н. Семенова, 1, Россия
b Институт органического катализа и электрохимии им. Д.В. Сокольского
050010 Алматы, ул. Кунаева, 142, Республика Казахстан
c Институт полимерных материалов и технологий
050019 Алматы, мкр. Атырау 1, 3/1, Республика Казахстан
d Казахский национальный исследовательский технический университет им. К.И. Сатпаева,
Лаборатория инженерного профиля
050013 Алматы, ул. Сатпаева, 22, Республика Казахстан
e Южный федеральный университет
344006 Ростов-на-Дону, ул. Большая Садовая, 105/42, Россия
* E-mail: dzhardim@icp.ac.ru
Поступила в редакцию 04.08.2019
После доработки 04.08.2019
Принята к публикации 10.09.2019
Аннотация
Представлены последние достижения в области получения полимер-иммобилизованных кластеров и наночастиц металлов и их применения в катализе многих органических реакций. Рассмотрены основные типы полимер-иммобилизованных катализаторов, включая кластерсодержащие полимеры, полимер-протектированные наночастицы металлов, закрепленные на неорганических носителях, гель-иммобилизованные катализаторы. Отдельное внимание уделяется роли полимерной матрицы, выполняющей функцию не только стабилизирующего агента, предотвращающего агрегацию наночастиц и их вымывание в реакционную среду, но также являющейся лигандом в координационной сфере металлоцентра, позволяющего сочетать преимущества гомогенных и гетерогенных катализаторов.
СОДЕРЖАНИЕ
3. Гель-иммобилизованные катализаторы
3.1. Катализаторы на основе наночастиц металлов, иммобилизованных в матрицу гидрогелей
3.2. Катализаторы на основе макропористых криогелей с иммобилизованными наночастицами металлов
ВВЕДЕНИЕ
Наночастицы металлов, именуемые также нанокластерами, наноструктурами, являются объектами интенсивных исследований благодаря широким возможностям их применения в различных областях, таких как катализ, электроника, запись и хранение информации, сенсоры, медицина [1–4]. Растущий интерес к использованию наночастиц металлов в катализе обусловлен следующими причинами. С одной стороны, они обладают развитой удельной поверхностью, что определяет их эффективность, аналогичную гомогенным катализаторам, а с другой – характеризуются высокой долей атомов металлов на поверхности, которые могут выполнять роль потенциальных активных центров и значительно превышают по числу обычные гетерогенные катализаторы. Разработка новых каталитических систем, сочетающих возможность рециклинга гетерогенных катализаторов с широким диапазоном катализируемых реакций и высокой селективностью гомогенных катализаторов, является сегодня актуальной задачей.
Нанокатализ как активно развивающееся направление современного катализа, в частности, инкапсулированные в полимеры наночастицы металлов, часто рассматривают как граничную область между гомогенным и гетерогенным катализом [5–7]. В случае полимер-иммобилизованных металлокомплексов тенденции к сочетанию достоинств и исключению недостатков традиционных гомогенных и гетерогенных катализаторов уже привели к созданию гибриднофазных каталитических систем [8–13]. Их отличительными признаками являются возможность легкого отделения от реакционной массы и многократного использования подобно типичным гетерогенным катализаторам; высокая эффективность и лучшая воспроизводимость, характерные для гомогенных катализаторов, в то время как структура гетерогенных катализаторов существенно зависит от метода их синтеза и обработки; меньшая чувствительность их свойств к следам кислорода и влаги благодаря гидрофобному характеру полимерной матрицы и т.д. Стремление к мультифункциональности наноматериалов также может обеспечиваться дизайном сложных гибридных наноструктур за счет соответствующих ассоциаций металлического ядра и стабилизирующей оболочки из органического лиганда, полимера, ионной жидкости, неорганических соединений, носителя и т.д. Эти ассоциации и взаимодействия весьма важны для тонкого управления свойствами поверхности наноструктуры и, следовательно, каталитической эффективностью гибридного нанокомпозита. При этом роль неметаллического компонента в таких системах может быть двоякой. С одной стороны, полимерная матрица, например, стабилизирует высокодисперсные, легко окисляемые металлические наночастицы, предотвращая их окисление и вымывание в окружающую среду, а с другой – может выступать как сокатализатор: ее свойства можно изменять определенным образом для того, чтобы напрямую управлять реакционной способностью и селективностью металлического остова. Так, размер наночастиц металлов, полученных в матрице полиамидоамина (PAMAM), зависит от числа функциональных групп в дендримере и его структуры [14]. Число внутренних аминогрупп определяет максимальное число ионов металлов, которые могут инкапсулироваться в полимерную матрицу, и тем самым контролируются размеры наночастиц. В реакции циклопропанирования в присутствии гибридного катализатора дендример-инкапсулированные наночастицы Au/SiO2 селективность по отношению к цис-диастероизомеру возрастала в 5 раз по сравнению с гомогенным аналогом, что было объяснено стерическими эффектами дендримерной матрицы [15].
Многочисленные экспериментальные данные и теоретические расчеты свидетельствуют о том, что различия в электронной и геометрической структурах нанесенных отдельных атомов, металлических кластеров и наночастиц благодаря размеру, характеру взаимодействия с носителем и реагентами и возможности эволюции их природы в ходе каталитической реакции существенным образом влияют на селективность и активность каталитического процесса [16]. При размере наночастиц <1 нм свыше 90% атомов локализуются на поверхности кластера, что может приводить к уменьшению координационного числа, укорочению связи металл–металл, более легкому окислению и т.д. Эти свойства субнанометровых металлических частиц, а также высокие значения поверхностной энергии и соотношения поверхность/объем обеспечивают их исключительную каталитическую активность и селективность во многих органических реакциях. Нанокластеры Pd (с размерами <2 нм), стабилизированные сополимером PEG–PNIPAM, демонстрируют весьма высокую активность в реакции Сузуки (Suzuki) при взаимодействии йод- или хлоробензола с фенилбороновой кислотой (TOF = 4.3 × 104 ч−1) [17]. Нанокластеры Cu30 в матрице дендримера PAMAM−OH(G6) проявляют 100% селективность в гидрировании С=О-связи до спиртовой молекулы в присутствии С=С-связи в гидрируемом соединении [18]. Наличие индукционного периода в некоторых Au-катализируемых реакциях, как, например, в реакциях бромирования и гидратации алкинов, когда в качестве исходных катализаторов используют моноядерные соединения (HAuCl4 или AuCl), связывают с in situ формированием каталитически активных кластеров Au [19]. Размернозависимые каталитические свойства кластеров в этих реакциях подтверждены на примере специально синтезированных кластеров Au5 и Au8, инкапсулированных в дендримерную молекулу поли(аминоамида) (PAMAM). При этом их активность была несколько ниже, чем в случае in situ формируемых систем, в которых структуры и координационные окружения кластеров оптимальны.
В гетерогенном катализе принцип создания монодисперсных каталитических систем – основополагающий. Большинство катализаторов, используемых в химической промышленности, энергетике, экологических системах, являются нанесенными: активные частицы размером 1–100 нм располагаются обычно на внутренней поверхности пористого носителя. Это обусловлено тем, что свойства нанесенных катализаторов зависят от структуры и размеров активной фазы, и при снижении ее содержания уменьшение размеров частиц наносимых металлов способствует увеличению числа активных центров одного состава, участвующих в каталитическом процессе, что приводит к повышению скорости и избирательности реакции.
За последние десятилетия достигнуты значительные успехи в применении полимеров качестве стабилизаторов наночастиц металлов-катализаторов многочисленных процессов органического синтеза [20–26]. Наряду со стабилизацией каталитических активных центров эффективно осуществляется стратегия выделения и рециклинга катализаторов. Использование, например, растворимых полимеров позволяет избежать диффузионных ограничений, лимитирующих скорость каталитической реакции. Наглядным является недавно развитый подход, основанный на применении полимерных носителей с ковалентно связанными молекулами ионных жидкостей, что позволяет использовать такие преимущества последних, как эффективная стабилизация и активация катализаторов, значительное увеличение скорости и селективности реакции и возможность эффективного рециклинга каталитических систем. Имеются многочисленные примеры успешного применения этой стратегии в иммобилизации каталитически активных наночастиц металлов [27–30], включая, например, хемоселективную гидрогенизацию непредельных альдегидов и кетонов наночастицами Pd, стабилизированных фосфинсодержащим полимер-иммобилизованным имидазолий-ионом (PdNP@PPh2-PIILP) и его PEG-модифицированным производным (PdNP@PPh2-PEGPIILP) [31] (рис. 1, табл. 1).
Рис. 1.
Фрагмент структуры PdNP@PPh2-PIILP (а) и схема каталитической реакции гидрирования циннамальдегида (б). Количественные параметры реакции в присутствии полимер-иммобилизованных наночастиц Pd приведены в табл. 1.
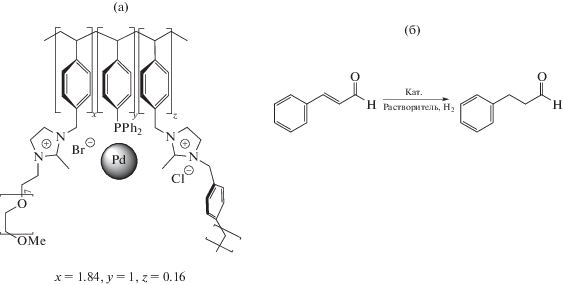
Таблица 1.
Селективное гидрирование циннамальдегида до гидроциннамальдегида*
| Основание | Конверсия, % | TOF, ч–1 | Селективность, % |
|---|---|---|---|
| NaOH | >99 | 200 | 100 |
| K2CO3 | >99 | 200 | 100 |
| K3PO4 | 100 | 200 | 100 |
| NEt3 | 90 | 180 | 88 |
Модификация носителя молекулами PEG способствует повышению эффективности действия катализатора как в отношении конверсии циннамальдегида, так и селективности гидрирования С=С-связи, достигающей 100%.
Эта же каталитическая система с незначительной сшивкой, обусловленной присутствием группы трис-4-винилфенилфосфина, обнаруживает высокую эффективность в реакции восстановления ароматических нитросоединений в умеренных условиях и при низкой загрузке катализатора с величинами TON, равными, соответственно, 3600 (TOF – 2580 ч−1) и 274 000 (TOF – 17 125 ч−1) для реакций гидрогенизации и гидрирования с переносом [32]. Кроме того, Pd-иммобилизованные катализаторы рассматриваемого типа проявляют активность в тандемном процессе кросс-присоединения Сузуки−Мияура (Suzuki−Miyaura) и восстановления нитроаренов с образованием биариламинов с высокими выходами.
Целью настоящей работы было краткое рассмотрение синтетической стратегии получения полимер-иммобилизованных кластеров и наночастиц металлов, особенностей их строения, структуры и каталитических свойств в реакциях основного органического синтеза.
1. КЛАСТЕРСОДЕРЖАЩИЕ ПОЛИМЕРЫ И ИХ КАТАЛИТИЧЕСКИЕ СВОЙСТВА
Интерес к кластерам переходных и благородных металлов обусловлен не только огромным многообразием способов связывания лиганд−металл и конфигураций лиганда на поверхности кластера, но и подвижностью самого металлического каркаса [33, 34]. Благодаря разнообразию химического состава и чрезвычайно интересным структурам, полимер-иммобилизованные кластеры являются потенциальными компонентами для получения различных полимерных материалов с необычными свойствами. Особенно значительный интерес для катализа кластерсодержащие полимеры представляют с точки зрения как фундаментальных аспектов управления составом и строением и моделей поверхности металлических катализаторов, так и практического применения в качестве селективных и высокоэффективных катализаторов многих реакций органического синтеза. Общие способы синтеза кластерсодержащих полимеров основаны на методах физического или химического инкапсулирования кластерных частиц в полимеры или на реакциях (со)полимеризации кластерсодержащих мономеров [35–39]. Интересны подходы, позволяющие включать кластерсодержащие фрагменты в основную цепь полимерной молекулы с образованием кластерсодержащих полимеров, например, {[{Pt3(μ-dppm)3}(μ-1,4-CNC6R4NC)][PF6]}n [40], {Ru6(μ6-C)(CO)15(Ph2PC2PPh2)}n [41], в том числе и биметаллических структур типа [−O(CH2)2(η-C5H4){Mo2Ir2(CO)10}(η-C5H4)(CH2)2OC(O)NHRNHC(O)−]n [42]. Для получения кластерсодержащих мономеров разработаны два основных подхода: введение полимеризационноспособных лигандов в полиядерные комплексы (замещение собственных лигандов на аналоги с кратными связами, их окислительное присоединение, присоединение в мягких условиях по кратным связам M−M и др.) или же достройка соответствующих лигандов кластерами [36, 43, 44]. С высокими выходами синтезированы карбоксилаты на основе трехъядерных карбонильных кластеров Os3(CO)12 и его производных Os3(CO)11(CH3CN), (μ-H)Os3(CO)10(μ-OR) (R = H, Ph), а также кластерный мономер (μ-H)Os3(μ-OCNMe2)(CO)9PPH2CH2CH=CH2 [45–47]:
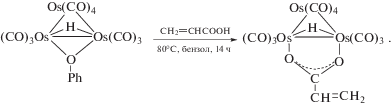
Принцип внешнесферного замещения был положен в основу ряда проведенных последовательных синтезов, направленных на получение Mo6-кластерных карбоксилатов [48, 49]:
Последующая (со)полимеризация металлосодержащих мономеров рассматриваемого типа приводит к формированию термически стабильных кластерсодержащих полимеров с контролируемым составом и распределением последовательностей в полимерной цепи [50–53]. Результаты специальных исследований показали, что путь сополимеризации кластерсодержащих мономеров более предпочтителен, чем модификация функционализированного полимера кластерными молекулами [54]. Взаимодействие кластера Co2(CO)8 c (со)полимерами, имеющими дитиокарбоксилатные или тиоамидные функциональные группы, сопровождалось формированием побочных серосодержащих кобальторганических соединений или коллоидного сульфида кобальта, которые было трудно отделить от целевого продукта. В то же время реакцией сополимеризации мономерных алкилидентрикобальт нонакарбонильного или сульфидотрикобальт гептакарбонильного кластеров:
синтезированы сополимеры ММА, стирола или N-[трис-1,1,1 (гидроксиметил)метил]акриламида (среднечисленная масса полимера ${{\bar {M}}_{n}}$ = = 6400–24 600) с количественным выходом, высокой степени чистоты и со строго определенными кластерными фрагментами.
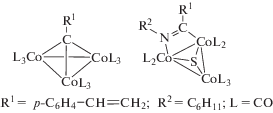
Мультифункциональный диблочный сополимер, содержащий молекулярный кластер Co6Se8, получен метатезисной полимеризацией кластерсодержащего мономера с раскрытием цикла, что является успешным примером синтеза линейного сополимера на основе халькогенидных кластеров [55]. В принципе, интеграция молекулярных кластеров в блок-сополимеры дает возможность создавать на их основе высокоорганизованные ансамбли для их применения в катализе. Множественные центры связывания лиганда на поверхности кластера часто приводят к формированию сетчатых структур, в отдельных случаях удается получить растворимые полимеры, как было показано для дендримерных молекул на основе кластеров Re6Se8 [56]. Интересно, что полимер-иммобилизованные гетерометаллические кластеры [ClOs3Au(CO)10(Ph3P] и [Co2Pt2(CO)8(Ph3P)2] были значительно стабильнее их аналогов − гомогенных или закрепленных на носителе SiO2 − и обнаруживали высокую активность в гидрировании олефинов [57]. Для смешанного металлокластера [Co2Pt2(CO)8(Ph3P)2] в гомогенном варианте наблюдали его быструю трансформацию в монометаллический кластер [Pt5(CO)6(PPh3)4]. В то же время при использовании полимерных аналогов кластеров (OC)9Co3CC(O)OCH2CH2OC(O)-C(CH3)=CH2, (OC)9Co3CC6H4C(O)C(CH3)=CH2 и (OC)9Co3CC6H4CH=CH2 в качестве катализаторов реакции гидроформилирования было показано, что активными частицами являются моно- или биядерные кобальт-карбонильные частицы [58].
Каталитическая активность полимер-иммобилизованных кластеров Os3, полученных сополимеризацией (CO)10Os3(μ-H)(N-4-винилпиридина) [59] или [(μ-H)Os3(μ-OCNMe2)(CO)9{P(CH2CH=CH2)Ph2}] [38] со стиролом, исследована в реакции окисления циклогексена:
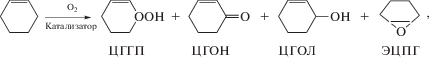
где ЦГГП − циклогексенилгидропероксид; ЦГОН − циклогексенон; ЦГОЛ − циклогексенол; ЭПЦГ − эпоксициклогексан.
Кинетические кривые поглощения O2 при окислении циклогексена имеют автокаталитический характер: время достижения максимальной скорости реакции составляет 40−108 мин и определяется удельным содержанием металла (табл. 2) [59]. Показано, что автокаталитический характер процесса обусловлен накоплением гидропероксида и эффективной генерацией радикалов по реакции:
Таблица 2.
Состав продуктов каталитического окисления циклогексена молекулярным О2 в присутствии Os3-содержащих полимеров [59]*
| Катализатор | [Os]0 × 105, г-атом/л | Время, мин | Селективность, % | |||
|---|---|---|---|---|---|---|
| ЦГОЛ | ЦГОН | ЭПЦГ | ЦГГП | |||
| Os3-ПС I | 1.31 | 65 | 2.2 | 2.2 | 0.03 | 95.5 |
| Os3-ПС II | 8.20 | 44 | 7.5 | 2.3 | 0.03 | 89.4 |
| Os3-ПС III | 6.52 | 192 | 5.0 | 52.4 | 0.05 | 41.8 |
| Os3-ПС IV | 7.84 | 204 | 4.5 | 51.2 | 0.05 | 43.3 |
| Os3-ПС V | 0.63 | 57 | 7.2 | 16.3 | 0.2 | 71.7 |
| Os3-ПС VI | 0.73 | 42 | 12.2 | 7.3 | 2.2 | 77.8 |
| Os3-ПС VII | 6.13 | 108 | 22.1 | 23.8 | 1.0 | 52.2 |
Os3-сополимеры рассматриваемого типа могут быть легко выделены из реакционной среды и использованы в повторных циклах без потери активности, как показано было в каталитической реакции окислительного дегидрирования 2,3,6-триметил-1,4-гидрохинона (TMHQ) с формированием 2,3,6-триметил-1,4-хинона (TMQ) – промежуточного продукта в синтезе α-токоферола (витамина E) (рис. 2, табл. 3) [38].
Рис. 2.
Фрагмент структуры кластерсодержащего полимера [(μ-H)Os3(μ-OCNMe2)(CO)9{P(CH2CH=CH2)Ph2}/стирол (а) и схема каталитической реакции окислительного дегидрирования TMHQ (б). Количественные параметры реакции в присутствии полимер-иммобилизованных наночастиц Os3 приведены в табл. 3.
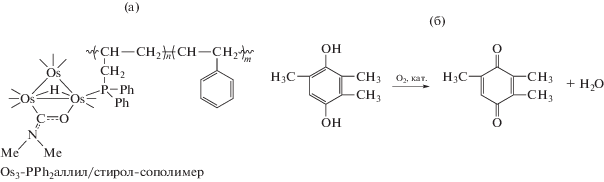
Таблица 3.
Каталитическое окислительное дегидрирование TMHQ в присутствии Os3-PPh2аллил/стирол-сополимера
| Цикл | Начальная скорость реакции, моль л–1 ч–1 | Удельная активность, ${\text{моль}}\,\,{\text{г - атом}}_{{{\text{Os}}}}^{{ - 1}}\,\,{{{\text{ч}}}^{{ - 1}}}$ |
|---|---|---|
| 1 | 0.32 ± 0.02 | 188 |
| 2 | 0.31 ± 0.02 | 182 |
| 3 | 0.30 ± 0.02 | 177 |
| 4 | 0,30 ± 0.02 | 177 |
| 5 | 0.29 ± 0.02 | 177 |
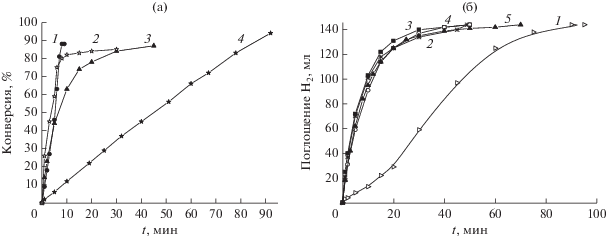
Эффективность каталитического действия полимер-иммобилизованных кластерных комплексов Rh в реакции гидрогенизации была сопоставлена с традиционными гетерогенными катализаторами на примере Rh/C (содержание Rh 1%), начальная удельная активность которого составляет 0.1 ${\text{мол}}{{{\text{ь}}}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ ${\text{г - атом}}_{{{\text{Rh}}}}^{{ - 1}}\,\,{{{\text{с}}}^{{ - 1}}}$ [60]. Показано, что исследуемые комплексы по своей активности находятся на уровне лучших гетерогенных катализаторов (рис. 3а). Начальная скорость гидрирования в присутствии катализатора на основе Rh6(CO)15CH3CN, иммобилизованного на сополимерах стирола с дифенилфосфоаллилом, с содержанием Rh 4.16% была невысокой (0.02 ${\text{мол}}{{{\text{ь}}}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ ${\text{г - атом}}_{{{\text{Rh}}}}^{{ - 1}}\,\,{{{\text{с}}}^{{ - 1}}}$); в дальнейшем, по мере увеличения числа циклов, скорость реакции повышалась до 0.4 ${\text{мол}}{{{\text{ь}}}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ ${\text{г - атом}}_{{{\text{Rh}}}}^{{ - 1}}\,\,{{{\text{с}}}^{{ - 1}}}$ (рис. 3б).
Рис. 3.
а – Кривые поглощения водорода при гидрировании циклогексена в присутствии катализаторов: 1 – Rh/C; 2 – сополимер стирола с Rh6(CO)14(4-VPy)2; 3 – Rh6(CO)15CH3CN, иммобилизованного на сополимере стирола с дифенилфосфоаллилом (4.16% Rh); 4 – сополимера стирола с Rh6(CO)15(4-VPy). Условия: Т = 40°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 1 атм, растворитель – изопропиловый спирт, 3.9 × 10−5 г-атом Rh в катализаторе, 4.5 ммоль субстрата. б – Кривые поглощения водорода в пяти последовательных циклах гидрирования циклогексена в присутствии Rh6(CO)15CH3CN, иммобилизованного на сополимере стирола с дифенилфосфоаллилом (4.16% Rh). Условия: Т = 40°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 1 атм, растворитель – изопропиловый спирт, 3.9 × 10−5 г-атом Rh в катализаторе, 6.5 ммоль субстрата. Номера кривых совпадают с номером цикла.
Аналогичное поведение характерно для полимер-иммобилизованных комплексов и в других реакциях [12] и связано с “разработкой” катализатора. Высказано предположение, что в ходе гидрирования происходит фрагментация кластера с образованием в низких концентрациях высокоактивных частиц, обладающих, возможно, большей нуклеарностью (наноразмерных), которые и являются истинными катализаторами. Такие выводы согласуются и с данными других работ. Так, например, в [61] было показано, что активность полимер-иммобилизованных моноядерных комплексов Rh возрастала с повышением степени декарбонилирования Rh и формированием металлоцентров, включающих ${\text{Rh}}_{n}^{0}$. Важно, что гетерогенизация металлокластеров позволяет выделить и исследовать некоторые их каталитически активные интермедиаты, например, используя РФЭ- и ИК-спектроскопию.
2. ПОЛИМЕР-ИММОБИЛИЗОВАННЫЕ НАНОЧАСТИЦЫ МЕТАЛЛОВ
Иммобилизация наночастиц металлов на поверхности твердых носителей представляет интерес не только для изучения фундаментальных вопросов взаимосвязи состава, строения и свойств каталитической системы “наночастица металла−полимер”, характера взаимодействия и специфической роли макролиганда в катализе гетерогенизированными металлокомплексами и кластерными частицами, но также важна с точки зрения практического применения катализаторов рассматриваемого типа. На практике чаще всего применяют полимерные носители двух основных групп [12]. Первую составляют жестко сшитые микропористые смолы с большой удельной поверхностью. Лигандные группы этих смол локализованы преимущественно на поверхности и могут контактировать с компонентами реакционной среды. Вторую группу составляют линейные или слабо сшитые сетчатые микропористые полимеры или их композиты. Они растворяются или набухают в растворителях, каталитические активные центры находятся не только на поверхности полимера, но и в его объеме. Иммобилизация металлокластеров может осуществляться за счет электростатических сил притяжения между заряженными нанокластерами и ионными группами носителя, посредством формирования прочных ковалентносвязанных или хелатных комплексов, а также при межмолекулярных взаимодействиях различных типов [62].
Повышенный интерес исследователей к наночастицам металлов, стабилизированных водорастворимыми полимерами, обусловлен возможностью контролировать кинетику роста, размеры и распределение наночастиц, что в конечном итоге определяет их каталитические, магнитные, электрические, оптические, оптоэлектронные, медико-биологические и другие свойства [63–69].
Среди методов получения наночастиц металлов широкое распространение получило восстановление ионов металлов в воде, органических растворителях и водно-органических растворах в присутствии функциональных полимеров и поверхностно-активных веществ, играющих роль как восстановителей, так и стабилизаторов [70–76]. Ионы металлов, адсорбированные в многослойные полиэлектролиты, дендримеры, блок-сополимеры, микрогели, липосомы, полимерные частицы и пленки или гидрогели, которые удерживаются в растворе благодаря функциональным группам гидрофильной структуры, могут быть легко восстановлены до соответствующих наночастиц металлов, кластеров или оксидов металлов с помощью подходящих восстановителей, таких как NaBH4, AlBH4, H2, NH2NH2, цитрат и т.п. [77, 78].
Стабилизирующий эффект наночастиц металлов полимерами осуществляется двумя различными способами – путем физической (процессы, обусловленные силами Ван-дер-Ваальса, дипольными взаимодействиями или слабыми, легко разрушающимися водородными связями) или химической адсорбции [65]. Нековалентное взаимодействие наночастиц с макромолекулой весьма слабо (порядка 10–4 Дж/м2); эффективность хемосорбции определяется числом полярных групп адсорбированного полимера на единицу поверхности независимо от конформации макромолекул – развернутой или глобулярной. Разумеется, что при этом важно не только наличие в полимере определенных функциональных групп, но и их интенсивное взаимодействие с поверхностными атомами наночастицы, например, в качестве доноров электронов [71]. Эффективность полимеров как доноров электронов значительно повышается, если они содержат центры специфических контактов, действующих одновременно по механизму зарядовой и стерической стабилизации. В работе [79] были синтезированы стабильные коллоидные растворы золота в условиях in situ восстановлением HAuCl4 в присутствии водорастворимых полимеров. Наиболее стабильные коллоиды получены с участием полимеров, имеющих гидрофобные хребты и гидрофильные боковые группы, связанные с ионами золота.
В работах [80–87] восстановление золота и серебра проводили в одну стадию кипячением смеси HAuCl4 (или AgNO3) и полимеров в присутствии KOH. В качестве полимеров, играющих роль как стабилизаторов, так и восстановителей, использованы следующие синтетические гидрофильные полимеры: поли-N-винилпирролидон (ПВПД), полиэтиленгликоль (ПЭГ), полиэтиленимин (ПЭИ), поли-N,N-диметил-N,N-диаллиламмоний хлорид (ПДМДААХ), поли-N-винилбензил-N,N,N-триметиламмоний хлорид (ПВБТМАХ), катионный полиэлектролит – JR-400, полиакриловая кислота (ПАК), природный полисахарид – геллан, β-циклодекстрин (ЦД). Наночастицы серебра, стабилизированные ПЭГ, ПВПД и ЦД, получены также “сухим” методом – механохимической активацией [88, 89] – и “влажным” способом – в этаноле, диметилформамиде и диметилсульфоксиде [82]. В работе [80] была исследована скорость образования наночастиц (НЧ) золота при нагревании в присутствии ПЭИ, ПДМДААХ и ЦД. Установлено, что ее значение в присутствии гидрофильных полимеров изменяется в следующей последовательности: ПДМДААХ > ПЭИ > > ПВПД > JR-400.
В литературе отсутствуют сведения о влиянии молекулярных масс полимеров на размеры НЧ. В табл. 4 представлены результаты измерения размеров НЧ золота и серебра, стабилизированных ПВПД различной молекулярной массы, методом динамического лазерного светорассеяния (ДЛС) [82, 86]. Как видно из данных табл. 4, с увеличением молекулярной массы полимеров размеры НЧ золота возрастают.
Таблица 4.
Изменение размеров НЧ золота и серебра в зависимости от молекулярной массы ПВПД
| Молекулярная масса ПВПД, Мw × 10–3, Дальтон |
Размеры НЧ золота, нм | Размеры НЧ серебра, нм |
|---|---|---|
| 10 | ~10 | ~6.5 |
| 40 | ~15 | ~12 |
| 350 | ~25 | ~44 |
Поверхностный заряд НЧ золота, стабилизированных ПВПД, отрицателен и составляет –2.03 мВ, тогда как поверхностные заряды НЧ золота, стабилизированных катионными полиэлектролитами (ПЭИ и JR-400), имеют положительные значения: +31.1 и +4.39 мВ соответственно. Дзета-потенциалы НЧ серебра, полученных в присутствии ПВПД и ЦД, в ДМСО имеют положительное значение, а в ДМФ – отрицательное [82]. Эти результаты свидетельствуют о хемосорбции неионогенного и катионного типа макромолекул на поверхности отрицательно заряженных НЧ золота.
Известны работы японских ученых [90–95], в которых было проведено исследование коллоидных катализаторов. Так, в [93] при взаимодействии солей Rh, Pd, Os, Jr и Pt в поливиниловом спирте в водно-метанольных растворах образовывались коллоиды со средними размерами частиц Os, Jr, Pt, Rh и Pd, равными 1.0; 1.4; 2.7; 4.0 и 5.3 нм соответственно. Авторы предполагают трехстадийную схему формирования коллоидной дисперсии: координирование полимера с ионами металла, восстановление металла метанолом до образования мелких частиц (например, 0.8 нм для Rh) и дальнейшее их укрупнение (до 4.0 нм). Показано, что коллоиды Rh, Pd, Os и Pt – активные и селективные катализаторы гидрирования олефинов при 30°С и атмосферном давлении водорода. Нанокластеры Au, стабилизированные ПВПД, являются эффективными катализаторами реакций окисления [96]. В реакции аэробного окислительного гомосочетания калий арилтрифторбората каталитическая активность Au/ПВПД зависит от размеров кластера [97]. Нанокластеры Au со средним диаметром dav = 1.3 ± 0.3 нм обладали высокой активностью в гомосочетании, в то время как система Au:PVP (dav = 9.5 ± 1.0 нм) была каталитически неактивной в изучаемой реакции. Полагают, что активные центры катализатора представляют собой положительно заряженные группы на поверхности нанокластера Au, генерируемые в ходе адсорбции молекулярного кислорода. Высокую хемоселективность в гидрировании α,β-непредельных альдегидов и кетонов проявляют биметаллические Ag–Au/ПВПД, которые предпочтительнее катализируют восстановление связи C=O, чем присоединение по C=C-связи [98]. Оптимизация реакции по соотношению ПВПД/металл для обеспечения эффективной стабилизации и по молярному соотношению Ag/Au, позволяющего сочетать высокую хемоселективность действия Au и активность Ag, дает возможность значительно повысить селективность формирования аллиловых спиртов.
Следует отметить появление, особенно в последнее десятилетие, большого количества работ по синтезу инкапсулированных в дендримерах наночастиц металлов, проявлявших каталитическую активность в различных процессах органического синтеза, включая гидрогенизацию многочисленных модельных соединений [99–107].
При взаимодействии растворов линейных полимеров с солями переходных металлов можно получать и гетерогенные катализаторы с равномерным распределением сферических наночастиц размером 4–6 нм, расположенных по всему объему полимерной матрицы (рис. 4а). Такие каталитические системы были исследованы в реакции гидрирования 2-пропен-1-ола [108–111]. В результате восстановления комплекса Pd с поли-2-винилпиридином (П2ВП) в условиях катализа (в этаноле или воде при 20–30°С), согласно данным РФЭС, образуются частицы нульвалентного металла [108].
Рис. 4.
Микрофотографии комплекса П2ВП-Pd, восстановленного водородом (а) и после гидрирования 30 порций 2‑пропен-1-ола (б) в воде при 25°С.

Укрупнение частиц происходит после проведения процесса гидрогенизации (рис. 4б). Однако при более детальном рассмотрении восстановленных и отработанных катализаторов видно, что они представляют собой агломераты более мелких исходных НЧ с размерами 4–6 нм. Можно предположить, что в условиях жидкофазной гидрогенизации полимер-металлические комплексы (ПМК) набухают, увеличиваются в размерах, частицы металла, инкапсулированные в них, становятся доступными для активации непредельных органических соединений. После высушивания такие образцы вновь сжимаются и за счет гидрофобных взаимодействий сегментов полимерных цепей объединяются в более крупные агрегаты. Повторное использование систем в катализе не приводит к снижению их активности. Более того, в некоторых случаях на “разработанном” комплексе П2ВП-Pd скорость реакции заметно возрастает [108]. Это, вероятно, связано с тем, что в реакционной среде ПМК вновь набухает, межмолекулярные гидрофобные взаимодействия ослабевают.
Для снижения влияния диффузионного фактора перспективным является создание катализаторов, получаемых путем иммобилизации полимерметаллических комплексов на твердые неорганические носители. При этом сохраняются как технологические параметры гетерогенных катализаторов (легкость отделения от продуктов реакции, возможность регенерации и многократного использования), так и, в значительной мере, активность, присущая гомогенным системам, за счет формирования на поверхности носителей металлокомплексов с полимерными лигандами, позволяющими сохранить относительную подвижность активных центров в реакционной среде.
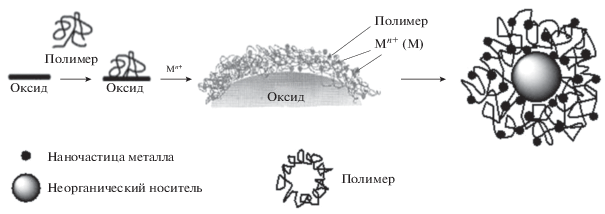
В качестве стабилизаторов наночастиц, а также при нанесении их на оксиды для синтеза катализаторов активно используется поливинилпирролидон [112–114]. Предлагаемые авторами катализаторы готовили закреплением полимер-протектированных наночастиц на носитель с последующими обычными стадиями высокотемпературного прокаливания и восстановления, что приводило к разрушению полимерной оболочки и сохранению наночастиц металла на носителях. Так, полимер-протектированные платиновые наночастицы были внедрены капиллярным способом в мезопористые каналы силикагеля SBA-15 с размерами пор 9 нм [114]. Последующая обработка ультразвуком позволяет локализовать наночастицы Pt преимущественно в порах силикагеля (рис. 5).

После окислительной и восстановительной обработки катализаторов для удаления полимера-стабилизатора, окружащего наночастицы, можно формировать катализаторы с различными размерами активной фазы [112]. Полученные системы были исследованы в гидрировании этилена и циклогексена. Обнаружено, что размеры частиц металлов, закрепленных на силикагеле, влияют на селективность процесса гидрирования цилогексана и этилена, однако характер этой зависимости достаточно сложный и требует дополнительных исследований. Вместе с тем авторы уверены, что, используя разработанные подходы к созданию катализаторов, можно регулировать размеры наночастиц, закрепленных на носителе, а, следовательно, и направленно изменять их каталитические свойства.
Аналогичный подход был применен в работе [100] при приготовлении катализаторов на основе дендримерных комплексов металлов, которые наносили на оксид кремния, а затем прокаливали и восстанавливали водородом при высоких температурах. При этом дендримерная оболочка выжигалась, а на поверхности носителя формировался равномерный слой наночастиц платины (схема 1 ):
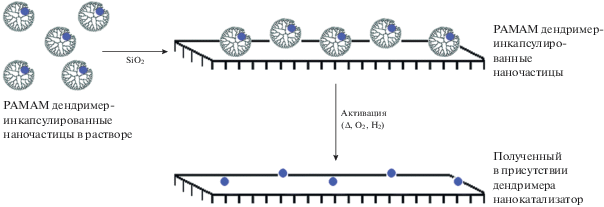
Схема 1 . Дендример-темплатный путь формирования нанокатализатора.
Приготовленные таким образом каталитические системы показали высокую активность в процессе гидрогенизации толуола и окисления СО.
Полимер-иммобилизованные наночастицы Pd активно исследуют в реакциях кросс-сочетания Сузуки-Мияуры арил- и винилборных кислот с арил- или винилгалогенидами [115–118]. Так, наночастицы Pd (d = 3.5 ± 1.5 нм), стабилизированные поли(4-стиролсульфоновая кислота-со-малеиновой кислотой) (PSSA-co-MA) проявляют высокую каталитическую активность в реакции р-бромацетофенона или йодбензола с фенилборной кислотой с величинами TOF 1980 и 5940 ч−1 соответственно [118]. Следует отметить, что Pd/PSSA-co-MA и Ru/PSSA-co-MA, in situ синтезируемые в ходе гидролиза борана аммиака (NH3BH3), являются эффективными катализаторами гидролитического дегидрирования NH3BH3, который рассматривается как перспективный материал для хранения и получения водорода [117]. Аналогичные свойства в этой реакции проявляют наночастицы Ni, стабилизированные N-поливинилпирролидоном [119]. Полимер-иммобилизованный катализатор демонстрировал хорошую воспроизводимость без потери активности в 5 циклах, что объяснено отсутствием агрегации металлических частиц благодаря наличию полимерной матрицы.
3. ГЕЛЬ-ИММОБИЛИЗОВАННЫЕ КАТАЛИЗАТОРЫ
В последние годы совместными усилиями химиков, физиков и материаловедов созданы различные типы наномасштабных структур, включающих в состав строительных блоков наночастицы металлов в комбинации с функциональными полимерами. Перспективными и пока малоисследованными полимерными матрицами для иммобилизации наночастиц металлов являются нано-, микро- и макропористые гидрогели неионного, анионного, катионного и амфотерного характера [120]. Пространственно (трехмерно) сшитые макромолекулы, содержащие функциональные группы различной природы, могут изменять размеры, форму и морфологию непрерывным образом или по принципу “все или ничего” (явление коллапса) при варьировании внешних факторов, в число которых входят температура, рН среды, ионная сила раствора, смесь водно-органических растворителей. Это, в свою очередь, позволяет тонко регулировать структуру и свойства иммобилизованных в матрицу гидрогелей наночастиц и контролировать их архитектуру. Кроме того, матрица гидрогеля может служить своего рода “микрореактором”, с помощью которого удается осуществлять обменные, окислительно-восстановительные, каталитические и другие типы реакций.
В настоящее время на основе гидро- и криогелей и благородных металлов получены различные типы полимер-иммобилизованных нанокатализаторов, обладающих высокой селективностью и стабильностью действия. Иммобилизация наночастиц металлов, стабилизированных водорастворимыми полимерами, в матрицу гидро- и криогелей представляет большой интерес с точки зрения создания каталитических систем, моделирующих действие металлоферментов, а также гетерогенизированных гомогенных катализаторов, способных ускорять реакции разложения, гидрирования, окисления, изомеризации и т.д.
Необходимость стабилизации и иммобилизации наночастиц металлов в матрицу гидрогелей продиктована тем, что зачастую наночастицы, имеющие большую величину избыточной поверхностной энергии, могут мгновенно пассивироваться за счет адсорбционных процессов либо коагуляции.
3.1. Катализаторы на основе наночастиц металлов, иммобилизованных в матрицу гидрогелей
Гидрогели, содержащие полимер-протектированные наночастицы металлов, представляют собой сложные системы, где в порах и междоузельном пространстве находятся как линейные макромолекулы в клубкообразном состоянии, так и наночастицы металлов. При этом линейные макромолекулы играют роль стабилизаторов, предотвращающих агрегацию активных наночастиц металлов, а роль гидрогелевой матрицы заключается в ограничении диффузии полимер-протектированных наночастиц внутри сетки и за ее пределы.
Полимер-протектированные наночастицы золота, серебра и палладия могут быть иммобилизованы в матрицу стимулчувствительных гидрогелей двумя путями (рис. 6) [121–130]. В первом случае предварительно высушенные образцы полиакриламидного геля (ПААГ) набухшие в водном растворе золотохлористоводородной и тетрахлорпалладиевой кислот или нитрата серебра, восстанавливают раствором борогидрида натрия (рис. 7).
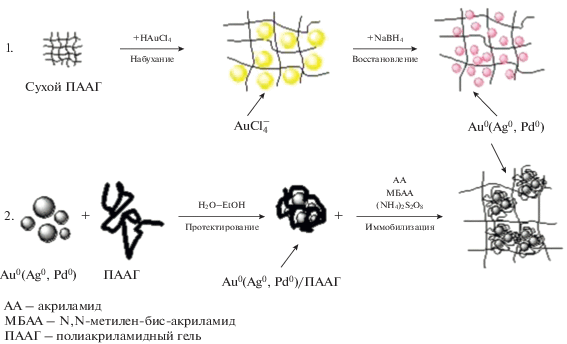
Рис. 7.
Восстановление НAuCl4 раствором NaBH4 в объеме ПААГ: а – высушенные (1А, 2А) и набухшие в растворе НAuCl4 (1Б, 2Б) образцы ПААГ (1А, 1Б) и ПААГ/ПЭИ (2А, 2Б); б – образцы ПААГ (1Д) и ПААГ/ПЭИ (2Д), обработанные раствором NaBH4 в течение 30 мин (1Д, 2Д). [HAuCl4] = 0.5 ммоль/л; [NaBH4] = 0.1 моль/л.

Во втором случае предварительно растворенный в коллоидных растворах металлов мономер, сшивающий агент и инициатор полимеризуют при 70°С в течение 30 мин. Образцы гидрогелей, в объеме которых содержатся стабилизированные полимером наночастицы металлов, являются однородными и имеют характерную окраску, соответствующую полимер-протектированным наночастицам металлов (рис. 8). Средние размеры наночастиц металлов, определенные методом СЭМ, представлены в табл. 5.

Таблица 5.
Средние размеры наночастиц металлов, стабилизированных гидрофильными полимерами и иммобилизованными в матрицу ПААГ
| Система | ПВПД-Pd(0) | ПЭИ-Pd(0) | ПЭИ-Au(0) |
|---|---|---|---|
| Размер наночастиц, нм | 60 | 10 | 10–50 |
Как видно из табл. 5, средние размеры наночастиц металлов, стабилизированных гидрофильными полимерами, составляют 10–60 нм, тогда как размеры частиц золота, сформированные в матрице ПААГ путем предварительной адсорбции HAuCl4 и последующего восстановления NaBH4, лежат в пределах 100 нм (рис. 9). Эти данные свидетельствуют о том, что наночастицы металлов, стабилизированные гидрофильными полимерами, имеют более узкое распределение по размерам, чем иммобилизованные в матрицу ПААГ методом адсорбции и восстановления.

Образцы гидрогеля поли(N-изопропилакриламида) (ПНИПААМГ), в матрицу которого иммобилизован полимер-протектированный палладий (ПВПД-Pd0), т.е. ПНИПААМГ/ПВПД-Pd0, обладают ярко выраженной термочувствительностью в интервале 25–40°С [131]. Причем при повышении температуры до 40°С наблюдается сжатие гидрогеля и выход иммобилизованного полимер-протектированного палладия ПВПД-Pd0 во внешнюю среду, сопровождаемый резким помутнением окружающего гидрогель водного раствора (рис. 10). При понижении температуры до 25°С образец набухает, а окружающий гидрогель водный раствор становится прозрачным, что связано с поглощением ПВПД-Pd0 гидрогелевой матрицей.
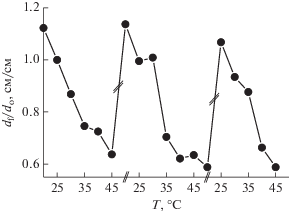
Рис. 10.
Изменение размера образца ПНИПААМГ/ПВПД-Pd0 при 25 и 40°С; dо и df – размеры образца набухшего и коллапсированного геля соответственно.

Каталитическая активность ПНИПААМГ/ ПВПД-Pd0 исследована в реакции гидрирования 2-пропен-1-ола [127]. Как и следовало ожидать, с повышением температуры скорость реакции резко возрастает за счет выхода каталитически активной фазы ПВПД-Pd0 из гидрогелевой матрицы в окружающий раствор (рис. 11). Снижение температуры, наоборот, приводит к стремительному падению скорости реакции гидрирования субстрата в результате поглощения ПВПД-Pd0 матрицей гидрогеля. Резкое ускорение или торможение каталитической реакции при периодическом изменении температуры объясняется тем, что при Т = 25°С, что ниже НКТР (нижней критической температуры растворения), ПНИПААМГ находится в равновесно-набухшем состоянии и выход катализатора ПВПД-Pd0 в окружающий раствор минимален (поры сетки закрыты). При Т = 40°С, что выше НКТР, ПНИПААМГ резко сжимается (коллапсирует) и активные центры кооперативно диффундируют из объема гидрогеля в окружающий раствор (поры сетки открыты). Таким образом, циклическое изменение температуры выше или ниже НКТР приводит к сжатию или набуханию ПНИПААМГ (рис. 12), что позволяет дозировать гомогенный катализатор в окружающий раствор и тем самым контролировать скорость каталитической реакции.
Рис. 11.
Температурная зависимость каталитической активности ПНИПААГ/ПВПД-Pd0 в реакции гидрирования 2-пропен-1-ола в нескольких циклах.
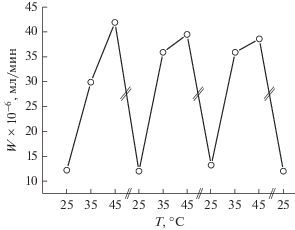
В работах [129, 130] гель-иммобилизованные НЧ золота были исследованы в реакции разложения перекиси водорода и окисления циклогексана пероксидом водорода. Кинетика разложения пероксида водорода на гель-иммобилизованных НЧ золота, полученных тремя способами в присутствии различных полимеров-стабилизаторов, представлена на рис. 13. Наиболее активными в разложении пероксида водорода оказались ПЭИ-протектированные и гель-иммобилизованные НЧ золота, полученные сорбционным методом (рис. 13, кривая 1). На этом катализаторе изучено влияние разных факторов (массы катализатора, температуры реакции и концентрации субстрата) на кинетику разложения пероксида водорода.
Рис. 13.
Разложение перекиси водорода гель-иммобилизованными наночастицами золота, полученными различными способами: метод сорбции, стабилизаторы ПЭИ (1), ПВП (2); метод in-situ, стабилизаторы ПЭИ (4) и ПВПД (5); боргидридный способ без стабилизатора (3).
Результаты исследования реакции окисления циклогексана (ЦГ) пероксидом водорода в присутствии гель-иммобилизованных НЧ золота показали [130], что данная система демонстрирует достаточно высокую селективность образования смеси циклогексанола (ЦОЛ) и циклогексанона (ЦОН) (табл. 6). При этом конверсия циклогексана не превышает 7%.
Таблица 6.
Окисление циклогексана пероксидом водорода на ПЭИ-протектированном и гель-иммобилизованном нанокатализаторах – наночастицах золота при 45°С [130]
| Катализатор | Конверсия ЦГ, % | Селективность, % | ||
|---|---|---|---|---|
| ЦОЛ | ЦОН | другие кислородсодержащие соединения | ||
| НЧ золота, стабилизированные ПЭИ и иммобилизованные в матрицу гидрогеля ПАА | 7.2 | 53.0 | 44.7 | 2.3 |
Таким образом, наибольшей каталитической активностью в реакциях разложения пероксида водорода и окисления циклогексана пероксидом водорода обладают ПЭИ-протектированные и гель-иммобилизованные НЧ золота, полученные сорбционным методом. Следует отметить, что при прочих равных условиях каталитическая активность гель-иммобилизованных НЧ золота ниже, чем у гетерогенизированных путем нанесения на неорганические носители полимер-протектированных НЧ золота. Вероятно, это связано с низкой стабильностью гель-иммобилизованных НЧ из-за подверженности полимерной сетки к деструкции под действием окислителя – пероксида водорода.
3.2. Катализаторы на основе макропористых криогелей с иммобилизованными наночастицами металлов
Среди большого количества полимерных материалов особое место занимают макропористые криогели, которые находят широкие применение в катализе, биотехнологии, биомедицине, биосепарации и в других сферах техники и технологии [132].
Систематические исследования, посвященные получению криогель-иммобилизованных наночастиц металлов и их использованию в качестве каталитических систем, были проведены авторами [133–140]. Такие системы рассматриваются как своего рода микрореакторы.
Так, в работе [140] полиакриламидный (ПАА) криогель модифицировали гидроксиламином по схеме (схема 2 ).
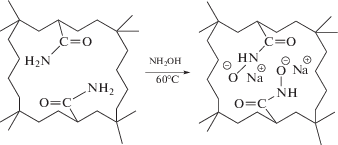
Схема 2 . Схема химической модификации ПАА-криогеля.
В результате был приготовлен амидо-ПАА-криогель, содержащий функциональные группы гидроксамовых кислот, которые придают криогелю возможность связывать ионы металлов по электростатическому механизму. После восстановления ионов металлов (Co, Ni, Cu) борогидридом натрия методом in situ получены наночастицы меди, кобальта и никеля, иммобилизованные в матрицу криогеля (схема 3 ).
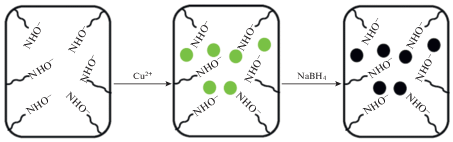
Схема 3 . Схема образования наночастиц меди в матрице макропористого амидо-ПАА-криогеля методом in situ [140].
В последующем эти нанокомпозиты были использованы в качестве катализаторов в процессе восстановления 2-нитрофенола (2-НФ). Установлено, что наибольшей каталитической активностью обладают композиты на основе наночастиц меди.
В работах [141–143] представлены результаты применения макропористого амфотерного криогеля на основе метакриловой кислоты (МАК) и N,N-диметиламиноэтилметакрилата (ДМАЭМ), сшитого метиленбисакриламидом (МБАА), с иммобилизованными наночастицами золота – ДМАЭМ-МАК/НЧЗ – в качестве катализатора восстановления 4-нитрофенола (4-НФ) и каталитического реактора проточного типа (рис. 14). Показано [141], что все криогелевые наносистемы проявляют высокую каталитическую активность при комнатной температуре и атмосферном давлении. Так, конверсия субстрата на криогелевых носителях неионогенной, анионной и катионной природы после гидрирования 10 порции 4-НФ составляет свыше 90%. Причем наибольшую каталитическую активность проявляют наночастицы золота, иммобилизованные в матрицу амфотерного криогеля (табл. 7).
Рис. 14.
Схематическое изображение каталитического реактора проточного типа на основе криогеля ДМАЭМ-МАК/НЧ(Au).
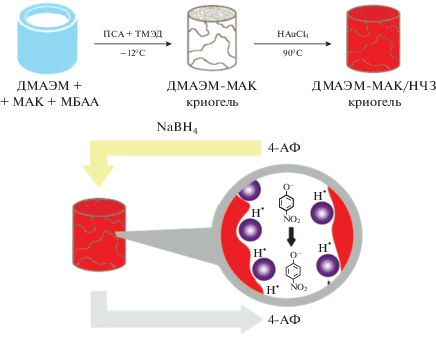
Таблица 7.
Влияние природа макропористых криогелей на конверсию 4-НФ
| Криогели | Природа криогелей | Конверсия, % |
|---|---|---|
| Поли(акриламид) | Неионогенный | 93 |
| Поли(акриламид-со-метакриловая кислота) | Анионный | 91 |
| Поли(акриламид-со-N,N-диметиламиноэтилметакрилат) | Катионный | 92 |
| Поли(метакриловая кислота-со-N,N-диметиламиноэтилметакрилат) | Амфотерный | 98 |
Получены следующие термодинамические параметры процесса гидрирования 4-НФ на наиболее активном криогелевом катализаторе МАК-ДМАЭМ/Au: Еа = 7.52 кДж моль–1, ΔHa = = 4.91 кДж моль–1, ΔSa = 284.34 Дж моль–1 К–1. Авторами [144–148] был исследован процесс гидрирования пара-нитробензойной кислоты (ПНБК) на неподвижной фазе макропористого криогеля (ДМАЭМ-МАК), содержащего наночастицы золота и палладия, при комнатной температуре и атмосферном давлении.
При гидрировании ПНБК на катализаторе ДМАЭМ-МАК/Au кроме пара-аминобензойной кислоты (ПАБК) образуются 2 побочных продукта. Один из них – пара-азодибензоат, продукт конденсации нитрозо-соединения с гидроксиламином, образование которого подтверждается Раман-спектроскопией, результаты которой находятся в удовлетворительном согласии с данными авторов [149] по фотовосстановлению ПНБК наночастицами серебра (схема 4 ).
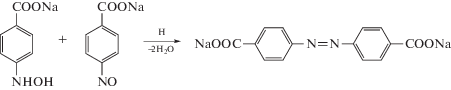
Схема 4 . Механизм образования пара-азодибензоата согласно [148].
Другим продуктом превращения ПНБК является натрий 4-(4-аминобензамидо)бензоат, формирующийся в результате конденсации концевых карбоксильных и аминных групп ПНБК, катализируемой наночастицами золота (схема 5 ).
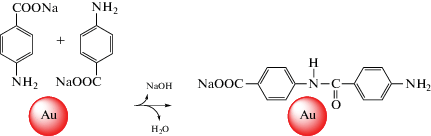
Схема 5 . Механизм образования натрий 4-(4-аминобензамидо)бензоата согласно [149].
Конденсационная полимеризация алкилированных производных ПНБК, приводящая к образованию алкиламинобензамидобензоатов, была показана авторами [150].
Таким образом, продуктами гидрирования ПНБК на криогелевом катализаторе ДМАЭМ-МАК/Au являются ее амино-, азо- и амидопроизводные.
В случае гидрирования ПНБК на палладий-криогелевом катализаторе ДМАЭМ-МАК/Pd побочных продуктов не наблюдается. Конверсия ПНБК в ПАБК составляет 40% при соотношении [ПНБК] : [NaBH4] = 1 : 50, моль/моль, 84% при [ПНБК] : [NaBH4] = 1 : 100, моль/моль и 100% при [ПНБК] : [NaBH4] = 1 : 200, моль/моль.
4. КАТАЛИЗАТОРЫ НА ОСНОВЕ ПОЛИМЕР-ПРОТЕКТИРОВАННЫХ НАНОЧАСТИЦ МЕТАЛЛОВ, НАНЕСЕННЫХ НА НЕОРГАНИЧЕСКИЕ НОСИТЕЛИ
Разработка нанокатализаторов, полученных путем иммобилизации полимер-протектированных наночастиц металлов на твердые неорганические носители (рис. 15), является перспективным направлением исследований. При нанесении наночастиц металлов на неорганические носители сохраняются как технологические параметры гетерогенных катализаторов (легкость отделения от продуктов реакции, возможность регенерации и многократного использования), так и, в значительной мере, активность, присущая гомогенным системам, за счет формирования на поверхности носителей наночастиц металлов, позволяющего сохранить относительную подвижность активных центров в реакционной среде.
Рис. 15.
Схематическое представление иммобилизации полимер-протектированных наночастиц металлов на твердые неорганические носители.
Известно [69, 151–153], что на неорганических подложках, в частности оксидах, макромолекулы адсорбируются практически необратимо из-за кооперативных взаимодействий сегментов полимерных цепей с носителем. Этим решается одна из наиболее сложных проблем закрепленных комплексов – предотвращение вымывания активной фазы.
Авторы работ [154, 155] для повышения селективности гидрирования ацетиленовых соединений предложили применять в качестве катализаторов полимерные палладийсодержащие мицелярные металлокомплексы. В работе [155] была исследована каталитическая активность мицеллярных катализаторов на основе Pd и Pd–Au, закрепленных на полистирол-поли-4-винилпиридиновой матрице и нанесенных на γ-Al2O3 в процессе гидрирования дегидролиналоола (2,6-диметилоктаен-6-ин-1-ол-3) в линалоол (2,6-диметилоктадиен-1,6-ол-3). Отмечалось, что проведение реакции в диффузной области обеспечивает наивысшую селективность процесса, но данных об условиях проведения гидрирования, к сожалению, приведено не было.
Сульман Э.М. с соавт. [156, 157] использовали катализатор Pd/Al2O3 в гидрогенизации сложных ацетиленовых спиртов с добавлением KOH и пиридина. Однако катализируемые реакции имели зачастую низкие выходы целевых продуктов из-за их загрязнения модифицирующими соединениями, такими как пиридин, хинолин, соли металлов и др. Поэтому в более поздних исследованиях [158–162] этот же процесс проводили в присутствии нанесенных на оксид алюминия палладиевых и родиевых катализаторов, модифицированных азотсодержащими полимерами. При этом были достигнуты высокие показатели выходов олефиновых соединений. По данным электронной микроскопии на поверхности оксида алюминия формировались наночастицы металла с размерами 2–3 нм.
Простой способ закрепления тонкого слоя ПМК на поверхности неорганических сорбентов (природные и синтетические цеолиты, оксиды металлов) был предложен в работе [163]. Процесс приготовления катализаторов включает в себя последовательное нанесение (адсорбцию) полимера, а затем ионов металла на поверхность неорганического оксида.
Отличительной особенностью метода является исключение стадий высокотемпературного прокаливания и восстановления катализаторов. В процессе гидрирования широкого спектра непредельных соединений [164–166] при температурах, не превышающих 40°С, такие системы проявляют высокую активность, селективность и стабильность при многократном использовании. Образующийся на поверхности твердого носителя полимер-металлический композит (комплекс) сохраняет гибкость и подвижность, не вымывается в процессе катализа из-за кооперативных взаимодействий функциональных групп макромолекул с твердыми неорганическими оксидами. При правильном подборе растворителя набухший поверхностный слой представляет собой трехмерный микрореактор, внутри и на поверхности которого, как в случае гидрогелей, осуществляется каталитический процесс.
Разработанные катализаторы тестировали в реакции гидрирования таких сложных ацетиленовых соединений как 3,7,11-триметилдодецин-1-ол-3 (ацетиленовый спирт С15), 3,7,11,15-тетраметилгексадецин-1-ол-3 (ацетиленовый спирт С20), 9-гексадецин-1-ол и 11-гексадецин-1-ол [167, 168]. Так, например, результаты сравнения обычного катализатора 1%Pd/ZnO, приготовленного адсорбционным методом, с ПЭГ-содержащим образцом в реакции гидрирования ацетиленового спирта С20 также свидетельствовали о значительных преимуществах последнего по трем основным параметрам – активности, селективности и стабильности (табл. 8). Было показано, что скорость образования и выход 3,7,11,15-тетраметилгексадецен-1-ола-3 на модифицированном полимером катализаторе выше, чем на обычной системе Pd/ZnO. Селективность по целевому продукту на нем увеличилась на 11.2%, TON возросло в 3 раза по сравнению с образцом без ПЭГ. Значение TOF на полимерсодержащем катализаторе, равное 670 ч−1, говорит о том, что за 5 мин конверсия ацетиленового спирта достигала 86%.
Таблица 8.
Результаты гидрирования ацетиленовых соединений на 1%Pd-ПЭГ/ZnO и 1%Pd/ZnO*
| Ацетиленовый спирт | W × 10–6, моль/с | SC=C | TON | TOF, ч−1 |
|---|---|---|---|---|
| Pd/ZnO | ||||
| 3,7,11,15-тетраметилгексадецин-1-ол-3 | 9.7 | 76.5 | 5100 | 160 |
| 1%Pd-ПЭГ/ZnO | ||||
| 3,7,11,15-тетраметилгексадецин-1-ол-3 | 16.3 | 87.7 | 15 400 | 670 |
| 11-Гексадецин-1-ол | 16.3 | 99.1 | 7600 | 400 |
| 9-Гексадецин-1-ол | 12.1 | 99.7 | 4000 | 240 |
Было установлено, что независимо от природы катализаторов в ацетиленовых спиртах С15 и С20 присоединение водорода осуществляется как по тройной, так и по двойной связи. При восстановлении 9- и 11-гексадецин-1-олов процесс резко замедлялся после поглощения первого моля водорода. Селективность по олефиновому производному практически на всех катализаторах достигала 99%.
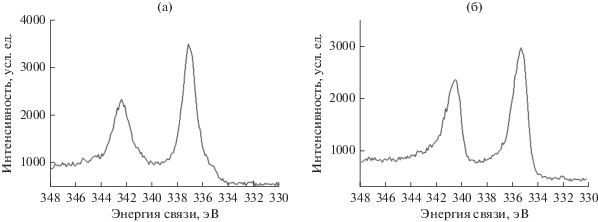
По данным РФЭС палладий в 1%Pd-ПЭГ/ ZnO находится в окисленном состоянии (Pd2+) с энергией связи Pd3d5/2 около 337 эВ (рис. 16а). Обработка катализаторов водородом в реакторе при 40°С приводит к полному восстановлению палладия до нуль-валентного состояния. В РФЭ-спектре Pd3d5/2 (рис. 16б) имеется пик, соответствующий энергии связи палладия в нуль-валентном состоянии (~335.2 эВ). Аналогичная картина наблюдалась при исследовании катализатора 1%Pd/ZnO.
Введение полимера в состав катализатора оказывает существенное влияние на размеры частиц активной фазы и их распределение по поверхности носителя. В образце 1% Pd/ZnO на оксиде цинка формируются частицы металла с размерами 30–50 нм (рис. 17а). Палладиевый катализатор, приготовленный с участием ПЭГ, характеризуется образованием равномерно локализованных мелких частиц металла размером 2–3 нм (рис. 17б), что отчетливо видно при снятии микрофотографии при большем увеличении (рис. 17в). Равномерное распределение частиц, вероятно, связано с наличием полимерной пленки на поверхности носителя, препятствующей их агрегации. Так, по данным просвечивающей электронной микроскопии ZnO представлен четко ограненными кристаллитами (рис. 18а). При обработке полиэтиленгликолем на его поверхности образуется прозрачный слой полимера (рис. 18б).


Таким образом, активность, селективность и стабильность модифицированного ПЭГ катализатора Pd/ZnO выше, чем приготовленного без него. Наибольшее влияние обработка полимером оказывает на стабильность (TON), которая возрастает в 2–4 раза.
Разработанные катализаторы, по сути, являются гетерогенными и легко отделяются от продуктов реакции. Гибкость, подвижность и способность к набуханию полимерных цепей полимерметаллических соединений, закрепленных на носителях, способствуют сохранению относительной подвижности активных центров в реакционной среде. Функции полимеров заключаются в формировании и фиксации наночастиц металлов на поверхности носителя, предотвращении их агломерации в процессе катализа, повышении стабильности активных центров. Полученные катализаторы показали высокую активность, селективность и стабильность в процессах гидрирования сложных ацетиленовых спиртов до олефиновых производных в мягких условиях.
Аналогичные закономерности обнаружены в случае стабилизированных ПВПД нанокатализаторов золота, нанесенных на оксиды алюминия и цинка (рис. 19) [81, 82, 85, 86]. Была изучена каталитическая активность полимер-стабилизированных наночастиц золота и серебра, нанесенных на оксиды металлов, в реакции разложения перекиси водорода и найдены оптимальные условия ее проведения: масса катализатора mкат = 30 мг, концентрация субстрата [H2O2] = 30% и температура T = 318 K (рис. 20) [86]. В работе [169] исследована каталитическая активность полимер-стабилизированных наночастиц золота, нанесенных на оксид цинка, в реакции окисления циклогексана пероксидом водорода. Результаты показали, что на Au-ПВПД/ZnO циклогексан окисляется до циклогексанола и циклогексанона с большей конверсией по сравнению с Au-ПАК/ZnO и Au-ПЭИ/ZnO (табл. 9).

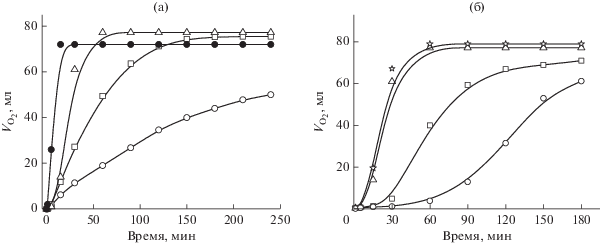
Таблица 9.
Результаты окисления циклогексана (ЦГ) пероксидом водорода в присутствии нанесенных наночастиц золота, стабилизированных различными полимерами
| Катализатор | Конверсия ЦГ, % | Выход продуктов, % | ||
|---|---|---|---|---|
| ЦОЛ | ЦОН | неидентифицированные продукты | ||
| Au-ПВПД/ZnO | 29.5 | 44.0 | 55.2 | Следы |
| Au-ПАК/ZnO | 3.4 | 8.8 | 18.7 | 72.5 |
| Au-ПЭИ/ZnO | 2.9 | 8.4 | 13.0 | 78.6 |
Таким образом, гетерогенизированные путем закрепления на поверхности носителя полимер-протектированные наночастицы золота достаточно эффективны в реакции парциального окисления циклогексана при мягких условиях. На активность и селективность получаемых катализаторов оказывает влияние природа полимера-стабилизатора и носителя, а размеры наночастиц благородных металлов зависят как от природы полимеров-стабилизаторов, так и от их молекулярных масс.
Разработан новый подход к созданию полимер-иммобилизованных наночастиц на поверхности неорганического носителя посредством фронтальной полимеризации акриламидных комплексов нитратов благородных металлов в присутствии SiO2, Al2O3, C, ZnO, SnO2 и т.д. [165, 170–173]. Полученные полимер-гибридные нанокомпозиты имеют достаточно развитую поверхность и пористую структуру (табл. 10), что обеспечивает доступность реагентов к активным центрам катализатора и их высокую активность в исследуемых реакциях.
Таблица 10.
Удельная поверхность и размеры пор в полимер-гибридных нанокомпозитах на основе Pd(CH2=CHCONH2)2(NO3)2(PdAAm) и [(H2C=CHCONH2)2RhOH](NO3)2 ⋅ H2O (RhAAm) [173]
| Образец | Удельная поверхность, м2/г | Объем пор, см3/г | Средний радиус пор, Å |
|---|---|---|---|
| Поли-PdAAm | 18.2 | 0.10 | 113.2 |
| Поли-PdAAm/SiO2 | 146.8 | 0.28 | 37.8 |
| Поли-PdAAm/Al2O3 | 60.2 | 0.17 | 55.0 |
| Поли-RhAAm/SiO2 | 210.9 | 0.385 | 36.5 |
| Поли-RhAAm/Al2O3 | 78.4 | 0.23 | 59 |
| Поли-RhAAm/C | 83.82 | 0.412 | 98.2 |
Комплексы PdAAм/SiO2 (Al2O3, C) проявляют значительную активность в модельной реакции гидрирования циклогексена. Так, в сопоставимых условиях начальная скорость реакции в присутствии катализатора поли-PdAAм/Al2O3 была почти в 3 раза выше, чем для стандартного Pd/C [165]. Важно, что исследуемые комплексы сохраняют каталитическую активность в рециклах, при этом иммобилизованная форма позволяет легко выделять их из реакционной среды и использовать многократно. Полимер-иммобилизованные наночастицы Pd (поли-PdAAм/SiO2) проявляют не только достаточную эффективность, но и селективность действия в важной с практической точки зрения реакции гидрирования 2,4-динитро- и 2,4,6-тринитротолуолов – скорость гидрирования второй нитрогруппы почти на порядок ниже, чем первой [170]. Избирательный характер каталитического процесса позволяет выделять целевые продукты на соответствующих стадиях, т.е. продукты частичного или полного гидрирования, а также, что не менее важно, каталитические интермедиаты. В интермедиате наряду с Pd0 (335.5 эВ) имеются атомы Pd c частично положительным зарядом Pdδ+ (337.0 эВ), способствующие координации молекул субстрата, что влияет на активность катализатора и, вероятно, преимущественному гидрированию одной нитрогруппы. На второй стадии происходит сдвиг значения энергии связи в РФЭ-спектре Pd3d5/2 в высокоэнергетическую область (337.7 эВ). Вероятно, наблюдаемая высокая селективность действия изученных гибридных нанокомпозитов прежде всего связана со стерическими факторами, в том числе, обусловленными и наличием полимерной матрицы, что приводит к сильной дифференциации координации нитрогрупп субстрата.
Предложен способ гетерогенизации гомогенных наночастиц металлов Pt, Pd, Rh, Au, Cu, стабилизированных дендримерами, на мезопористом силикагеле, основанный на последовательных реакциях комплексообразования ионов металлов с третичными аминогруппами РАМАМ, восстановления ионов металлов с формированием наночастиц и стадии закрепления гомогенного катализатора на неорганическом носителе методом импрегнации [174–176]. Полученные гетерогенные катализаторы были эффективны в гидрогенизационной изомеризации метилциклопентана. Так, Pt-нанокластеры с размерами 1.2–1.9 нм наряду со значительной активностью гидрирования при 200–225°C (TOF = 334 ч−1) проявляют высокую селективность (до 99.6%) в изомеризации метилциклопентана с раскрытием цикла при конверсии 94% [175]. Можно полагать, что решающую роль в характере каталитического действия Pt-нанокластеров играют молекулы дендримера и создаваемое ими локальное окружение каталитических центров. В то же время Pt-нанокластеры (1.5 нм), стабилизированные поливинилпирролидоном и закрепленные на этом же силикагеле (SBA-150), проявляли в реакции изомеризации метилциклопентана значительно меньшую активность и селективность (TOF = 19 ч−1 при 280°C, селективность 70%) [177].
Резюмируя вышесказанное, можно отметить, что рассмотренные подходы для создания полимер-защищенных наночастиц металлов, закрепленных на неорганических носителях, дают возможность получать системы, обладающие преимуществами как гомогенных (высокая активность и селективность, мягкие условия проведения процесса), так и гетерогенных катализаторов (простота технологии приготовления и отделения катализатора от продуктов реакции).
Способ получения катализаторов с наноразмерными частицами активной фазы прост, не требует больших затрат электроэнергии на стадиях прокаливания и восстановления, обязательных при приготовлении обычных нанесенных каталитических систем для процессов каталитической гидрогенизации. Процесс осуществляется при комнатной температуре с использованием экологически чистых водных растворов. Приготовление катализаторов включает в себя следующие стадии: 1) прочную адсорбцию растворимого полимера на поверхности носителя вследствие кооперативных взаимодействий сегментов полимерных цепей с подложкой; 2) добавление активной фазы (например, соли палладия для гидрогенизации). В результате на поверхности носителя формируются наночастицы активной фазы, протектированные полимерами.
Таким образом, при обработке поверхности традиционных оксидных подложек полимерами происходит их прочная или необратимая адсорбция. Ионы переходных металлов при их введении на такой носитель локализуется вокруг функциональных групп полимера, формируя наноразмерные частицы активной фазы. Полимерная “прослойка” способствует равномерному распределению активной фазы, предотвращению ее агломерации, а неорганический носитель – упрощению стадии отделения катализатора от продуктов реакции, устойчивости к действию растворителя.
5. ЗАКЛЮЧЕНИЕ
Благодаря развитой в последние годы синтетической методологии и возможности контролируемого получения необходимых свойств функционального материала, полимер-иммобилизованные кластеры и наночастицы металлов представляют значительный интерес для катализа. Иммобилизация гомогенных металлокластерных наночастиц на твердую поверхность без потери их каталитической активности крайне важна для практического использования катализаторов. Гетерогенизированные полимер-иммобилизованные нанокластеры металлов являются высокоактивными катализаторами гидрогенизации олефинов [178]. Сведения об электронной и геометрической структуре отдельных атомов, нанокластеров и наночастиц, в том числе закрепленных на полимерных носителях, и их каталитических свойствах в различных органических реакциях, включая окисление СО, селективное гидрирование, электро- и фотокаталитические реакции, были обобщены в недавнем обзоре [179]. Благодаря подвижной геометрической структуре полимерной молекулы становится возможным преодолевать стерические ограничения традиционных твердых неорганических носителей. И в этом случае, например, дендримерная полимерная матрица, которая инкапсулирует наночастицы металлов, может активировать гетерогенный катализатор таким образом, что в его присутствии начинают идти реакции, которые обычно протекают только с участием гомогенных катализаторов, например, реакции активации π-связей или альдольной конденсации. Следует отметить, что дендример-инкапсулированнные кластеры Au проявляют меньшую каталитическую активность, чем in situ сформированные нанокластеры, благодаря их оптимизированной структуре и координационному окружению, наиболее благоприятным для катализа [180]. Строгий контроль размера и формы наночастиц, в том числе и субнанометровых, можно осуществлять при использовании амфифильных (со)полимеров и мицеллов на их основе [181, 182]. Такой подход представляется весьма перспективным для создания каталитических систем различной наноархитектуры (сферических, стержнеобразных, 3D-сшитых). Нанокластеры Pd (средний размер ≈0.7 нм), стабилизированные полимерной мицеллой амфифильного сополимера стирола, 2-[(2-фенилаллилокси)метил]оксирана и тетраэтиленгликоль-моно-2-фенил-2 пропенилового эфира, проявляют высокую активность в реакциях гидрирования и Хека [181].
Активно развиваются исследования в области гибриднофазных катализаторов. Гибридные катализаторы на примере рассмотренных выше полимер-протектированных наночастиц металлов, закрепленных на неоганических оксидных носителях, демонстрируют улучшение их производительности (активности, селективности, продолжительности работы) и возможность рециклинга. Кроме того, им присущи дополнительные функции, такие как микроокружение и конфигурация, для реализации различных реакционных маршрутов и каскадных реакций для продуктов, которые проблематично получить другими способами. Интересны также подходы к разработке высокостабильных каталитических мезоструктур, в которых полимерная матрица, покрывающая поверхность металлической наноструктуры, может замещаться металлоксидным лигандом, выступающим в роли сокатализатора [183]. Ожидается, что такие структуры будут эффективны в каталитических реакциях с высокой энергией активации. В целом, принято считать, что лиганды, как в случае гомогенных катализаторов, оказывают значительное влияние на конверсию и селективность катализируемых реакций. Однако ввиду огромного числа типов лигандов (полимеры с разными функциональными группами, включая фосфор-, серу-, азотсодержащие и др.) и различных параметров, таких как длина цепи, конфигурация, стерические ограничения и др., общие заключения об их роли требуют более детальных систематических исследований.
Смешанные металлокластеры, в том числе и кластерсодержащие полимеры, являются уникальными молекулярными прекурсорами для получения би- и мультиметаллических катализаторов. Пространственная близость металлических центров обеспечивает мультицентровые взаимодействия с субстратом, приводя к активации молекул и тем самым создавая благоприятные условия для повышения каталитической активности. Cвязывание субстрата с М1 оказывает влияние на взаимодействие этого металла с М2, который в свою очередь выступает как “лиганд” для М1 и модифицирует его электронные взаимодействия с субстратом [57]. Полимер-иммобилизованные биметаллические наноструктуры, такие как Pt–Ru, Ru–Co, Pd–Ag, Pt–Ni, активны в реакциях получения водорода гидролизом аминоборанов [184], восстановлении нитроароматических соединений [185, 186], окисления формальдегида [187].
В последние годы привлекает внимание еще один способ гетерогенизации гомогенных катализаторов посредством внедрения молекулярных комплексов в структуру металл-органических каркасов (Metal organic frameworks, MOFs) или координационных полимеров, которые, как и цеолиты, из-за их уникальной пористой кристаллической структуры с развитой удельной поверхностью и большим внутренним объемом пор представляют значительный интерес для катализа [188–190]. Однородные по размеру поры и каналы в существенной степени обуславливают их каталитическую селективность, особенно в области энантиоселективного катализа [191]. В полости металлокаркасных структур могут быть получены и стабилизированы моноцентровые катализаторы на основе переходных металлов для реакций гидрирования алкенов, иминов, карбонилов и гетероциклов [192, 193]. Более того, сочетание синергетического и стерических эффектов, возникающих при введении Pt-SnOx в полость координационного Zn-имидазольного полимера (ZIF-8), способствовало дизайну высокоселективного катализатора гидрогенизации ненасыщенных альдегидов [194]. Среди других концептуальных приложений MOFs в катализе можно выделить каркасную стабилизацию активной частицы, сочетание катализа с химическим разделением, пост-синтетическое внедрение каталитических металлоцентров, субстрат-селективный катализ [195]. В недавнем обзоре [196] металлорганические каркасные структуры были проанализированы на предмет использования их как носителей для иммобилизации ферментов. MOFs с иерархической структурой можно рассматривать в качестве мультиэнзимного биореактора для ступенчатого закрепления ферментов в подходящих по размеру порах и каналах.
Гибридные катализаторы демонстрируют лучшую производительность и устойчивость их действия, обеспечивающих хорошую воспроизводимость и возможность масштабирования [197]. Дизайн молекулярной архитекуры полимерной матрицы с инкапсулированными наночастицами металлов способствует высокой селективности продуктов, в том числе диастерео- и энантоселективности. Однако задачи сочетания высокой активности и стабильности, 100%-ой селективности по целевому продукту и возможности рециклинга остаются еще открытыми и ждут своего решения, направленного на поиск оптимального баланса между ними. Разработка модульных каталитических систем, совмещающих в себе органические полимерные компоненты и неорганические нано- и субнаноразмерные частицы, в которых путем изменения различных параметров можно тонко регулировать их каталитические свойства, является сегодня актуальным и перспективным направлением.
Список литературы
Schmid G. Nanoparticles. From theory to application. Weinheim: Wiley, 2004. 533 p.
Nanotechnology in catalysis. Eds. Zhou B., Han S., Raja R., Somorjai G. New York: Kluwer Academic / Plenum Publisher, 2003. 342 p.
Pomogailo A.D., Dzhardimalieva G.I. Nanostructured Materials Preparation via Condensation Ways. Heidelberg: Springer, 2014. 460 p.
Uflyand I.E., Dzhardimalieva G.I. Nanomaterials Preparation by Thermolysis of Metal Chelates. Cham: Springer, 2018. 549 p.
Gross E., Toste F. D., Somorjai G.A. // Catal. Lett. 2015. V. 145. P. 126.
Somorjai G.A., Frei H., Park J.Y. // J. Am. Chem. Soc. 2009. V. 131. P. 16589.
Philippot K., Lignier P., Chaudret B. // Top. Organomet. Chem. 2014. V. 48. P. 319.
Pomogailo A.D. // React. Polym. 1989. V. 9. P. 109.
Савостьянов В.С., Помогайло А.Д., Пономарев А.Н. // Кинетика и катализ. 1989. Т. 30. № 6. С. 1414.
Pomogailo A.D., Uflyand I.E. // J. Mol. Catal. 1989. V. 55. P. 429.
Джардималиева Г.И., Помогайло А.Д. // Кинетика и катализ. 1998. Т. 39. № 6. С. 893.
Помогайло А.Д. // Кинетика и катализ. 2004. Т. 45. №1. С. 67.
Помогайло А.Д. // Высокомол. соедин. А. 2008. Т. 50. № 12. С. 2090.
Borodko Y., Thompson C.M., Huang W., Yildiz H.B., Frei H., Somorjai G.A. // J. Phys. Chem. C. 2011. V. 115. P. 4757.
Johansson M.J., Gorin D.J., Staben S.T., Toste F.D. // J. Am. Chem. Soc. 2005. V. 127. P. 18002.
Liu L., Corma A. // Chem. Rev. 2018. V. 118. P. 4981.
Chen Z., Liang Y., Jiaa D.S., Cui Z.M., Song W.G. // Chin. J. Catal. 2017. V. 38. P. 651.
Maity P., Yamazoe S. Tsukuda T. // ACS Catal. 2013. V. 3. P. 182.
Oliver-Meseguer J., Cabrero-Antonino J. R., Dominguez I., Leyva-Perez A., Corma A. // Science. 2012. V. 338. P. 1452.
Crespo-Quesada M., Andanson J.-M., Yarulin A., Lim B., Xia Y.N., Kiwi-Minsker L. // Langmuir. 2011. V. 27. P. 7909.
Zhao Y., Baeza J.A., Koteswara Rao N., Calvo L., Gilarranz M.A., Li Y.D., Lefferts L. // J. Catal. 2014. V. 318. P. 162.
Dong Y., Jin Y., Wang J., Zhang M. // Chem. Eng. J. 2017. V. 324. P. 303.
Albuquerque B.L., Denicourt-Nowicki A., Mériadec C., Domingos J.B., Roucoux A. // J. Catal. 2016. V. 340. P. 144.
Evangelisti C., Panziera N., D’Alessio A., Bertinetti L., Botavina M., Vitulli G. // J. Catal. 2010. V. 272. P. 246.
Chavda N., Trivedi A., Thakarda J., Agrawal Y. K., Maity P. // Catal. Lett. 2016. V. 146. P. 1331.
Dai Y., Yu P., Zhang X., Zhuo R. // J. Catal. 2016. V. 337. P. 65.
Gruttadauria M., Liotta L. F., Salvo A. M. P., Giacalone F., La Parola V., Aprile C., Noto R. // Adv.Synth. Catal. 2011. V. 353. P. 2119.
Liu X., Zhao X., Lu M. // Catal. Lett. 2015. V. 145. P. 1549.
Burguete M. I., García-Verdugo E., Garcia-Villar I., Gelat F., Licence P., Luis S. V., Sans V. // J. Catal. 2010. V. 269. P. 150.
Moreno-Marrodan C., Barbaro P., Catalano M., Taurino A. // Dalton Trans. 2012. V. 41. P. 12666.
Doherty S., Knight J.G., Backhouse T., Abood E., Alshaikh H., Fairlamb I.J.S, Bourne R.A., Chamberlain T.W., Stones R. // Green Chem. 2017. V. 19. P. 1635.
Doherty S., Knight J.G., Backhouse T., Bradford A., Saunders F., Bourne R.A., Chamberlain T.W., Stones R., Clayton A., Lovelock K. // Catal. Sci. Technol. 2018. V. 8. P. 1454.
Gonzalez-Moraga G. Cluster Chemistry. Berlin, Heidelberg: Springer–Verlag, 1993. 304 p.
Губин С.П. Химия кластеров. Основы классификации и строение. Москва: Наука, 1987. 263 с.
Bar-Sela G., Warchawsky A. // React. Polym. 1983. V. 1. P. 149.
Помогайло А.Д., Савостьянов В.С. Металлосодержащие мономеры и полимеры. Mосква: Химия, 1988. 384 с.
Kubeil M., Stephan H., Pietzsch H.-J., Geipel G., Appelhans D., Voit B., Hoffmann J., Brutschy B., Mironov Y.V., Brylev K.A., Fedorov V.E. // Chem. Asian J. 2010. V. 5. P. 2507.
Pomogailo S.I., Ershova V.A., Shilov G.V., Virovets A.V., Pogrebnyak V.M., Podberezskaya N.V., Golovin A.V., Dzhardimalieva G.I., Pomogailo A.D. // J. Organomet. Chem. 2005. V. 690. P. 4258.
Efremova O.A., Brylev K.A., Kozlova O., White M.S., Shestopalov M.A., Kitamura N., Mironov Y.V., Bauer S., Sutherland A.J. // J. Mater. Chem. C. 2014. V. 2. P. 8630.
Bradford A.M., Kristof E., Rashidi M., Yang D.-S., Payne N.C., Puddephatt R.J. // Inorg. Chem. 1994. V. 33. P. 2355.
Johnson B.F.G., Sanderson K.M., Shephard D.S., Ozkaya D., Zhou W., Ahmed H., Thomas M.D.R., Gladden L., Mantle M. // Chem. Commun. 2000. P. 1317.
Lucas N. T., Humphrey M. G., David Rae A. // Macromolecules. 2001. V. 34. P. 6188.
Pomogailo A.D. // Platinum Metals Rev. 1994. V. 38. №. 2. P. 60.
Помогайло А.Д. // Успехи химии. 1997. Т. 66. № 8. С. 750.
Ершова В.А., Головин А.В., Шелудяков Л.Ф., Семянников П.П., Помогайло С.И., Помогайло А.Д. // Изв. РАН. Сер. хим. 2000. № 8. С. 1455.
Максаков В.А., Кирин В.П., Конченко С.Н., Бравая Н.М., Помогайло А.Д., Вировец А.В., Подберезская Н.В., Барановская И.Г., Ткачев С.В. // Изв. АН. Серия хим. 1993. С. 1293.
Bravaya N.M., Pomogailo A.D. / In book: Metal-Containing Polymeric Materials. Eds. Pittman C.U., Jr., Carraher C.E., Jr., Zeldin M., Culberston B. New York: Plenum Publ. Corp., 1996. P. 51.
Голубева Н.Д., Адаменко О.А., Бойко Г.Н., Петрова Л.А., Ольхов Ю.А., Помогайло А.Д. // Неорг. матер. 2004. Т. 40. № 3. С. 363.
Адаменко О.А., Лукова Г.В., Голубева Н.Д., Смирнов В.А., Бойко Г.Н., Помогайло А.Д., Уфлянд И.Е. // Докл. Акад. Наук. 2001. Т. 381. № 3. С. 360.
Бравая Н.М., Помогайло А.Д., Максаков В.А., Кирин В.П., Белов Г.П., Соловьева Т.И. // Изв. РАН. Сер. хим. 1994. № 3. С. 423.
Бравая Н.М., Помогайло А.Д., Максаков В.А., Кирин В.П., Грачев В.П., Кузаев А.И. // Изв. Акад. Наук. Cер. хим. 1995. № 6. С. 1102.
Wong W.-T., Wong W.-Y., Yip C.-W. // J. Cluster Sci. 1995. V. 6. №. 2. P. 311.
Liang W., Li D., Lau P.-L., Xu L., Yang C., Wei M., Chan W.T.-K., Wong W.-Y. // J. Organomet. Chem. 2018. V. 870. P. 8.
Gressier J.C., Levesque G., Patin H. // Polym. Bull. 1982. V. 8. P. 55.
Voevodin A., Campos L. M., Roy X. // J. Am. Chem. Soc. 2018. V. 140. № 16. P. 5607.
Wang R., Zheng Z. // J. Am. Chem. Soc. 1999. V. 121. P. 3549.
Buchwalter P., Rose J., Braunstein P. // Chem. Rev. 2014. V. 115. № 1. P. 28.
Withers H.P., Seyferth D. // Inorg. Chem. 1983. V. 22. P. 2931.
Холуйская С.Н., Помогайло А.Д., Бравая Н.М., Помогайло С.И., Максаков В.А. // Кинетика и катализ. 2003. Т. 44. № 6. С. 831.
Помогайло С.И., Дорохов В.Г., Ляхович А.М., Михайлова С.С., Джардималиева Г.И., Помогайло А.Д. // Кинетика и катализ. 2006. Т. 47. № 5. С. 742.
Bhaduri S., Khwaja H. // J. Chem. Soc. Dalton Trans. I. 1983. № 2. P. 419.
Помогайло А.Д., Джардималиева Г.И. Металлополимерные гибридные нанокомпозиты. Москва: Наука, 2015. 494 с.
Hodge P., Sherrington D. Polymer-Supported Reactions in Organic Synthesis. New York: Wiley. 1980. 484 p.
Бектуров Е.А., Кудайбергенов С.Е. Катализ полимерами. Алма-Ата: Наука, 1988. 182 с.
Помогайло А. Д. Полимерные иммобилизованные металлокомплексные катализаторы. Москва: Наука, 1988. 303 с.
Bekturov E.A., Kudaibergenov S.E. Catalysis by Polymers. Heidelberg: Huthig and Wepf Verlag Zug., 1996. 153 p.
Бектуров Е.А., Кудайбергенов С.Е., Жармагамбетова А.К., Искаков Р.М., Ибраева Ж.Е., Шмаков С.Н. Полимер-протектированные наночастицы металлов. Алматы: Ин-т органического катализа и электрохимии им. Д.В. Сокольского, 2010. 274 с.
Ибраева Ж.Е., Кудайбергенов С. Е., Бектуров Е.А. Стабилизация наночастиц металлов гидрофильными полимерами. Saarbrücken: Lambert Academic Publishing, 2013. 376 p.
Помогайло А.Д., Розенберг А.С, Уфлянд И.Е. Наночастицы металлов в полимерах. Москва: Химия, 2000. 672 p.
Ибраева Ж.Е., Жармагамбетова А.К., Кетц И., Бектуров Е.А., Кудайбергенов С.Е. // Хим. журн. Казахстана. 2008. Т. 1. № 19. С. 245.
Закарина Н.А., Бектуров Е.А. // Изв. НАН РК. Сер. хим. 2008. № 1. С. 3.
Bergbreiter D.E., Case B.L., Liu Y-S., Caraway J.W. // Macromolecules. 1998. V. 31. P. 6053.
Mohan Y.M., Lee K., Premkumar T., Geckeler K.E. // Polymer. 2007. V. 48. P. 158.
François N.J., Allo S., Jacobo S.E., Daraio M.E. // J. Appl. Polym. Sci. 2007. V. 105. P. 647.
Metal clusters in catalysis. Eds. B.C. Gates, L. Guezi, H. Knosinger. Amsterdam: Elsevier, 1986. 234 p.
Shan J., Tenhu H. // Chem. Commun. 2007. V. 44. P. 4580.
Wang Y., Yan R., Zhang J.Z., Zhang W.Q. // J. Mol. Catal. A. 2010. V. 317. P. 81.
Park S., Murthy P.S.K., Park S., Mohan Y.M., Koh W.G. // J. Ind. Eng. Chem. 2011. V. 17. P. 293.
Mayer A.B.R., Mark J.E. // Eur. Polym. J. 1998. V. 34. P. 103.
Kudaibergenov S.E., Tatykhanova G.S., Baigaziyeva E. // Proc. Int. Conf. Nanomaterials: Applications and Properties, NAP-2012, Alushta, Crimea. 2012. V. 1. P. 3.
Yesmurzayeva N., Selenova B., Kudaibergenov S. // J. Am. Nanomaterials. 2013. V. 1. P. 1.
Kudaibergenov S.E., Baigaziyeva E.K., Yesmurzayeva N.N., Nurakhmetova Zh.A., Selenova B.S. / Proc. Int. Conference. Nanomaterials: Applications and Properties. Crimea. September 16–21, 2013. V. 2. № 2. P. 02PCN03.
Baygazieva E.K., Yesmurzayeva N.N., Tatykhanova G.S., Mun G.A., Khutoryanskiy V.V., Kudaibergenov S.E. // Int. J. Biol. Chem. 2014. V. 7. № 1. P. 14.
Kudaibergenov S.E., Tatykhanova G.S. // Int. J. Biol. Chem. 2014. V. 6. № 2. P. 40.
Ibrayeva Zh., Baigaziyeva E., Yesmurzayeva N., Tatykhanova G., Yashkarova M., Kudaibergenov S. // Macromol. Symp. 2015. V. 351. P. 51.
Yesmurzayeva N.N., Selenova B.S., Kudaibergenov S.E. // Supramol. Catal. 2015. V. 2. № 2. P. 1.
Nurgaziyeva E.K., Tatykhanova G.S., Mun G.A., Khutoryanskiy V.V., Kudaibergenov S.E. // Proc. Int. Conf. Nanomaterials: Applications and Properties. 2015. V. 4. P. 42.
A.C. № 69560, 2011.
Tatykhanova G., Mukazhanova Zh., Baigazieva E., Yashkarova M., Orazzhanova L., Abdullin Kh., Kudaibergenov S. // Proc. Int. Conf. Nanomaterials: Applications and Properties (NAP-2011). 2011. V. 1. Part 1. P. 165.
Hirai H., Ohtaki M., Komiyama M. // Chem. Lett. 1987. P. 149.
Toshima N. // Macromol. Symp. 2000. V. 156. P. 45.
Toshima N. // J. Macromol. Sci. Chem. 1990. V. 27. № 9–11. P. 1225.
Hirai H., Nakao Y., Toshima N. // J. Macromol. Sci. Chem. 1979. V. A13. № 6. P. 727.
Hirai H. // J. Macromol. Sci. Chem. 1979. V. A13. № 5. P. 633.
Toshima N., Wang Y. // Langmuir. 1994. V. 10. P. 4574.
Tsunoyama H., Ichikuni N., Tsukuda T. // Langmuir. 2008. V. 24. P. 11327.
Sakurai H., Tsunoyama H., Tsukuda T. // J. Organomet. Chem. 2007. V. 692. P. 368.
Mertens P.G.N., Vandezande P., Ye X., Poelman H., Vankelecom I.F.J., De Vos D.E. // Appl. Catal. A. Gen. 2009. V. 355. P. 176.
Karakhanov E., Maximov A., Zolotukhina A., Mamadli A., Vutolkina A., Ivanov A. // Catalysts. 2017. V. 7. P. 12.
Lang H., May R.A., Iversen B.L., Chandler B.D. // J. Am. Chem. Soc. 2003. V. 125. № 48. P. 14832.
Scott R.W.J., Wilson O.M., Crooks R.M. // J. Phys. Chem. B. 2005. V. 109. № 2. P. 692.
Zhang W., Li L., Du Y., Wang X., Yang P. // Catal. Lett. 2009. V. 127. № 3–4. P. 429.
Ornelas C., Aranzaes J.R., Salmon L., Astruc D. // Chem. Eur. J. 2008. V. 14. P. 50.
Ornelas C., Ruiz Aranzaes J., Cloutet E., Alves S., Astruc D. // Angew. Chem. 2007. V. 119. P. 890.
Deraedt C., Salmon L., Astruc D. // Adv. Synth. Catal. 2014. V. 356. P. 2525.
Peng X., Pan Q., Rempel G.L. // Chem. Soc. Rev. 2008. V. 37. P. 1619.
Jin Z., Xiao H., Zhou W., Zhang D., Peng X. // R. Soc. Open Sci. 2017. V. 4. P. 1714.
Zharmagambetova A.K., Golodov V.A., Saltykov Yu.P. // J. Mol. Catal. 1989. V. 55. P. 406.
Zharmagambetova A.K., Mukhamedzhanova S.G., Bekturov E.A. // Reactive Polym. 1994. V. 24. P. 17.
Zharmagambetova A.K., Mukhamedzhanova S.G., Dusenbina B. // Reactive Polym. 1994. V. 24. P. 21.
Жармагамбетова А.К., Мухамеджанова С.Г., Бектуров Е.А. / Полимерные электролиты, гидрогели, комплексы и катализаторы. Алматы: Ин-т органического катализа и электрохимии им. Д.В. Сокольского, 2007. С. 224.
Somorjai G.A., Contreras A.M., Montano M., Rioux Robert M. // PNAS of the USA. 2006. V. 103. № 28. P. 10 577.
Hoefelmeyer J.D., Niesz K., Somorjai G.A., Don Tilley T. // Nano Lett. 2005. V. 5. № 3. P. 435.
Rioux R. M., Song H., Hoefelmeyer J. D., Yang P., Somorjai G.A. // J. Phys. Chem. B. 2005. V. 109. № 6. P. 2192.
Li Y., Hong X.M., Collard D.M., El-Sayed M.A. // Org. Lett. 2000. V. 2. P. 2385.
Narrayanan R., El-Sayed M.A. // J. Am. Chem. Soc. 2003. V. 125. P. 8340.
Metin O., Sahin S., Ozkar S. // Int. J. Hydrogen Energy. 2009. V. 34. P. 6304.
Metin O., Durap F., Aydemir M., Ozkar S. // J. Mol. Catal. A: Chem. 2011. V. 337. P. 39.
Umegaki T., Yan J.-M., Zhang X.-B., Shioyama H., Kuriyama N., Xu Q. // Int. J. Hydrogen Energy. 2009. V. 34. P. 3816.
Kudaibergenov S., Nueraje N., Khutoryanskiy V. // Soft Matter. 2012. V. 8. P. 9302.
Kudaibergenov S.E., Dolya N., Tatykhanova G., Ibraeva Zh. E., Musabaeva B.Kh., Yashkarova M.G., Bimendina L.A. // Euras. Chem. Technol. J. 2007. V. 9. № 3. P. 177.
Доля Н., Мусабаева Б.Х., Яшкарова М.Г., Бимендина Л.А., Кудайбергенов С.Е. // Вестн. НАН РК. 2007. № 2. С. 20.
Доля Н.А., Яшкарова М.Г., Мусабаева Б.Х., Жармагамбетова А.К., Кудайбергенов С.Е. // Изв. научно-технического общества “Кахак”. 2007. №17. С. 231.
Svetlichnyi D.S., Dolya N., Govenko P., Musabaeva B.Kh., Yashkarova M.G., Kudaibergenov S.E. // Euras. Chem. Technol. J. 2007. № 10. P. 41.
Доля Н.А., Светличный Д.С., Калиаскарова Б.А., Мусабаева Б.Х., Яшкарова М.Г., Жармагамбетова А.К., Кетц И., Кудайбергенов С.Е. // Докл. НАН РК. 2008. №3. С.67.
Kudaibergenov S. E., Ibraeva Zh. E., Dolya N. A., Musabaeva B.Kh., Zharmagambetova A.K., Koetz J. // Macromol. Symp. 2008. V. 274. P. 11.
Доля Н.А., Жармагамбетова А.К., Мусабаева Б.Х., Кудайбергенов С.Е. // Вестн. НАН РК. 2008. № 1. С. 55.
Доля Н.А., Мусабаева Б.Х., Яшкарова М.Г., Кудайбергенов С.Е. // Хим. журн. Казахстана. Спец. вып. 2008. № 21. С. 139.
Kudaibergenov S.E., Tatykhanova G.S. // Int. J. Biol. Chem. 2014. V. 6. № 2. P. 40.
Kudaibergenov S.E., Tatykhanova G.S., Selenova B.S. // J. Inorg. Organomet. Polym. Mater. 2016. P. 1.
Dolya N., Kudaibergenov S. / Temperature-Responsive Polymers: Chemistry, Properties and Applications. Ed. Khutoryanskiy V. London: John Wiley & Sons Ltd., 2018. P. 357.
Okay O., Lozinsky V.I. // Adv. Polym. Sci. 2014. V. 263. P. 103.
Sahiner N., Seven F. // Energy. 2014. V. 71. P. 170.
Sahiner N., Yildiz S. // Fuel Process. Technol. 2014. V. 126. P. 324.
Sahiner N., Seven F., Al-lohedan H. // Water Air and Soil Pollution. 2015. V. 226. № 4. P. 10.
Ajmal M., Demirci S., Siddiq M., Aktas N., Sahiner N. // Colloids and Surfaces. A: Physicochemical and Engineering Aspects. 2015. V. 486. P. 29.
Sahiner N., Yildiz S., Al-Lohedan H. // Appl. Catal. B: Env. 2015. V. 166. P. 145.
Sahiner N., Yasar A. O. // Fuel Process. Technol. 2016. V. 144. P. 124.
Yildiz S., Sahiner M., Sahiner N. // Eur. Polym. J. 2015. V. 70. P. 66.
Demirci S., Sahiner N. // Water Air and Soil Pollution. 2015. V. 226. № 3. P. 64.
Klivenko A.N., Tatykhanova G.S., Nuraje N., Kudaibergenov S. // Bull. Karaganda University. Ser. Chem. 2015. V. 80. № 4. P. 10.
Tatykhanova G.S., Klivenko A.N., Kudaibergenova G.M., Kudaibergenov S.E. // Macromol. Symp. 2016. V. 363. № 1. P. 49.
Kudaibergenov S. / Proc. IX Int. Bermzhan Symp. on Chemistry and Technology. Almaty, December 9–10, 2016. P. 21.
Klivenko A.N., Yergazieva E., Kudaibergenov S.E. / Proc. Int. Conf. Nanomaterials: Application & Properties (NAP), 2016. P. 02NSA03-1-02NSA03-5. https://doi.org/10.1109/NAP.2016.7757304
Kudaibergenov S., Aldabergenov M., Dauletbekova M., Kabdrakhmanova S., Ibrayeva Zh., Selenova B. / Proc. World Congress of Engineers and Scientists. WSEC-2017. Astana, 2017. V. 3. P. 32.
Aldabergenov M., Dauletbekova M., Toleutay G., Klivenko A., Kudaibergenov S. / Proc. 7th International Conference Nanomaterials: Application and Properties (NAP-2017). Zatoka, Odessa, September 10–15, 2017. P. 03NNSA27-1-03NNSA27-3. https://doi.org/10.1109/NAP.2017.8190273
Aldabergenov M., Dauletbekova M., Shakhvorostov A., Toleutay G., Klivenko A., Kudaibergenov S. // J. Chem. Techn. Metallurgy. 2018. V. 53. № 1. P. 17.
Kudaibergenov S., Dauletbekova M., Toleutay G., Kabdrakhmanova S., Seilkhanov T., Abdullin Kh. // J. Inorg. Organomet. Polym. 2018. V. 28. P. 2427.
Muniz-Miranda M. // J. Anal. Bioanal. Technol. 2015. V. 6. P. 1.
Shibasaki Y., Abe Y., Sato N., Fujimori A., Oishi Y. // Polymer J. 2010. V. 42. P. 72.
Липатов Ю.С., Сергеева Л.М. Адсорбция полимеров. Киев: Наукова думка, 1972. 195 с.
Парфит Г., Рочестер К. Адсорбция из растворов на поверхностях твердых тел. Москва: Мир, 1986. 488 с.
Липатов Ю.С. Межфазные явления в полимерах. Киев: Наукова думка, 1980. 260 с.
Усанов А.Е., Демиденко Г.Н., Михайлов И.А., Чернышов Д.М. / Тез. докл. 12-ой Международной конференции молодых ученых по химии и химической технологии – МКХТ-98. Москва, 1998. С. 4.
Усанов А.Е., Семагина Н.В., Демиденко Г.Н., Михайлов И.А. / Тез. Докл. 5-ой Международной конференции. Наукоемкие химические технологии. Ярославль, 1998. С. 190.
Санников О.Б., Попов О.С., Сульман Э.М., Анкудинова Т.В., Шахова М.К. / Вопросы кинетики и катализа: Межвузовский сборник, Ивановский хим-тех институт, 1980. С. 108.
Сульман Э.М., Попов О.С., Самохвалов Г.И. / Катализ и катал. процессы производства химико-фармокологических препаратов. Москва, 1985. Т. 2. С. 93.
Сидоров А.И., Сульман Э.М., Бронштейн Л.М., Анкудинова Г.В., Автушенко Ю.Е., Мирзоева Е.Ш., Баукова Е.Ю., Валецкий П.М. // Кинетика и катализ. 1993. Т. 34. № 1. С. 87.
Mirzoeva E.Sh., Bronstein L.M., Valetsky P.M., Sulman E.M. // Reactive Polymers. 1995. V. 24. P. 243.
Sulman E., Bodrova Y., Matveeva V., Semagina N., Cerveny L., Kurtc V., Bronstein L., Platonova O., Valetsky P. // Appl. Catal. A: Gen. 1999. V. 76. № 1. P. 75.
Bronstein L., Chernyshov D., Volkov I., Ezernitskaya M., Valetsky P., Matveeva V., Sulman E. // J. Catal. 2000. V. 196. № 2. P. 302.
Semagina N, Joannet E, Parra S., Sulman E., Renken A., Kiwi-Minsker L. // App. Catal. A: Gen. 2005. V. 280. P. 141.
Пaт. PK № 22029, 2009.
Жармагамбетова А.К. / В кн. Бектуров Е.А., Кудайбергенов С.Е., Жармагамбетова А.К., Искаков Р.М., Ибраева Ж.Е., Шмаков С.А. Полимер-протектированные наночастицы металлов. Алматы, 2010. С. 231.
Голубева Н.Д., Дюсеналин Б.К., Селенова Б.С., Помогайло А.Д., Жармагамбетова А.К., Джардималиева Г.И. // Кинетика и катализ. 2011. Т. 52. № 2. С. 250.
Жармагамбетова А.К., Сейткалиева К.С., Талгатов Э.Т., Ауэзханова А.С., Джардималиева Г.И., Помогайло А.Д. // Кинетика и катализ. 2016. Т. 57. №. 3. С. 362.
Жармагамбетова А.К., Заманбекова А.Т., Мухамеджанова С.Г., Тумабаев Н.Ж. // Изв. НАН РК. Сер. хим. 2009. № 4. С. 61.
Жармагамбетова А.К., Заманбекова А.Т., Дарменбаева А.С., Ауезханова А.С., Джумекеева А.И., Талгатов Э.Т. // Теорет. эксп. химия. 2017. Т. 53. № 4. С. 250.
Есмурзаева Н.Н., Байгазиева Э.К., Нурахметова Ж.А., Ержанова Д.С., Селенова Б.С., Кудайбергенов С.Е. // Тез. докл. II Российского конгресса по катализу. Самара, 2–5 октября, 2014. С. 55.
Джардималиева Г.И., Дорохов В.Г., Голубева Н.Д., Помогайло С.И., Ляхович А.М., Савченко В.И., Помогайло А.Д. // Изв. РАН. Сер. хим. 2009. № 10. С. 2007.
Pomogailo A.D., Dzhardimalieva G.I. // J. Catal. 2013. P. 12. http:/dx.doi.org/.https://doi.org/10.1155/2013/276210
Kalinina K.S., Golubeva N.D., Dzhardimalieva G.I., Pomogailo A.D. // Macromol. Symp. 2015. V. 351. № 1. P. 1.
Помогайло А.Д., Калинина К.С., Голубева Н.Д., Джардималиева Г.И., Помогайло C.И., Кнерельман Е.И., Протасова С.Г., Ионов А.М. // Кинетика и катализ. 2015. Т. 56. № 5. С. 704.
Huang W., Kuhn J. N., Tsung C. K., Zhang Y., Habas S. E., Yang P., Somorjai G. A. // Nano Lett. 2008. V. 8. P. 2027.
Deraedt C., Melaet G., Ralston W. T., Ye R., Somorjai G.A. // Nano Lett. 2017. V. 17. P. 1853.
Ye R., Yuan B., Zhao B., Ralston W.T., Wu C.-Y., Unel Barin E., Toste D.F., Somorjai G.A. // J. Am. Chem. Soc. 2016. V. 138. P. 8533.
Alayoglu S., Aliaga C., Sprung C., Somorjai G.A. // Catal. Lett. 2011. V. 141. P. 914.
Toshima N., Shiraishi Y., Teranishi T. // J. Mol. Catal. A: Chem. 2001. V. 177. P. 139.
Liu L., Corma A. // Chem. Rev. 2018. V. 118. P. 4981.
Oliver-Meseguer J., Leyva-Perez A., Al-Resayes S.I., Corma A. // Chem. Commun. 2013. V. 49. P. 7782.
Okamoto K., Akiyama R., Yoshida H., Yoshida T., Kobayashi S. // J. Am. Chem. Soc. 2005. V. 127. P. 2125.
Menezes W.G., Zielasek V., Dzhardimalieva G.I., Pomogailo S.I., Thiel K., Wohrle D., Hartwig A., Baumer M. // Nanoscale. 2012. V. 4. P. 1658.
An K., Alayoglu S., Musselwhilte N., Plamthottam G., Malaet G., Lindeman A.E., Somorjai G.A. // J. Am. Chem. Soc. 2013. V.135. P. 16689.
Rakap M. // Appl. Catal. A: Gen. 2014. V. 478. P. 15.
Udumula V., Tyler J.H., Davis D.A., Wang H., Linford M.R., Minson P. S., Michaelis D. J. // ACS Catal. 2015. V. 5. P. 3457.
Ranjith K.S., Celebioglu A., Uyar T. // Nanotechnol. 2018. V. 29. 245602. https://doi.org/10.1088/1361-6528/aab9da
He F.-G., Du B., Sharma G., Stadler F.J. // Polymers. 2019. V. 11. P. 674.
Wang Z., Chen G., Ding K. // Chem. Rev. 2008. V. 109. P. 322.
Corma A., Garcia H., Xamena F.X.L. // Chem. Rev. 2010. V. 110. P. 4606.
Chizallet C., Lazare S., Bazer-Bachi D., Bonnier F., Lecocq V., Soyer E., Quoineaud A., Bats N. // J. Am. Chem. Soc. 2010. V. 132. P. 12 365.
Ma L., Abney C., Lin W. // Chem. Soc. Rev. 2009 V. 38. P. 1248.
Ji P., Manna K., Lin Z., Urban A., Greene F.X., Lan G., Lin W. // J. Am. Chem. Soc. 2016. V. 138. P. 12 234.
Cohen S.M., Zhang Z., Boissonnault J.A. // Inorg. Chem. 2016. V. 55. P. 7281.
Lan X., Xue K., Wang T. // J. Catal. 2019. V. 372. P. 49.
Lee J.Y., Farha O.K., Roberts J., Scheidt K.A., Nguyen S.T., Hupp J.T. // Chem. Soc. Rev. 2009. V. 38. P. 1450.
Drout R.J., Robison L., Farha O.K. // Coord. Chem. Rev. 2019. V. 381. P. 151.
Kudaibergenov S. // Gels. 2019. V. 5. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ