

Кинетика и катализ, 2020, T. 61, № 2, стр. 151-167
Необычный механизм реакции смешанных фосфониево-иодониевых илидов с ацетиленами
Т. Д. Некипелова a, *, Т. А. Подругина b
a ФГБУН Институт биохимической физики им. Н.М. Эмануэля РАН
119334 Москва, ул. Косыгина, 4, Россия
b ФГБОУ ВО Московский Государственный Университет им. М.В. Ломоносова, Химический факультет
119991 Москва, ГСП-1, Ленинские горы, 1, стр. 3, Россия
* E-mail: nekip@sky.chph.ras.ru
Поступила в редакцию 27.09.2019
После доработки 27.09.2019
Принята к публикации 28.10.2019
Аннотация
Реакции смешанных фосфониево-иодониевых илидов с нитрилами и ацетиленами позволяют синтезировать труднодоступные и новые гетероциклические соединения в простой one-pot, metal-free системе. В обзоре представлены результаты исследования механизма реакции фосфониево-иодониевых илидов с ацетиленами методами спектрофотометрии, ЭПР- и ЯМР-спектроскопии, которые позволили объяснить некоторые необычные закономерности протекания этих реакций: период индукции, кислотный катализ, эффект химической поляризации ядер (ХПЯ), наблюдаемый в некоторых системах, и другие. Однозначно установлены радикальный характер инициирования реакции в результате кислотного автокатализа распада исходных илидов на радикалы и участие радикальных интермедиатов в образовании всех целевых продуктов. Предложен механизм образования радикальных пар при наблюдении ХПЯ и установлена роль микрогетерогенности растворов илидов в хлористом метилене. На основании проведенного исследования механизма реакции могут быть оптимизированы условия реакции для увеличения выходов новых гетероциклических соединений.
ВВЕДЕНИЕ
Структурные особенности илидов обуславливают их большие синтетические возможности и позволяют активно использовать эти соединения в тонком органическом синтезе. Класс смешанных илидов – соединений, содержащих два положительно заряженных гетероатома у отрицательно заряженного углеродного центра, по-прежнему остается наименее изученным. Наличие нескольких реакционных центров в смешанных илидах объясняет возможность их эффективного применения в структурном дизайне различных классов органических соединений. Фосфониево-иодониевые илиды впервые были получены О. Нейландом и Г. Ванагом в 1964 г. [1] и 20 лет спустя Р. Мориарти (R. Moriarty) с сотр. [2] окислением стабилизированных фосфоранов с помощью фенилиодозоацетата с последующим введением таких противоионов, как галогены или борфторид анион. До начала 2000-х гг. изучение фосфониево-иодониевых илидов носило фрагментарный характер и ограничивалось единичными примерами [3, 4]. В начале 2000-х гг. под руководством академика Н.С. Зефирова на Химическом факультете МГУ им. М.В. Ломоносова были начаты систематические исследования по разработке методов синтеза и изучению свойств смешанных фосфониево-иодониевых илидов, общая формула которых представлена ниже [5–7]:
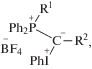
где R1 = Ph,  (X = S, NH, O; Y = N); R2 = = CO(Ph), CO(OMe), CO(OEt), PO(OEt)2, CN, S(O)2(p-MeC6H4).
(X = S, NH, O; Y = N); R2 = = CO(Ph), CO(OMe), CO(OEt), PO(OEt)2, CN, S(O)2(p-MeC6H4).
При сравнении свойств смешанных илидов было выявлено, что их стабильность зависит от характера акцепторных групп у илидного атома углерода, уменьшаясь при переходе от карбоалкокси- к бензоил- и далее к цианозамещенному илиду, и от природы противоиона: тетрафторборат анион в наибольшей степени стабилизирует молекулу [5]. В результате проведенных исследований было показано неаддитивное сочетание свойств фосфониевого и иодониевого фрагментов. Налицо синергия свойств “ониевых” заместителей в молекулах смешанных илидов, приводящая к появлению новых процессов и созданию новых классов соединений на их основе, обладающих необычными и неизвестными ранее свойствами [6–10, 12, 13]. Поскольку фенилиодониевый фрагмент является хорошо уходящей группой для процессов нуклеофильного замещения, фосфониево-иодониевые илиды легко вступают в реакцию нуклеофильного замещения, открывая путь к труднодоступным фосфоранам. В качестве нуклеофилов могут выступать галогениды, N-, О-, S-, Se-нуклеофилы [5, 7, 4].
Структура смешанных илидов, содержащих карбонильные и карбоксильные группы, участвующие в делокализации отрицательного заряда у илидного атома углерода, описывается несколькими резонансными структурами (схема 1):
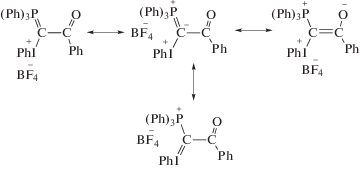
Схема 1.
Данные рентгеноструктурного анализа (РСА) подтверждают существенную двоесвязность между илидным атомом углерода и атомом углерода стабилизирующего заместителя [8]. Исследование зависимости характера спектров ЯМР смешанных илидов от температуры показало существование динамического равновесия между двумя геометрическими изомерами, что наряду с данными РСА говорит в пользу значительного вклада структуры с зарядом, локализованным на атоме кислорода. Это объясняет тот факт, что смешанные илиды с карбонильными и карбоксильными заместителями успешно выступают в роли О-нуклеофилов в реакциях алкилирования, силилирования и ацилирования [9]. Иодониевая группа, как еще один акцептор у илидного атома углерода, не позволяет смешанным илидам вступать в реакцию Виттига с карбонильными соединениями. Но для них возможна тандемная реакция замещения и Виттига, приводящая к α-замещенным сопряженным сложным эфирам или кетонам [7, 9].
Наиболее интересными и важными с точки зрения использования в тонком органическом синтезе являются недавно открытые фотохимические реакции гетероциклизации смешанных илидов с соединениями, содержащими С≡С- и С≡N-связи. Смешанные фосфониево-иодониевые илиды с карбоалкоксильной и карбонильной группами в условиях УФ-облучения реагируют с алифатическими, ароматическими и функционально замещенными нитрилами, образуя оксазолилзамещенные фосфониевые соли (схема 2). Эта реакция рассматривается как формальное [3 + 2] циклоприсоединение илидов по связи C≡N [10, 11].
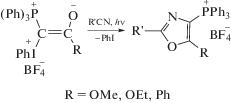
Схема 2.
Смешанные фосфониево-иодониевые илиды взаимодействуют с ацетиленами, не содержащими акцепторные заместители, образуя два типа гетероциклических соединений: фосфорсодержащие гетероциклические соединения (λ5-фосфинолины (3)) и фосфонийзамещенные фураны (4) (схема 3) [13, 14].
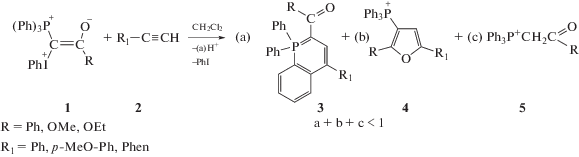
Схема 3.
Эта реакция была первоначально открыта при фотолизе смеси светом с длиной волны λ ≤ 400 нм, однако в дальнейшем было показано, что для некоторых пар илид–ацетилен взаимодействие происходит без облучения после смешения реагентов. Схема 3 приведена для наиболее исследованных карбонил- и карбоксилзамещенных трифенилфосфониевых илидов; в этой и последующих схемах противоион ${\text{BF}}_{4}^{ - }$ опущен для простоты, поскольку продуктов, содержащих B или F, не было обнаружено. Фосфониевая соль (5), являющаяся основным продуктом при фотолизе собственно илидов, в смесях с ацетиленами становится минорным продуктом.
Соотношение гетероцикличеких продуктов для конкретного илида зависит от заместителя при тройной связи в ацетилене: алкины с алкильными и фенильными заместителями образуют преимущественно λ5-фосфинолины, а ацетилены с электронодонорными заместителями (активированный гетероцикл, OCH3, полиядерные ароматические системы) наряду с фосфинолинами образуют фосфонийзамещенные фураны, причем в приведенном ряду соотношение продуктов увеличивается в пользу фуранового производного (табл. 1).
Таблица 1.
Выходы продуктов фотолиза смеси бензоильного илида 1a с ацетиленами R1С≡СH [13, 14]
| R1 | Продукты | |
|---|---|---|
| 3 | 4 | |
| Ph | 60 | Cледы |
| C8H17 | 50 | Cледы |
| Тиофен | 60 | 7 |
| п-Метоксифенилацетилен | 30–40 | 40–30 |
| п-Метоксинафтилацетилен | 10 | 60 |
| 9-Этинилфенантрен | 5 | 80 |
На основании зависимости выхода гетероциклических продуктов от энергии ионизации ацетиленов был предположен электрофильный характер фотохимической гетероциклизации и предложена возможная схема такого процесса (схема 4) [14]. Этот механизм предполагает гетеролитический разрыв связи C–I+Ph с отщеплением PhI при облучении и образование λ5-фосфинолина 3 и производного фурана 4 из промежуточного карбкатиона или карбена (A1) в параллельных реакциях (a) и (b). Однако участие карбена было отвергнуто ввиду отсутствия циклопропанового производного при фотолизе и термолизе илида со стиролом [13].
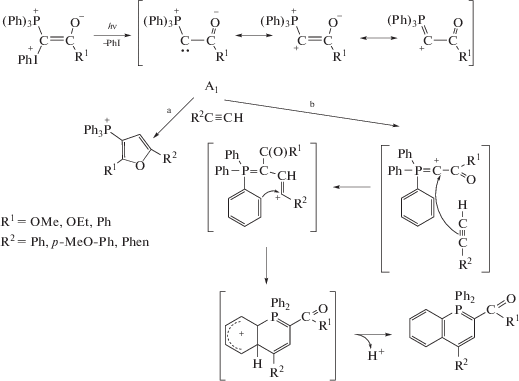
Схема 4.
При замене в фосфониевом фрагменте смешанного илида одного из бензольных колец на электроноизбыточный гетероцикл происходит циклизация с участием гетероцикла, приводящая к новому классу конденсированных гетероароматических систем – фосфининофуранам и фосфининотиофенам, что также логично укладывается в схему электрофильного механизма этого процесса [14–16].
Обычно механизм реакций органического синтеза постулируют на основании анализа продуктов, большого объема эмпирических закономерностей, соотношений структура–реакционная способность и исследования взаимодействий отдельных компонентов реакции. В этом смысле реакции между смешанными фосфониево-иодониевыми илидами и соединениями с тройной связью, ацетиленами и нитрилами, являются одним из немногочисленных примеров, когда оказывается возможным непосредственно следить за ходом реакции синтеза несколькими физико-химическими методами. Надо отметить, что помимо чисто фундаментального интереса к необычному с точки зрения кинетических закономерностей процессу в этой реакции удается достаточно легко в one-pot, metal-free системе при комнатной температуре синтезировать фосфорсодержащие гетероциклы. В последние годы резко возросло количество публикаций по синтезу соединений с атомом фосфора в цикле в связи с возможностью их использования в качестве функциональных материалов для органической электроники и для визуализации и анализа в биологических системах [17–20]. Производные фурана и оксазола представляют интерес как фармакофорные соединения [21].
В настоящем обзоре представлены результаты исследования механизма взаимодействия смешанных илидов 1a–1f, структура которых представлена ниже, с фенилацетиленом (2a), п-метоксифенилацетиленом (2b) и 9-этинилфенантреном (2c) методами спектрофотометрии, ЭПР- и ЯМР-спектроскопии с привлечением масс-спектрометрии и динамического рассеяния света.
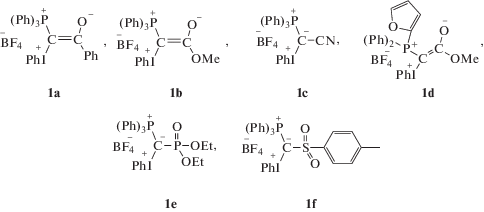
ИССЛЕДОВАНИЕ МЕХАНИЗМА ВЗАИМОДЕЙСТВИЯ СМЕШАННЫХ ФОСФОНИЕВО-ИОДОНИЕВЫХ ИЛИДОВ С АЦЕТИЛЕНАМИ
Экспериментальные противоречия предложенному механизму
Эффект растворителя. К сожалению, предложенный в [14] механизм (схема 4) не объясняет экспериментально наблюдаемые особенности протекания реакции. Эти особенности не типичны для гомогенных реакций органического синтеза в растворах. Первая из них – это уникальность растворителя: взаимодействие ацетиленов с фосфониево-иодониевыми илидами с образованием целевых гетероциклических соединений происходит только в хлористом метилене при концентрациях илида, превышающих 0.005–0.01 моль/л (рис. 1). При этих концентрациях раствор илида в CH2Cl2 не является истинно гомогенным: он представляет собой устойчивую суспензию со средним диаметром частиц, определенным методом динамического рассеяния света, около 800 нм при концентрациях илида 0.010–0.15 моль/л [22]. Критическая зависимость реакции образования целевых гетероциклов от концентрации илида совпадает с резким увеличением размера его частиц в растворителе при концентрации илида около 0.01 моль/л. Следует отметить, что в других растворителях (спирты, насыщенные и ароматические углеводороды, C2H4Cl2) илиды плохо растворяются, не образуют микросуспензию и выпадают в виде белого кристаллического осадка, в то время как суспензия в CH2Cl2 окрашена в светло-желтый цвет, хотя кристаллы илида белые. Илиды хорошо растворяются в ДМФА и ацетонитриле, при этом концентрированные растворы в этих растворителях также светло-желтые. Однако реакция с ацетиленами в ДМФА не протекает. Причины этого будут обсуждены далее. При фотолизе смеси илидов с ацетиленами в ацетонитриле происходит конкурентная реакция с тройной связью растворителя с образованием производного оксазола (схема 2) [10, 11].
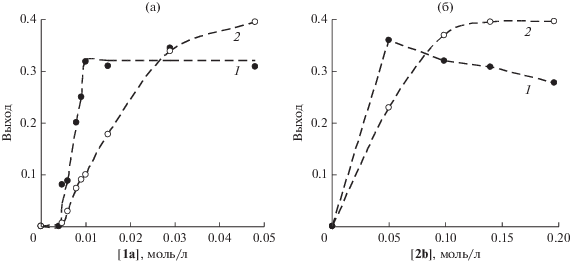
Рис. 1.
Зависимости выходов λ5-фосфинолина 3 (1) и фуранового производного 4 (2) от концентрации 1a при постоянной концентрации 2b (0.14 моль/л) (а) и от концентрации 2b при постоянной концентрации 1a (0.05 моль/л) (б).
При фотолизе бензоильного илида 1а в фенилацетилене, который был одновременно реагентом и растворителем (навеска соответствовала общей концентрации илида 0.02 моль/л, однако бóльшая часть илида присутствовала в виде белого осадка), после периода индукции (около 1 ч) осадок спонтанно растворялся, и раствор приобретал коричневую окраску. Анализ конечных продуктов показал, что выход λ5-фосфинолина 3, продукта присоединения фенилацетилена, был существенно ниже, чем при аналогичных концентрациях илида в CH2Cl2.
Таким образом, реакция взаимодействия смешанных илидов с ацетиленами происходит в CH2Cl2 при концентрациях реагентов, обеспечивающих образование относительно крупных частиц.
Концентрационная зависимость выхода гетероциклических продуктов. Предложенная ранее схема взаимодействия между исследуемыми илидами и ацетиленами предполагает образование целевых гетероциклических соединений в параллельных реакциях взаимодействия карбкатиона с ацетиленом (схема 4). Это означает, что соотношение выходов λ5-фосфинолина 3 и фуранового производного 4 должно определяться соотношением констант скорости конкурирующих реакций (a) и (b). Однако экспериментальные данные о концентрационных зависимостях выходов продуктов в системе илид 1a–ацетилен 2b демонстрируют разные закономерности для 3 и 4 (рис. 1). При постоянной избыточной начальной концентрации ацетилена ([2b]0 = 0.14 моль/л) и меняющейся концентрации 1a в диапазоне 0.005–0.05 моль/л выход 3 достигает максимального значения (32%) уже при [1a]0 = 0.01 моль/л и далее практически не изменяется. При этом выход 4 растет с увеличением концентрации илида. Если зафиксировать постоянную начальную концентрацию илида ([1a] = 0.05 моль/л), то при повышении концентрации 2b выход 3 слегка уменьшается, а выход 4 сначала возрастает и достигает предельного значения при соотношении концентраций [2b]0/[1a]0 = 3.
Этот результат показывает, что гетероциклы 3 и 4 не могут образовываться в параллельных реакциях из общего предшественника. Однако он имеет практическое значение, поскольку, меняя соотношение концентраций исходных веществ, можно изменять соотношение выходов целевых гетероциклических продуктов.
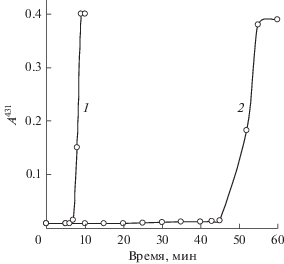
Период индукции. Важной особенностью реакции является наличие периода индукции [23], что также не находит объяснения в рамках предложенного ранее механизма. В случае смеси илида 1а с фенилацетиленом 2а через 5–20 мин после начала фотолиза (350 < λвозб < 450 нм) происходит спонтанное развитие реакции, которая начинается локально либо на гетерогенной частице илида 1а, либо на стенке кюветы, либо на поверхности раствора, что проявляется в появлении пятна яркой коричневой окраски. В течение 1–2 мин реакция распространяется на весь объем раствора. Конечный раствор имеет коричневый цвет и представляет собой практически гомогенную смесь продуктов реакции. К этому моменту илид полностью израсходовался, хотя продолжаются превращения промежуточных продуктов, и через 1–2 ч раствор становится желтым. Интересно отметить, что в конце периода индукции облучение может быть отключено, и реакция происходит в его отсутствии [23]. Если в момент выхода из периода индукции раствор перемешать, то реакция останавливается и наступает новый период индукции. Если фотолиз проводить при постоянном перемешивании, то период индукции увеличивается (рис. 2). Необычная зависимость от перемешивания означает, что вокруг частицы илида в периоде индукции формируется реакционная среда, которая разрушается при перемешивании. Продолжительность периода индукции зависит также от интенсивности облучения и чистоты илида и растворителя.
Рис. 2.
Изменение поглощения (λрег = 431 нм) смеси 1a (0.012 моль/л) + 2a (0.07 моль/л) при фотолизе (λвозб = 400 нм) без перемешивания (1) и с перемешиванием (2), растворитель CH2Cl2; для снятия спектров поглощения пробы раствора разбавляли в 100 раз спиртом.
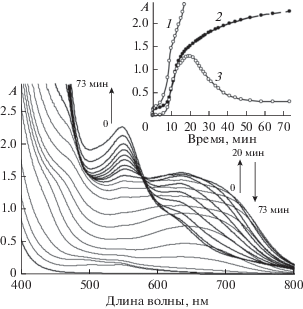
Интересные результаты были получены для смеси илида 1a с п-метоксифенилацетиленом 2b. Для вышеуказанной пары реакция происходит не только при облучении, но и без облучения смеси реагентов сразу после их смешения [24]. При этом состав продуктов практически не зависит от облучения, а период индукции уменьшается от нескольких минут без облучения до 0.2–0.5 мин при облучении. После смешения реагентов первоначально светло-желтый раствор сначала розовеет, затем становится изумрудно-зеленым, который переходит в коричневый цвет конечного раствора. Изменение окраски в процессе реакции было зарегистрировано спектрофотометрически (рис. 3). Из рис. 3 видно, что процесс образования конечных продуктов происходит в результате сложных превращений интермедиатов. Данный вопрос более детально будет рассмотрен ниже. Однако и в этом случае есть период автоускорения реакции. Периоды индукции наблюдали также для других пар смешанных илидов с ацетиленами.
Рис. 3.
Эволюция спектров поглощения смеси илид 1a (0.01 моль/л) + п-метоксифенилацетилен (0.05 моль/л), растворитель CH2Cl2. Вставка: кинетика изменения поглощения на длинах волн, нм: 450 (1), 548 (2), 700 (3).
Кислотный автокатализ радикального распада илидов. Одна из особенностей исследуемой реакции заключается в увеличении кислотности раствора в процессе реакции [23]. Сопоставление рН водных экстрактов начального и конечного растворов показывает заметное повышение кислотности реакционной смеси в ходе реакции (рН 6.0 и 2.0 соответственно) как при фотолизе одного илида в CH2Cl2, так и в его реакции с ацетиленами. Проблема образования кислоты и ее влияния на процесс была детально изучена в [22, 25, 26].
Первоначально при рассмотрении необычного развития реакции илидов с ацетиленами обсуждалась возможность автокаталитического или цепного механизмов, а также природа продуктов, обеспечивающих автокатализ или продолжение и разветвление цепей [23]. Следует иметь в виду, что реакция между 1a и фенилацетиленом 2a протекает при облучении светом с 350 < λвозб < 450 нм, что означает, что 2a вступает в реакцию в основном состоянии, а первичной активной частицей является именно илид 1a в возбужденном состоянии. Поскольку при образовании нейтрального λ5-фосфинолина 3 из катиона 1a происходит элиминирование протона (схема 3), то было предположено, что в данной реакции мы имеем дело с кислотным автокатализом. Проведенное формально-кинетическое исследование влияния добавок трифторуксусной кислоты (ТФК) на период индукции реакции 1a + 2a показало, что эти добавки уменьшают период индукции при концентрациях добавленной кислоты, не превышающих исходную концентрацию илида ([ТФК] ≤ [1a]) [25]. При больших концентрациях кислоты целевой λ5-фосфинолин 3 не образуется. Было также обнаружено, что с добавками кислоты реакция не требует облучения и переходит в режим термической.
Однако наблюдаемое увеличение кислотности среды при фотолизе илида без добавок ацетиленов свидетельствует о том, что источником кислоты является не только и не столько реакция образования λ5-фосфинолина. Смешанные фосфониево-иодониевые илиды содержат иодониевую группу. Поэтому аналогично диарилиодониевым солям, которые широко используются как инициаторы катионной полимеризации [27, 28], под действием света в них может происходить не только гетеролитический, но и гомолитический разрыв связи C–I+Ph с образованием радикалов и катион-радикалов (реакция (I)) [11, 23, 29]:
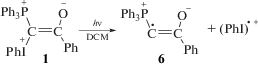
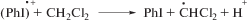
В [27, 28] было показано, что при взаимодействии катион-радикала (PhI)•+ с растворителем образуется H+. Аналогично гомолитический распад илидов должен приводить к появлению кислоты при взаимодействии катион-радикала (PhI)•+ с CH2Cl2 (реакция (II)). Дополнительно при фотолизе в CH2Cl2 может образовываться хлористый водород в результате превращений другого продукта реакции (II), радикала C•HCl2 [30, 31]. Мы предположили, что автокатализ кислотой происходит за счет протонирования исходного илида с образованием дикатиона 7 (реакция (III)). Дикатион 7 является более активным в распаде на радикальные интермедиаты, чем исходный илид [22, 26], и его образование in situ при взаимодействии 1 с H+ приводит к возрастанию скорости генерирования радикалов (реакция (IV)):
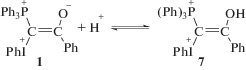
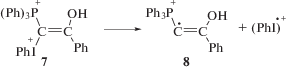
Теоретический термохимический анализ реакций (I) и (IV) с учетом сольватации CH2Cl2 подтвердил предположение о более легком по сравнению с илидом 1 распаде дикатиона 7 на радикальные и ион-радикальные интермедиаты [26]. Расчеты показывают, что при 298 К (25°С) в реакциях (I) и (IV) изменение свободной энергии Гиббса (ΔG298) составляет 42.6 ккал/моль (178 кДж) и 21.2 ккал/моль (89 кДж) соответственно, а изменение энтальпии – 58.1 ккал/моль (244 кДж) и 38.5 ккал/моль (161 кДж) соответственно. Таким образом, дикатион 7 менее устойчив к распаду (реакция (IV)) по сравнению с исходным илидом 1 (реакция (I)). Дополнительными свидетельствами в пользу бóльшей склонности 7 к распаду является удлинение связи между атомом иода и атомом углерода (С2–I), связанным с PPh3-группой, от 2.063 Å в 1 до 2.094 Å в 7, тогда как длина связи между атомом иода и атомом углерода фенильной группы в 7 короче (2.128 Å), чем в 1 (2.137 Å). Заряды (по Малликену) на атоме иода в 7 и 1 равны 0.594 и 0.606 соответственно. Следует отметить, что эти расчеты были проведены для термического распада илида и его протонированной формы, т.е. в отсутствие облучения протонированная форма распадается гораздо легче, чем илид, с чем связан переход от фотолитической к темновой реакции в присутствии кислоты и в развившемся процессе.
В результате последовательности реакций (I)–(IV) концентрация H+ удваивается, т.е. должен происходить автокатализ распада исходного илида (схема 5).
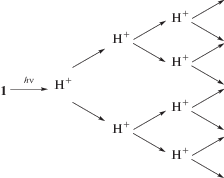
Схема 5.
На самом деле автоускорение реакции фотолиза собственно илида ниже, поскольку H+ не только обеспечивает ускорение реакции, но и расходуется на образование соли 5 (см. ниже реакция (V)) и некоторые побочные реакции (схема 6).
Квантово-химический расчет не отвечает на следующие вопросы: 1) почему катализ кислотой происходит только при низких концентрациях кислоты и 2) участвуют ли радикалы 6 и 8 в образовании конечных продуктов.
На основании сопоставления фотолиза илида в CH2Cl2 [22] и его темновой реакции в присутствии различных концентраций ТФК в CH2Cl2 и ацетонитриле [26] была предложена схема взаимодействия илида 1a с кислотами (схема 6):
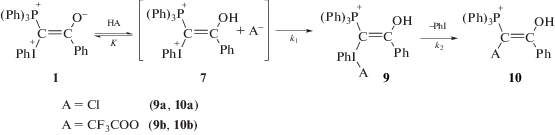
Схема 6.
Масс-спектрометрический анализ продуктов фотолиза илида в CH2Cl2 и в темновой реакции в присутствии различных концентраций добавленной ТФК в CH2Cl2 и ацетонитриле показал наличие продуктов 9 и 10, причем при фотолизе в CH2Cl2 были идентифицированы 9a и 10a, а в присутствии ТФК в обоих растворителях – только 9b и 10b. Образование соединений, подобных 9 и 10, было предложено в [32] при изучении SN2‑замещения в алкилвинил(фенил)иодониевых солях. В этой работе было показано, что соединение, формирующееся при присоединении аниона (A–) к иодониевой группе является неустойчивым, выбрасывает PhI и быстро превращается в продукт присоединения A к атому углерода. Оценки, которые были даны в [26], свидетельствуют, что константа равновесия между илидом и его протонированной формой невелика, и повышение концентрации кислоты в системе должно приводить к необратимому выводу илида из целевой реакции согласно схеме 6. Именно с этим связан тот факт, что в экспериментах с добавками ТФК в реакционную смесь 1a + 2a выход фосфинолина 3 ниже, чем при фотолизе в отсутствие добавленной кислоты [25]. При фотолизе нейтрального раствора концентрация образующейся в реакции кислоты не может быть сильно выше начальной концентрации илида, а в случае добавленной кислоты она начинает превышать концентрацию илида на более ранних стадиях реакции, приводя к его непродуктивному расходованию.
Участие первичных радикалов, генерируемых при фотолизе илидов или при распаде его протонированной формы, в образовании конечных продуктов было установлено с использованием стабильного нитроксильного радикала 2,2,4,4-тетраметил-3-гидроксипиперидил-1-оксила (ТЕМПОЛ) [33]. Эти эксперименты были проведены на системе илид 1a–п-метоксифенилацетилен 2b с мониторингом кинетики образования продуктов по спектрам ЯМР 31P в резонаторе ЯМР-спектрометра и расходования ТЕМПОЛа методом ЭПР. Система 1a + 2b очень удобна для исследования методом ЯМР, поскольку, как указывалось выше, реакция в ней начинается после смешения реагентов с небольшим периодом индукции (1.5–3 мин) без облучения, при этом методом ЯМР 31P регистрируются все соединения, содержащие фосфор: 1, 3–5 и минорные продукты. Было показано, что без добавок ТЕМПОЛа 4 и 5 образуются за время расходования илида (10–20 мин), а образование λ5-фосфинолина 3 продолжается еще в течение 1 ч из нескольких интермедиатов. При добавлении ТЕМПОЛа (2 мол. %) к смеси 1a + 2b методом ЯМР 31P наблюдается период индукции (60 мин), во время которого никакие фосфорсодержащие продукты не регистрируются, а ТЕМПОЛ полностью расходуется. По окончании периода торможения следует быстрое расходование 1a, при этом закономерности образования продуктов такие же, как и в случае отсутствия ингибитора. Эти эксперименты однозначно свидетельствуют о том, что синтез всех конечных продуктов инициируется радикальным распадом исходного илида или его протонированной формы.
В заключение этого раздела следует отметить, что кислотный автокатализ генерирования радикалов также подтверждается тем, что реакция между илидами и ацетиленами тормозится не только акцептором радикалов ТЕМПОЛом, но и основанием (Et)3N [26]. Этим же объясняется отсутствие реакции в оснόвном растворителе ДМФА.
Радикалы при распаде илидов и в их реакциях с ацетиленами
Экспериментальные результаты по торможению ТЕМПОЛом реакций илидов с ацетиленами свидетельствуют о том, что смешанные фосфониево-иодониевые илиды под действием света или в результате кислотного катализа распадаются гомолитически на радикалы и ион-радикалы. Первичные радикалы 6, (PhI)•+ и C•HCl2 (реакции (I), (II), (IV)) являются короткоживущими и не регистрируются методом ЭПР при комнатной температуре в реакционной среде. Однако облучение растворов илидов 1a, 1c и 1d в CH2Cl2 при 77 K полным светом ртутной лампы (λ > 190 нм) в течение 4–8 мин или светом с длиной волны λ > 360 нм в течение 15–30 мин приводит к возникновению радикалов, регистрируемых методом ЭПР [33]. Спектры ЭПР представляют собой не очень хорошо разрешенный дублет с СТВ-константой аР = 21–23 Гс и g-фактором 2.0030–2.0032. Согласно квантово-химическому расчету [29] неспаренный электрон в подобных радикалах локализуется преимущественно на углероде, так что все другие константы малы и маскируются огибающей линией, что и наблюдается экспериментально. Повышение температуры до 293 K приводит к гибели радикалов в растворах илидов 1c и 1d, в то время как в растворе илида 1a зарегистрирован радикал с хорошо разрешенным сигналом ЭПР (рис. 4, кривая 1). Удовлетворительная симуляция спектра ЭПР этого радикала была получена в предположении расщеплений с константами СТВ на ядре фосфора 31Р (аР ~ 23.66 Гс), на одном протоне (аH ~ 1.25 Гс), трех протонах (аH ~ 4.47 Гс) и двух протонах (аH ~ 1.6 Гс). Стабильность вышеуказанного радикала по сравнению с радикалами из илидов 1c и 1d обусловлена более эффективным экранированием радикального центра дополнительным фенильным кольцом в илидном фрагменте молекулы.
Рис. 4.
Спектры ЭПР катион-радикалов, генерированных при фотолизе илида 1a: 1 – 8 после размораживания раствора, облученного в CH2Cl2 при 77 K; 2 – 11 при облучении при комнатной температуре.
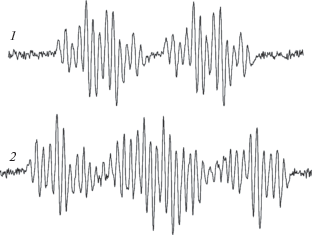
При фотолизе растворов илида 1a в CH2Cl2 при комнатной температуре наблюдали спектр ЭПР радикала 11 (время жизни 10 мин), который является вторичным продуктом фотолиза (реакция (V)) [29]: катион-радикал 11 появляется в результате взаимодействия фотогенерированного радикала 6 (реакция (I)) с H+, образовавшимся из растворителя в реакции (II). Реакция катион-радикала 11 с растворителем дает фосфониевую соль 5, основной продукт фотолиза илидов (реакция (VI)). Таким образом, регистрация спектра ЭПР радикала 11 позволила установить порядок реакций присоединения H+ (реакция (V)) и отрыва H от растворителя (реакция (VI)) при образовании фосфониевой соли 5 в CH2Cl2.
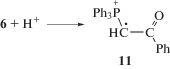
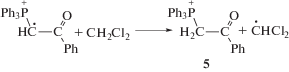
Сопоставление спектра ЭПР катион-радикала 11 (рис. 4, кривая 2) со спектром радикала, наблюдаемым при размораживании облученного раствора илида, демонстрирует, с одной стороны, близость параметров СТВ, которые относятся к взаимодействию неспаренного электрона с атомом фосфора и протонами фенильного кольца бензоильной группы, а с другой стороны – показывает наличие взаимодействия неспаренного электрона с удаленным протоном с СТВ ~ 1.25 Гс в первичном радикале вместо 16.4 Гс для протона при радикальном центре для радикала 11. Мы полагаем, что это различие обусловлено тем, что в первом случае мы регистрируем катион-радикал 8, образовавшийся из дикатиона 7 (реакция (IV)), в котором протонирован атом кислорода бензоильной группы, а во втором – его изомер, вторичный катион-радикал 11, в котором протонирован атом С с неспаренным электроном.
Следует отметить, что при УФ-облучении растворов илидов 1c и 1d в CH2Cl2 при комнатной температуре вторичных радикальных интермедиатов зарегистрировано не было. Как указывалось выше, для этих илидов также не обнаружены радикалы при размораживании растворов с первичными радикалами. Это подтверждает нестабильность именно первичных радикалов. В случае других исследованных илидов вторичные радикалы образуются при облучении в растворе, при этом для илидов 1b и 1f спектры ЭПР были зарегистрированы только при облучении УФ-светом, а для 1e, как и для 1a – видимым светом с λ ≤ 400 нм [33].
В смесях илидов 1a–1f с ацетиленами 2a–2c в CH2Cl2 относительно стабильные радикальные интермедиаты образуются для всех пар. При этом для большинства систем радикалы были зарегистрированы при облучении видимым светом с λ ≥ 380 нм, а для некоторых – без облучения после смешения реагентов. Время жизни этих радикалов больше, чем для радикалов из соответствующего илида, а их спектры ЭПР свидетельствуют о том, что они содержат фрагменты илида и ацетилена [29, 33]. Анализ условий образования радикалов показывает, что чем ниже потенциал ионизации ацетилена и выше сродство к электрону илида, тем легче генерируются радикалы. Форма спектра ЭПР определяется наличием атома фосфора и структурой ацетилена. Константы СТВ aP для радикалов из илидов, выше, чем для их смесей с ацетиленами, что свидетельствует о перераспределении электронной плотности неспаренного электрона на остаток ацетилена.
Следует отметить некоторые особенности радикалов для разных пар илидов и ацетиленов. При фотолизе смеси илида 1a с фенилацетиленом 2a генерируются два радикала в соизмеримых концентрациях, причем один из этих радикалов содержит атом иода [29], т.е. он образуется в комплексе фенилацетилена с иодониевой группой илида до элиминирования фенилиодониевой группы. Для других пар реагентов при симуляции спектров ЭПР не требовалось учитывать СТВ на атоме иода. Это означает, что радикалы регистрируются после отщепления фенилиодониевой группы. Для илидов 1b–1f радикал в смеси с 2a имеет наименьшую концентрацию, его спектр ЭПР представляет собой дублет и практически неразрешенную тонкую структуру, кроме СТВ на 31P. В случае илида 1e, содержащего два атома фосфора, для самого илида спектр ЭПР моделируется с достаточно высокой константой СТВ aP = 28.9 Гс для двух атомов Р, а в случае радикалов с ацетиленами наблюдаются две константы СТВ (18–20 и 12–13 Гс), которые могут быть отнесены к атомам фосфора, что свидетельствует о бόльшей удаленности одного из ядер P от радикального центра [33].
Спектры радикалов, полученные для смесей ацетиленов с илидом 1d с фурильным заместителем при атоме фосфора, претерпевают превращения в процессе наблюдения. Первоначальные спектры представляют собой сложные дублеты с константой СТВ aP = 20–21 Гс [33], превращаясь постепенно в течение 20–60 мин в сложные триплеты с константой СТВ aP = 9–10 Гс. Для этого илида наряду с образованием соединений, аналогичных 3 и 4 (схема 3), известно образование продуктов, содержащих два атома фосфора за счет раскрытия фуранового кольца (схема 7) [34]. Вероятно, наблюдаемые радикалы являются предшественниками этих продуктов.
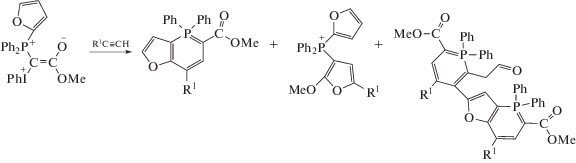
Схема 7.
Эффект ХПЯ в реакции илида 1a с п-метоксифенилацетиленом
При мониторинге кинетики реакции между илидом 1a и п-метоксифенилацетиленом (2b) по спектрам ЯМР 31P в резонаторе ЯМР-спектрометра наблюдали эффект химической поляризации ядер 31P (ХПЯ) в продуктах, а именно: усиленную абсорбцию для сигнала фосфониевой соли 5 и эмиссию для сигналов фуранового производного 4 и некоторых минорных продуктов (рис. 5б) [24]. Известно, что ХПЯ может возникнуть, если существует хотя бы одно промежуточное состояние реакции, которое состоит из спин-коррелированной радикальной пары [35, 36]. В механизме реакции между смешанными фосфониево-иодониевыми илидами и ацетиленами, предложенном ранее, нет места для радикальных пар (схема 3) [14], т.е. наблюдение ХПЯ также противоречит этому механизму. В литературе анализируются эффекты ХПЯ в термических реакциях иодониевых соединений [37, 38] и в термических и фотолитических реакциях фосфорсодержащих соединений [39–42]. Однако все эти примеры представляют собой специально выбранные реакции для изучения особенностей ХПЯ. Система илид 1a–ацетилен 2b представляет собой весьма редкий пример, когда случайно обнаруженный эффект ХПЯ позволяет пролить свет на механизм образования продуктов реакции. Другими словами, механизм реакции между илидом 1a и ацетиленом 2b на пути образования конечных продуктов должен включать спин-селективную реакцию, происходящую в спин-коррелированной радикальной паре. Было предположено, что одноэлектронный перенос (SET) от 2b к протонированной форме илида дикатиону 7, который присутствует в растворе в достаточных количествах к концу периода индукции, генерирует геминальную ион-радикальную пару RP1 (реакция (VII)). Это предположение базируется на зависимости выхода гетероциклических продуктов реакции от потенциала ионизации ацетилена: для данного илида чем ниже потенциал ионизации ацетилена, тем выше выход фуранового производного 4 и ниже выход λ5-фосфинолина 3 [14] (табл. 1). В случае ацетилена 2a фурановое производное обнаружено в следовых количествах, для 2b целевые гетероциклы образуются в соизмеримых количествах, а для 9-этинилфенантрена 2c выход фуранового производного составляет до 80% (табл. 1).
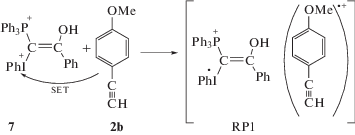
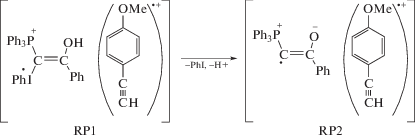
Протонирование илида 1a не только ускоряет скорость генерирования RP1, но и способствует гомолитической диссоциации дикатиона 7 в ней с образованием радикальной пары RP2 (реакция (VIII)). Радикалы в RP2 или вступают в реакцию между собой в паре, или выходят из нее. Вышедшие радикалы могут прорекомбинировать в результате встречи или вступить в реакции с нерадикальными исходными соединениями, растворителем или промежуточными продуктами. Как отмечалось выше, фосфинолин 3 образуется из промежуточных продуктов после израсходования илида. Это означает, что его предшественниками являются радикалы, вышедшие из пары или генерированные в системе независимо.
Возможны три пути взаимодействия радикалов в RP2 (схема 8): основной путь I – это образование фурана 4 (~80% от продуктов, образовавшихся в RP2) из E-изомера радикала 6 и катион-радикала 2b; путь II дает изомер λ5-фосфинолина 3i из Z-изомера радикала 6. Здесь следует отметить, что среди минорных продуктов, для которых наблюдали эмиссию в спектрах ЯМР 31P, есть соединение 3i (выход 3%) с химическим сдвигом 2.2 м. д., близким к химическому сдвигу (2.3 м. д.) λ5-фосфинолина 3 (выход 30%) (рис. 5). Мы полагаем, что 3i является региоизомером 3 (ср. строение 3 и 3i, схемы 3 и 8). Путь III предполагает образование катион-радикала 11 в результате переноса протона от катион-радикала ацетилена на радикал 6. Вышедший из пары катион-радикал 11 дает соль 5 в реакции с растворителем (реакция (VI)). Соль 5, образовавшаяся из катион-радикала 11, генерированного в RP2, должна иметь знак поляризации, противоположный продуктам, образовавшимся в результате рекомбинации в радикальной паре, т.е. иметь положительную поляризацию, что и наблюдается экспериментально (рис. 5б). Конечная концентрация соли 5 почти в пять раз меньше конечной концентрации фурана 4, т.е. основной реакцией в RP2 является образование фуранового производного.
Рис. 5.
Эволюция спектров ЯМР 31P в реакции илида 1a с ацетиленом 2b, время регистрации, мин: 0 (а), 5 (б), 13 (в), 150 (г). Отнесение сигналов: 1 – илид 1a, 2 – фосфониевая соль 5, 3 – фуран 4, 4 – фосфинолин 3, 5 – фосфинолин 3i, 6–9 – интермедиаты-предшественники фосфинолина 3; растворитель CH2Cl2.
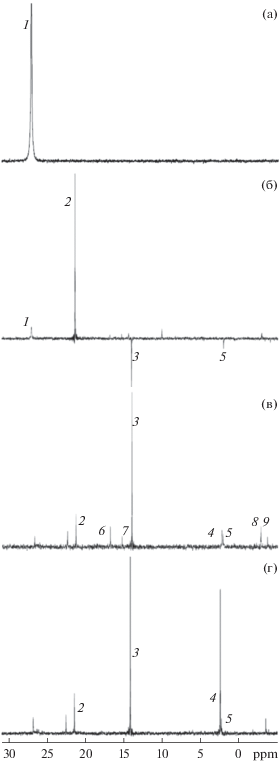
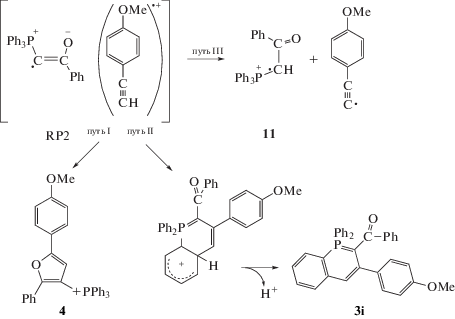
Схема 8.
Таким образом, ХПЯ в реакции между илидом 1a и п-метоксифенилацетиленом 2b с очевидностью демонстрирует участие радикальных пар в синтезе фурана 4 в этой системе. В периоде индукции образуется дикатион 7, что делает возможным SET от 2b к 7. Следует подчеркнуть, что SET между 7 и 2b облегчается за счет близости реагентов в результате их концентрирования в микрогетерогенном растворе в CH2Cl2. В случае фенилацетилена 2a реакция происходит при облучении с периодом индукции, фурановое производное обнаружено в следовых количествах, а λ5-фосфинолин 3 является основным гетероциклическим продуктом. Различие в составе продуктов для 2a и 2b объясняется более низким потенциалом ионизации для последнего примерно на 0.8 эВ. Это делает возможным SET от 2b к иодониевой группе илида, инициируя отщепление PhI и генерируя радикальную пару. В случае фенилацетилена этот процесс практически не имеет места, и λ5-фосфинолин образуется в реакциях фотогенерированных радикалов 6 и 8 через цепь промежуточных превращений подобно тому, как это происходит для 2b [24].
Нерешенные загадки образования λ5-фосфинолина
Мониторинг реакции илида 1a с ацетиленами 2a и 2b методом ЯМР 31P показывает сходный механизм образования λ5-фосфинолинов 3a и 3b из промежуточных соединений, независимо от синтеза фуранового производного. Для системы 1a + 2b на спектрах ЯМР 31P отчетливо наблюдается появление неполяризованных сигналов при 16.9, 15.4, –2.75 и –2.8 м. д. в момент наибольшего проявления ХПЯ (рис. 5б), которые достигают максимальной интенсивности по израсходованию исходного илида (рис. 5в, сигналы 6, 7, 8, 9). В этот момент времени в спектрах появляется сигнал, соответствующий λ5-фосфинолину 3 (сигнал 4). Далее интермедиаты расходуются, а сигнал фосфинолина усиливается (рис. 5г). Здесь следует отметить, что яркая цветная картина, полученная при спектрофотометрическом мониторинге этой реакции (рис. 3), относится именно к превращениям этих промежуточных продуктов.
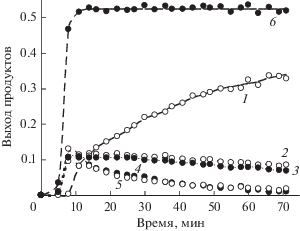
Аналогичная картина наблюдается при мониторинге методом ЯМР 31P реакции в смеси 1a + 2a (рис. 6). Реакция 1a с 2a начинается только после облучения. Выше было показано, что облучение необходимо для генерирования более активного в радикальном распаде дикатиона 7, поэтому в этом случае смесь реагентов в ампуле для ЯМР сначала облучали в течение 1 мин, а затем помещали в резонатор спектрометра. После периода индукции происходит быстрое расходование илида в течение 3 мин. За это время появляются сигналы фосфониевой соли 5 (сигнал 1), неидентифицированного соединения с химическим сдвигом 22.19 м. д., оба соединения в конечных концентрациях (выход 4 и 9% соответственно), а также сигналы промежуточных соединений с химическими сдвигами 16.4 (3), 14.9 (4), –2.94 (5) и –3.05 (6) м. д. и сигнал λ5-фосфинолина 3a при 2.20 м. д. (2) (рис. 6а). Удивительным образом сигналы интермедиатов при 16.4 и 3.05 м. д. и при 14.9 и –2.94 м. д. попарно имеют практически одинаковую интенсивность, и компоненты такой пары расходуются с одинаковой скоростью (рис. 7). Сигналы при 14.9 и –2.94 м. д. исчезают примерно за 1 ч, при этом интенсивности сигналов при 16.4 и 3.05 м. д. в течение часа практически не изменяются, а затем медленно в течение нескольких часов снижаются. Это означает, что интермедиаты, к которым относится первая пара сигналов, являются предшественниками соединений с сигналами при 16.4 и 3.05 м. д. Одновременно наблюдается рост выхода λ5-фосфинолина 3a. Сумма концентраций расходующихся интермедиатов и образующегося 3a остается постоянной, равной конечной концентрации 3a (рис. 7). Это однозначно свидетельствует о том, что вышеуказанные промежуточные продукты являются предшественниками 3a. Следует отметить, что в смеси 1a + 2b также наблюдали симбатное попарное расходование интермедиатов, при этом сумма концентраций интермедиатов и образующегося фосфинолина была равна конечной концентрации фосфинолина. К сожалению, выделить и идентифицировать эти интермедиаты не удается, поскольку при хроматографическом разделении промежуточной смеси они превращаются в конечный фосфинолин, а спектры ЯМР 1H реакционной смеси практически не поддаются расшифровке из-за большого количества близких сигналов ароматических протонов в слабом поле. Следует также отметить, что образование фосфинолина происходит быстрее в случае ацетилена 2b. Это коррелирует с временем жизни радикалов, фиксируемых методом ЭПР для смесей 1a + 2a и 1a + 2b [29]. В первом случае время жизни промежуточных радикалов выше. Оценка констант скорости первого порядка для первой (k1) и для второй пары интермедиатов (k2) дает значения k1a = (6 ± 1) × 10–3, k2a = (1.5 ± 0.2) × 10–3 с–1 и k1b = (1.5 ± 0.2) × 10–2, k2b = (6 ± 1) × 10–3 с–1 для ацетиленов 2a и 2b соответственно.
Рис. 6.
Спектр ЯМР 31P смеси 1a (0.05 моль/л) + 2a (0.15 моль/л) в CH2Cl2 через 3 мин (а) и через 5 ч (б) после окончания периода индукции: 1 – фосфониевая соль 5, 2 – λ5-фосфинолин 3a, 3–6 – интермедиаты.
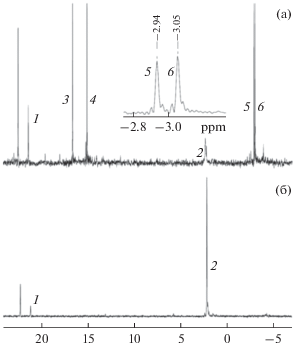
Рис. 7.
Кинетика образования λ5-фосфинолина (1) и образования и расходования интермедиатов с химическими сдвигами (м. д.) по спектрам ЯМР 31P смеси 1a (0.05 моль/л) + 2a (0.15 моль/л) в CH2Cl2: 16.4 (2), 3.05 (3), 14.9 (4) и 2.94 (5); сумма выходов λ5-фосфинолина и интермедиатов (6).
Гипотетически наблюдаемое явление может означать, что существуют две формы одного соединения, которые находятся в динамическом равновесии с константой равновесия, близкой к 1, при этом скорости прямой и обратной реакций достаточно велики по сравнению со скоростью реакции, т.е. наблюдается превращение одной квазиравновесной пары в другую, которая затем превращается в конечный продукт. При этом не так уж важно, превращается в продукт один из компонентов пары или оба (схема 9).
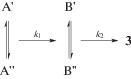
Схема 9.
С одной стороны, это предположение базируется на известном факте, что для илидов, содержащих карбонильные и карбоксильные стабилизирующие группы, а также для образующихся радикальных и других интермедиатов можно написать несколько резонансных и изомерных форм (см. схема 1). С другой стороны, в случае быстрого установления равновесия между двумя формами должен регистрироваться один усредненный сигнал ЯМР. Обычно для наблюдения методом ЯМР двух форм одного соединения необходимо регистрировать спектры в широком диапазоне температур, при этом слияние двух сигналов в один происходит при повышении температуры. К сожалению, при проведении реакции в CH2Cl2 нельзя повысить температуру существенно выше комнатной.
Однако нам представляется, что более адекватным является предположение о том, что одинаковые концентрации и симбатное кинетическое поведение двух сигналов ЯМР 31P означает, что они относятся к одному соединению с двумя атомами фосфора. Поскольку каждый из этих сигналов представляет собой синглет, а не дублет, как это должно быть для соединения с двумя атомами P, то, по-видимому, эти атомы фосфора находятся на достаточном удалении друг от друга, и JP–P не разрешается в условиях записи спектров. Выше мы отмечали, что в случае илида 1d с фурильным заместителем при атоме фосфора были выделены продукты с двумя атомами фосфора в одном соединении (схема 7), при этом в спектрах ЯМР 31P этих продуктов взаимодействие двух атомов P также не было зарегистрировано [34]. Для 1d и других илидов с фурильной группой эти соединения достаточно устойчивы. Появление лабильных соединений с двумя атомам фосфора возможно при рекомбинации или диспропорционировании промежуточных радикалов, содержащих в своем составе фрагменты илида и ацетилена. В настоящее время работа по расшифровке механизма образования λ5-фосфинолина из радикальных предшественников продолжается, в том числе с использованием спиновых ловушек.
ЗАКЛЮЧЕНИЕ
Реакция смешанных фосфониево-иодониевых илидов с ацетиленами, несмотря на экспериментальную простоту синтеза гетероциклических соединений – сложный процесс, который включает в себя такие явления, как реакция в микрогетерогенной системе, кислотный автокатализ радикального инициирования, взаимодействие радикальных, ион-радикальных и ионных интермедиатов, а также реакции переноса электрона при взаимодействии реагентов с образованием спин-коррелированной радикальной пары и реакции образования продуктов в результате рекомбинации радикалов или переноса протона между радикалами в такой паре. Детальное исследование каждого из этих процессов имеет самостоятельное фундаментальное значение. Использование современных физико-химических методов, таких как спектрофотометрия, ЭПР, ЯМР спектроскопия, масс-спектрометрия и ДРС, дает уникальную возможность изучения каждого из этих процессов в отдельности и их совокупности при установлении механизма реакции.
Детальное комплексное исследование кинетических закономерностей реакции смешанных фосфониево-иодониевых илидов с ацетиленами позволило установить механизм кислотного автокатализа в радикальном инициировании реакции за счет образование промежуточного дикатиона, более активного в радикальном распаде, чем исходный илид, и роль промежуточных радикальных интермедиатов в образовании целевых гетероциклических соединений. На основании проведенного исследования можно сделать практические выводы об оптимизации условий реакции с целью получения более высоких выходов целевых гетероциклов.
В заключение следует отметить, что при интерпретации результатов фотолиза илидов в ацетонитриле с образованием производных оксазолов было предположено [11], что в этом растворителе распад илида под действием света происходит одновременно по двум возможным механизмам: гетеролитическому и гомолитическому. При этом фосфониевая соль образуется при распаде илида по гомолитическому механизму из радикалов (реакции (I), (V), (VI)), а оксазол – при распаде илида по гетеролитическому механизму из карбкатиона. В настоящее время проводится более детальное исследование этого процесса, и предварительные результаты свидетельствуют о том, что в ацетонитриле реализуются оба возможных пути фоторазложения смешанного илида.
Список литературы
Нейланд О., Ванаг Г. // Докл. АН СССР. 1964. Т. 159. № 2. С. 373.
Moriarty R.M., Prakash I., Prakash O., Freeman W.A. // J. Am. Chem. Soc. 1984. V. 106. P. 6082.
Huang Z.-Zh., Yu X.-Ch., Huang X. // Tetrahedron Lett. 2002. V. 43. P. 6823.
Huang Z.-Zh., Yu X.-Ch., Huang X. // J. Org. Chem. 2002. V. 67. P. 8261.
Zhdankin V.V., Maydanovych O., Herschbach J., Bruno J., Matveeva E.D., Zefirov N.S. // Tetrahedron Lett. 2002. V. 43. P. 2359.
Zhdankin V.V., Maydanovych O., Herschbach J., Bruno J., Matveeva E.D., Zefirov N.S. // J. Org. Chem. 2003. V. 68. P. 1018.
Матвеева Е.Д., Подругина Т.А., Гришин Ю.К., Ткачев В.В., Жданкин В.В., Алдошин С.М., Зефиров Н.С. // ЖOрХ. 2003. Т. 39. С. 572.
Матвеева Е.Д., Подругина Т.А., Павлова А.С., Миронов А.В., Зефиров Н.С. // Изв. АН Сер. хим. 2008. С. 391.
Матвеева Е.Д., Подругина Т.А., Гришин Ю.К., Павлова А.С., Зефиров Н.С. // ЖOрХ. 2003. Т. 43. С. 209.
Matveeva E.D., Podrugina T.A., Pavlova A.S., Mironov A.V., Gleiter R., Zefirov N.S. // Eur. J. Org. Chem. 2009. V. 14. P. 2323.
Nekipelova T.D., Kuzmin V.A., Matveeva E.D., Gleiter R., Zefirov N.S. // J. Phys. Org. Chem. 2013. V. 26. P. 137.
Матвеева Е.Д., Подругина Т.А., Павлова А.С., Миронов А.В., Зефиров Н.С. // Изв. РАН. Сер. хим. 2008. С. 2195.
Matveeva E.D., Podrugina T.A., Pavlova A.S., Mironov A.V., Borisenko A.A., Gleiter R., Zefirov N.S. // J. Org. Chem. 2009. V. 74. P. 9428.
Matveeva E.D., Podrugina T.A., Taranova M.A., Borisenko A.A., Mironov A.V., Gleiter R., Zefirov N.S. // J. Org. Chem. 2011. V. 76. P. 566.
Matveeva E.D., Podrugina T.A., Taranova M.A., Ivanova A.M., Gleiter R., Zefirov N.S. // J. Org. Chem. 2012. V. 77. № 13. P. 5770.
Matveeva E.D., Podrugina T.A., Taranova M.A., Melikhova E.Y., Gleiter R., Zefirov N.S. // Tetrahedron. 2013. V. 69. № 35. P. 7395.
Baumgartner T. // Acc. Chem. Res. 2014. V. 47. P. 1613.
Duffy M.P., Delaunay W., Bouit P.A., Hissler M. // Chem. Soc. Rev. 2016. V. 45. P. 5296.
Regulska E., Romero-Nieto C. // Dalton Trans. 2018. V. 47. P. 10344.
Belyaev A., Chen Y.-T., Su S.-H., Tseng Y.-J., Karttunen A.J., Tunik S.P., Chou P.-T., Koshevoy I.O. // Chem. Commun. 2017. V. 53. P. 10954.
Zhang D., Song H., Cheng N., Liao W.-W. // Org. Lett. 2019. V. 21. P. 2745.
Levina I.I., Klimovich O.N., Bormotov D.S., Vinogradov D.S., Kononikhin A.S., Dement’eva O.V., Senchikhin I.N., Podrugina T.A., Nikolaev E.N., Kuzmin V.A., Nekipelova T.D. // J. Phys. Org. Chem. 2018. P. e3844. https://doi.org/10.1002/poc.3844
Некипелова Т.Д., Таранова М.А., Матвеева Е.Д., Подругина Т.А., Кузьмин В.А., Зефиров Н.С. // Докл. АН. 2012. Т. 447. № 3. С. 296.
Levina I.I., Tarasov V.F., Podrugina T.A., Nekipelova T.D. // RSC Adv. 2019. V. 9. P. 26 219. https://doi.org/10.1039/c9ra05697
Некипелова Т.Д., Таранова М.А., Матвеева Е.Д., Кузьмин В.А., Зефиров Н.С. // Кинетика и катализ. 2015. Т. 56. С. 411.
Некипелова Т.Д., Подругина Т.А., Виноградов Д.С., Демьянов П.И., Кузьмин В.А. // Кинетика и катализ. 2019. Т. 60. С. 45.
Dektar J.L., Hacker N.P. // J. Org. Chem. 1990. V. 55. P. 639.
Kampmeief J.A., Nalli T.W. // J. Org. Chem. 1994. V. 59. P. 1381.
Некипелова Т.Д., Каспаров В.В., Коварский А.Л., Воробьев А.Х., Подругина Т.А., Виноградов Д.С., Кузьмин В.А., Зефиров Н.С. // Докл. АН. 2017. Т. 474. № 6. С. 707.
Symons J.P., Yarwood A.J. // Trans. Faraday Soc. 1963. V. 59. P. 90.
Михеев Ю.А., Пустошный В.П., Топтыгин Д.Я. // Изв. Акад. наук СССР. Сер. хим. 1976. № 9. С. 2094.
Okuyama T., Takino T., Sato K., Ochiai M. // J. Am. Chem. Soc. 1998. V. 120. P. 2275.
Некипелова Т.Д., Мотякин М.В., Каспаров В. В., Дегтярев Е.Н., Левина И. И., Потапов И.Д., Подругина Т.А. // Хим. физика. 2019. Т. 38. № 12. С. 19.
Matveeva E.D., Vinogradov D.S., Podrugina T.A., Nekipelova T.D., Mironov A.V., Gleiter R., Zefirov N.S. // Eur. J. Org. Chem. 2015. P. 7324.
Kaptein R. // Adv. Free-Radical Chem. 1975. V. 5. P. 319.
Salikhov K.M., Molin Yu.N., Sagdeev R. Z., Buchachenko A.L. Spin Polarization and Magnetic Effects in Radical Reactions. Ed. Molin Yu. N., Amsterdam: Elsevier; Budapest: Akadémiai Kiadó, 1984.
Gragerov I.P., Levit A.F., Kiprianova L.A., Buchachenko A.L. // Org. Magn. Resonance. 1973. V. 5. P. 445.
Скрунц Л.К., Левит А.Ф., Киприанова Л.А., Грагеров И.П // Журн. Орг. Хим. 1980. Т. 16. С. 604.
Левин Я.А. // Докл. Акад наук СССР. 1976. Т. 230. С. 358.
Levin Ya.A., Il’yasov A.V., Goldfarb E.I., Vorkunova E.I. // Org. Magn. Resonance. 1973. V. 5. P. 487.
Pobedimsky D.G., Kirpichnikov P.A., Samitov Yu.Yu., Goldfarb E. I. // Org. Magn. Resonance. 1973. V. 5. P. 503.
Ananchenko G.S., Purtov P.A., Bagryanskaya E.G., Sagdeev R.Z. // J. Phys. Chem. A. 1999. V. 103. P. 3430.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ