

Кинетика и катализ, 2020, T. 61, № 2, стр. 278-286
Влияние промотирования Re и Al2O3 на стабильность работы кобальтовых катализаторов синтеза Фишера–Тропша
Р. Е. Яковенко a, *, И. Н. Зубков a, Г. Б. Нарочный a, О. П. Папета a, О. Д. Денисов a, А. П. Савостьянов a
a ФГБОУ ВО Южно-Российский государственный политехнический университет (НПИ) им. М.И. Платова
346128 Новочеркасск, ул. Просвещения, 132, Россия
* E-mail: jakovenko@lenta.ru
Поступила в редакцию 09.07.2019
После доработки 18.10.2019
Принята к публикации 25.10.2019
Аннотация
Представлены результаты исследований стабильности работы кобальтовых катализаторов на основе SiO2 и Al2O3, промотированных Re и Al2O3, в синтезе углеводородов из СО и Н2 в непрерывных испытаниях в течение 200–300 ч. Полученные катализаторы охарактеризованы методами просвечивающей электронной спектроскопии, термопрограммированного восстановления водородом, термопрограммированной десорбции СО, рентгенофлуоресцентого анализа и испытаны при температуре 200°С, давлении 0.1 МПа и ОСГ 100 ч–1. Определено, что наибольшей активностью обладает кобальтсиликагелевый катализатор, промотированный Al2O3. Установлено, что введение Al2O3 в кобальтсиликагелевый катализатор повышает степень превращения СО и селективность по углеводородам С5+, а также ингибирует агрегацию частиц Со в условиях синтеза Фишера–Тропша под действием реакционной среды. Показано, при что введении 0.1 мас. % рения в катализатор Со/γ-Al2O3 начальная степень превращения СО возрастает в 2 раза, однако при этом скорость его дезактивации повышается, что обусловлено практически двукратным увеличением размеров частиц кобальта в процессе синтеза за 300 ч работы.
ВВЕДЕНИЕ
Актуальность развития технологий получения синтетических углеводородов на основе синтеза Фишера–Тропша (ФТ) обусловлена, прежде всего, необходимостью переработки нетрадиционных углеродсодержащих ресурсов (попутный нефтяной газ, уголь, биомасса и др.) и ужесточением требований к получаемой продукции [1–3]. Синтез ФТ – это гетерогенный каталитический процесс, который в зависимости от применяемого катализатора и условий синтеза позволяет получать широкий спектр углеводородных продуктов из СО и Н2 [4, 5]. В промышленности наибольшее распространение получили нанесенные кобальтовые катализаторы на основе Al2O3, SiO2, ТiO2 [6–8]. При использовании носителей на основе Al2O3 и ТiO2 часть кобальта образует трудноостанавливаемые соединения с подложкой, которые не активны в синтезе ФТ [9, 10]. Снизить степень взаимодействия металл–носитель и повысить селективность в образовании целевого продукта возможно за счет ввода промоторов – благородных металлов Pt, Re, Ru [16–18].
Для применения катализаторов в промышленности с целью получения углеводородов С5+ важна их стабильная работа в длительном непрерывном режиме. Недавно была разработана технология и изготовлена на ЗАО “Самарский завод катализаторов” опытно-промышленная партия кобальтсиликагелевого катализатора, промотированного оксидом алюминия [19]. Представляет интерес определение стабильности работы этого катализатора и сравнение ее с известными образцами.
Поэтому целью настоящей работы было изучение стабильности работы кобальтовых катализаторов на различных носителях в синтезе углеводородов из СО и Н2 в длительных непрерывных испытаниях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализаторов синтеза углеводородов
Было приготовлено четыре катализатора синтеза ФТ: Co/SiO2, Co–Al2O3/SiO2, Co/γ-Al2O3, Co–Re/γ-Al2O3.
Катализаторы получали методом пропитки пористых носителей. В качестве носителей использовали силикагель марки КСКГ производства ООО “Салаватский катализаторный завод” (ГОСТ 3956-76) и γ-Al2O3 марки АН (ТУ 2163-142-60201897-2010) ООО “Новомичуринский катализаторный завод”.
Для приготовления катализаторов применяли пропиточный раствор нитрата кобальта (Co(NO3)2) концентрацией 55%. Гранулы носителя погружали в пропиточный раствор нитрата кобальта и выдерживали в течение 0.5 ч при температуре 70–80°С. Введение рения в катализаторы осуществляли совместной пропиткой нитрата кобальта и перрената аммония (NH4ReO4). Концентрацию перрената аммония в пропиточном растворе поддерживали таким образом, что в готовом катализаторе содержания Re составляло 0.1 мас. %. Методика приготовления катализатора Co–Al2O3/SiO2 включала в себя совместную пропитку носителя нитратами кобальта и алюминия [19].
После пропитки избыток пропиточного раствора сливали, гранулы влажного катализатора подвергали термообработке в термопрограммированном режиме: 80°С – 4 ч; 100°С – 1 ч, 120°С – 1 ч, 140°С – 1 ч, 400°С – 4 ч, скорость нагрева 10°С/мин.
Методики исследований катализатора
Содержание кобальта в образцах катализаторов определяли методом рентгенофлуоресцентого анализа (РФлА) на спектрометре ARLQUANT’X (“Thermo Scientific”, Швейцария) при следующих условиях: среда – воздух, тефлоновая подложка, эффективная площадь облучения 48.9 мм2.
Исследования свежеприготовленных катализаторов методом температурно-программированного восстановления Н2 (ТПВ-Н2) проводили с использованием анализатора ChemiSorb 2750 (“Micromeritics”, США) с детектором по теплопроводности (TCD). Образец массой 0.1–0.15 г помещали в кварцевый реактор, находящийся в термопрограммируемой печи. Перед началом ТПВ-Н2 образец катализатора выдерживали в токе He (20 мл/мин) в течение 2 ч при температуре 200°С для удаления влаги и других адсорбированных газов. Затем образец охлаждали до комнатной температуры и переключали поток на смесь 10% H2 и 90% N2 (20 мл/мин). Исследование процесса восстановления осуществляли в интервале температур 20–800°С со скоростью нагрева 5°С/мин.
Изучение катализаторов методом температурно-программированной десорбции СО (ТПД СО) проводили с использованием анализатора ChemiSorb 2750 (“Micromeritics”, США) с детектором по теплопроводности (TCD). Образец массой около 0.1 г помещали в U-образный кварцевый реактор и выдерживали в токе гелия (20 мл/мин) в течение 2 ч при температуре 200°С для удаления влаги и других адсорбированных газов. Восстановление вели водородно-азотной смесью (10% H2 и 90% N2) в течение 1 ч при температуре 400°С. Импульсную адсорбцию СО до насыщения поверхности образца осуществляли при 20°С в токе гелия (20 мл/мин). После насыщения образец продували гелием (20 мл/мин) в течение 1 ч при температуре 100°С для удаления физически адсорбированного газа. Десорбцию СО проводили, повышая температуру от 100 до 800°C со скоростью 20°С/мин.
Исследование катализаторов методом просвечивающей электронной микроскопии (ПЭМ) осуществляли на микроскопе Tecnai G2 Spirit BioTWIN (“FEI”, Нидерланды) с ускоряющим напряжением 120 кВ. Образец исходного катализатора предварительно восстанавливали азото-водородной смесью (10% Н2 + 90% N2) при линейном нагреве от комнатной температуры до 500°С в течение 1 ч. Образцы измельчали в токе СО2 и в виде суспензий (изопропиловый спирт-катализатор) наносили на медные сетки.
Для предварительно восстановленных катализаторов методом просвечивающей электронной микроскопии было определено распределение кристаллитов кобальта по размерам. Средний размер кристаллитов вычисляли по формуле (1) [20]:
(1)
${{d}_{{{\text{ср}}}}}({\text{C}}{{{\text{o}}}^{0}}) = \frac{{\sum {{{n}_{i}}} d_{i}^{3}}}{{\sum {{{n}_{i}}} d_{i}^{2}}},$По этим данным была рассчитана дисперсность кобальта (D, %) [21]:
где dср(Co0) – средний размер кристаллитов металлического кобальта, нм.Стандартное отклонение определяли по формуле (3) [30]:
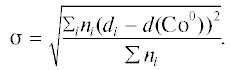
Удаление углеводородов из пор катализаторов после синтеза (“отмывку”) осуществляли в экстракторе Сокслета. Для этого в резервуар, находящийся в центре экстрактора, помещали фильтр Шотта с навеской образца катализатора. В качестве растворителей использовали 1,2-диметилбензол (о-ксилол) и н-гептан. Методика “отмывки” катализаторов включала в себя экстракцию углеводородов о-ксилолом (2 ч при температуре 144°C) и затем экстракцию н-гептаном в течение 1 ч при температуре 98°C.
Синтез углеводородов по методу Фишера–Тропша проводили в стальном проточном реакторе (dвн = 16 мм) со стационарным слоем катализатора (10 см3) при атмосферном давлении (0.1 МПа), объемной скорости газа (ОСГ) 100 ч–1, постоянной температуре (Т) 200°C. Предварительно образцы катализатора восстанавливали в токе Н2 при ОСГ = 3000 ч–1, Т = 400°С в течение 1 ч. Активацию катализаторов осуществляли синтез-газом путем ступенчатого подъема температуры (2.5°С/ч) от 150 до 200°С. После активации катализатора синтез вели в непрерывном режиме в течение 300 ч.
О скорости дезактивации катализаторов судили по параметру RДК (%/ч), показывающему уменьшение степени конверсии СО за 1 ч работы катализатора. RДК определяли по формуле (4):
(4)
${{R}_{{{\text{ДК}}}}} = \frac{{X_{{{\text{CO}}}}^{{\text{н}}} - X_{{{\text{CO}}}}^{{\text{к}}}}}{\tau },$Анализ состава синтез-газа и газообразных продуктов синтеза осуществляли методом газо-адсорбционной хроматографии на хроматографе марки Кристалл 5000 (“Хроматэк”, Россия) с детектором по теплопроводности, колонки Hayesep R (“Хроматэк”, Россия), и молекулярные сита NaX. Первую колонку использовали для анализа углеводородов С1–С5 и СО2 (газ-носитель – гелий, расход – 15 мл/мин), вторую – для анализа СО, Н2, N2 (газ-носитель – аргон, расход – 15 мл/мин); температурно-программированный режим, 80–240°С, скорость нагрева 8°С/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристики приготовленных и отработанных катализаторов приведены в табл. 1. Как видно из этих данных, содержание кобальта в исследуемых катализаторах варьируется в пределах 19.7–22.8%.
Таблица 1.
Физико-химические свойства исходных и отработанных катализаторов
| Образец | С(Со), % | Исходные | Отработанные | ||
|---|---|---|---|---|---|
| *dср, нм | *D, % | *dср, нм | *D, % | ||
| Co/SiO2 | 21.9 | 14.8 ± 2.8 | 6.5 | – | |
| Co–Al2O3/SiO2 | 22.8 | 8.8 ± 1.6 | 10.9 | 7.1 ± 2.0 | 13.5 |
| Co/γ-Al2O3 | 19.9 | 21.8 ± 5.9 | 4.4 | – | |
| Co–Re/γ-Al2O3 | 19.7 | 13.9 ± 3.6 | 6.9 | 27.6 ± 5.2 | 3.5 |
На рис. 1 представлены ПЭМ-изображения и диаграммы распределения по размерам кристаллитов кобальта для восстановленных катализаторов.
Рис. 1.
ПЭМ-микрофотографии восстановленных катализаторов и гистограммы распределения частиц кобальта по размеру: a – Co/SiO2, б – Co–Al2O3/SiO2, в – Co/γ-Al2O3, г – Co–Re/γ-Al2O3.
На поверхности Co/SiO2 катализатора (рис. 1a) имеются частицы кобальта размером 5–20 нм, средний размер частиц составляет 14.8 нм (табл. 1).
Добавка Al2O3 в силикагелевый катализатор (рис. 1б) существенно меняет дисперсность активного компонента – ее значение увеличивается с 6.5 до 10.9%. Введение оксида алюминия способствует уменьшению размеров частиц кобальта и сужению распределения частиц по размерам. Вероятно, добавка Al2O3 количестве 1.0 мас. % ингибирует агрегацию наночастиц металлического кобальта, что проявляется в наблюдаемом снижении среднего размера частиц. Распределение становится практически мономодальным, средний размер частиц кобальта составляет 8.8 нм, что считается оптимальным для катализаторов синтеза ФТ [22].
Частицы кобальта в катализаторе Co/γ-Al2O3 (рис. 1в) имеют средний размер 21.8 нм (табл. 1). Введение рения в состав Co/γ-Al2O3 (рис. 1г) приводит к значительному уменьшению средних размеров наночастиц кобальта с 21.8 до 13.9 нм (табл. 1) и, как следствие, увеличению дисперсности с 4.4 до 6.9%. Полученные данные о влиянии добавки рения в катализатор на основе γ-Al2O3 согласуются с результатами работ [23–27].
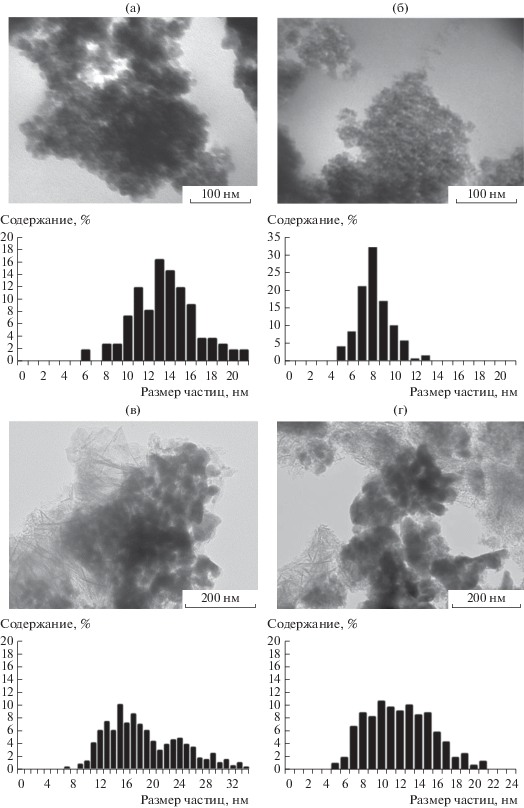
На рис. 2 и в табл. 2 представлены кривые ТПВ-Н2 образцов катализаторов и их характристика.
Рис. 2.
Кривые ТПВ-Н2 катализаторов: 1 – Co/SiO2, 2 – Co–Al2O3/SiO2, 3 – Co/γ-Al2O3, 4 – Co–Re/γ-Al2O3.
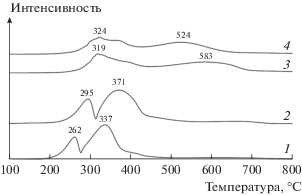
Таблица 2.
Характеристика профилей ТПВ-Н2 катализаторов
| Катализатор | Пик 1 | Пик 2 | S2/S1 | ||
|---|---|---|---|---|---|
| Т, °С | площадь S1, % | Т, °С | площадь S2, % | ||
| Co/SiO2 | 262 | 25.0 | 337 | 75.0 | 3.00 |
| Co–Al2O3/SiO2 | 295 | 25.0 | 371 | 75.0 | 3.00 |
| Co/γ-Al2O3 | 319 | 51.1 | 583 | 48.9 | 0.96 |
| Co–Re/γ-Al2O3 | 324 | 50.4 | 524 | 49.6 | 0.98 |
На кривых ТПВ-Н2 всех образцов присутствуют два интенсивных пика восстановления (рис. 2), относящиеся к переходам Co3+→ Co2+ в области 262–324°С и Co2+ → Co0 в области 337–583°С. Наблюдается некоторое смещение пиков 1 и 2 для катализатора Co–Al2O3/SiO2 в диапазон более высоких температур. Это может быть следствием взаимодействия между нанесенными частицами оксида кобальта и Al2O3 [28].
Отношение площадей S2/S1 двух основных пиков (табл. 2) катализаторов на основе SiO2, показывающее количество водорода, пошедшего на переход Co3+ → Co0, равно 3, что соответствует теоретически ожидаемому значению, исходя из стехиометрии реакций ступенчатого восстановления оксида кобальта Co3O4 до Со0 [29].
Добавка рения в катализатор Co/γ-Al2O3 приводит к смещетемпературного максимума пика 2 в сторону более низкой температуры, не оказывая в тоже время существенного влияния на температуру восстановления оксида кобальта Co3O4 до CoО (пик 1). Полученные данные о влиянии промотирования рением катализаторов на основе γ-Al2O3 на процесс восстановления CoО до Со0 согласуются с выводами авторов [23] о роли рения как промотора восстановления. Меньшие значения отношения площадей S2/S1 для этих катализаторов, по-видимому, указывают на то, что часть кобальта находится в трудно восстанавливаемой форме.
Адсорбция СО на восстановленных катализаторах была исследована с использованием метода TПД СО. Кривые TПД СО для восстановленных образцов приведены на рис. 3.
Рис. 3.
Профили ТПД СО катализаторов: 1 – Co/SiO2, 2 – Co–Al2O3/SiO2, 3 – Co/γ-Al2O3, 4 – Co–Re/γ-Al2O3.
Все образцы катализаторов имеют ярко выраженные профили температурно-программированной десорбции СО. Наблюдаются два основных пика десорбции. Первый – низкотемпературный с максимумом 207–220°C для катализаторов на основе SiO2 и 317–337°C для образцов на основе γ-Al2O3 – можно отнести к десорбции хемосорбированного CO. Второй – высокотемпературный с максимумом 672–772°C – по-видимому, появляется вследствие десорбции СО2, образовавшегося из хемосорбированного СО по реакции Будуара [30]. Введение промоторов Al2O3 и Re в катализаторы приводит к существенному увеличению количества десорбированного СО в области температур появления первого пика. Лапидусом А.Л. и соавт. [31] было показано, что первый пик в профиле ТПД СО соответствует слабосвязанной адсорбции СО в молекулярной форме на оксидной поверхности катализатора. Он характеризует бифункциональные активные центры поверхности, участвующие в полимеризационном процессе.
В табл. 3 приведены усредненные значения основных параметров каталитической активности образцов в синтезе углеводородов за 300 ч работы в непрерывном режиме.
Таблица 3.
Активность катализаторов в синтезе Фишера–Тропша*
| Образец | Конверсия СО, % | Селективность, % | Производительность по С5+, кг м–3 ч–1 | RДК, %/ч | |||
|---|---|---|---|---|---|---|---|
| СН4 | С2–С4 | С5+ | СО2 | ||||
| Co/SiO2 | 75.3 | 18.0 | 15.4 | 62.8 | 3.8 | 10.2 | 0.052 |
| Co–Al2O3/SiO2 | 78.6 | 16.5 | 15.4 | 64.2 | 3.9 | 11.3 | 0.047 |
| Co/γ-Al2O3** | 41.9 | 14.4 | 12.1 | 71.1 | 2.4 | 6.4 | 0.035 |
| Co–Re/γ-Al2O3 | 68.5 | 17.9 | 10.7 | 65.4 | 6.0 | 9.5 | 0.102 |
Все исследованные катализаторы активны в синтезе углеводородов из СО и Н2. В условиях экспериментов наиболее активен Co–Al2O3/SiO2: конверсия СО составляет 78.6%, селективность и производительность по углеводородам С5+ – 64.2% и 11.3 кг м–3 ч–1 соответственно. Промотирование оксидом алюминия за счет повышения конверсии СО и селективности по С5+ приводит к увеличению на 10.8% производительности катализатора. Кроме того, при введении оксида алюминия наблюдается снижение скорости дезактивации катализатора на 9.6%.
Введение рения в состав катализатора Co/γ-Al2O3 способствует увеличению степени конверсии с 41.9 до 68.5%. При этом селективность по углеводородам С5+ падает с 71.1 до 65.4% из-за более интенсивного образования газообразных продуктов реакции С1–С4 и СО2. Значительное повышение селективности по СО2 (в 2.5 раза) для промотированного образца вероятно связано с возрастанием скорости реакции водяного газа за счет облегчения диссоциативной адсорбции СО [32].
Промотирование рением Со/γ-Al2O3 катализатора приводит к увеличению скорости его дезактивации в 2.9 раза. Авторы [33, 34] также отмечали, что введение благородных металлов в состав кобальтовых катализаторов способствует их ускоренной деактивации.
Зависимость конверсии СО и селективности по СН4 от продолжительности синтеза показаны на рис. 4. В целом все исследуемые катализаторы демонстрируют закономерное снижение конверсии СО во времени [35]. Падение активности сопровождается уменьшением селективности по углеводородам C5+ за счет усиления образования метана и СО2.
Рис. 4.
Зависимости конверсии СО (а) и селективности по СН4 (б) от продолжительности синтеза: 1 – Co/SiO2, 2 – Co–Al2O3/SiO2, 3 – Co/γ-Al2O3, 4 – Co–Re/γ-Al2O3.
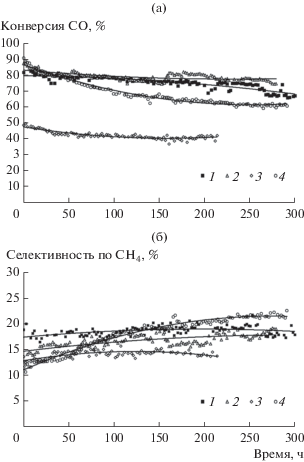
Рост селективности по метану во времени может быть следствием накопления в порах катализатора жидких углеводородов С5+, что, вероятно, приводит к сокращению количества доступных активных центров роста цепи [32]. С другой стороны, накопление продуктов синтеза в порах катализатора, как отмечают авторы [36], способствует увеличению мольного отношения H2/CO внутри гранулы катализатора из-за разной скорости диффузии реагентов. Действительно, в [37] было показано, что коэффициенты диффузии водорода и СО через пленку н-тетрадекана при 200°С составляют 1.6 × 10–4 и 2.2 × 10–4 м2/с соответственно. Это приводит к увеличению отношения Н2/СО на поверхности катализатора и росту селективности образования метана [38].
Первые 50 ч синтеза для всех образцов характеризуются наибольшей скоростью падения активности. Возможно, в этот период продолжается стабилизация активного компонента катализатора под действием реакционной среды.
Катализаторы Co/SiO2 и Co–Al2O3/SiO2 демонстрируют близкую начальную конверсию СО и селективность образования углеводородов C5+ и СН4. Однако после ~250 ч синтеза на образце Co/SiO2 наблюдается резкое падение активности, которое сопровождается увеличением доли образующегося метана и уменьшением производительности по углеводородам С5+.
Для катализаторов на основе γ-Al2O3 в начальный период отмечается резкое повышение степени конверсии СО с 48 до 90% при введении добавки рения (рис. 4а). Подобный эффект был обнаружен в работе [39], в которой также фиксировалось и быстрое падение активности катализатора Co–Re/γ-Al2O3 в сравнении с образцом без рения.
Процесс потери активности катализатора обычно вызван совокупностью факторов, которые оказывают различное влияние на поведение каталитической системы в процессе синтеза. Образование большого количества воды при высокой степени превращения СО принято считать одной из основных причин дезактивации кобальтовых катализаторов на основе оксидных носителей. Эффект окисления активного металла под воздействием Н2О приводит к снижению металлической поверхности кобальтового катализатора [11]. При этом скорость окисления активной фазы прямо пропорциональна парциальному давлению воды [40].
Для исследований катализаторов после синтеза методом ПЭМ их выгружали в токе азота и в целях удаления углеводородов из пор подвергали двухступенчатой отмывке о-ксилолом и н-гептаном. Как показали данные ПЭМ (рис. 5а, табл. 1), дисперсность и распределение частиц Со по размерам в проработавшем ~300 ч образце Co–Al2O3/SiO2 сопоставимы с показателями исходного восстановленного катализатора (рис. 1б). Вероятно, введение Al2O3 в кобальтсиликагелевый катализатор помимо повышения конверсии СО и селективности процесса по углеводородам С5+ ингибирует агрегацию наночастиц Со в условиях синтеза ФТ под действием реакционной среды, что способствует его стабильной работе.
Рис. 5.
ПЭМ-микрофотографии катализаторов после синтеза и гистограммы распределения частиц кобальта по размеру: a – Co–Al2O3/SiO2, б – Co–Re/γ-Al2O3.
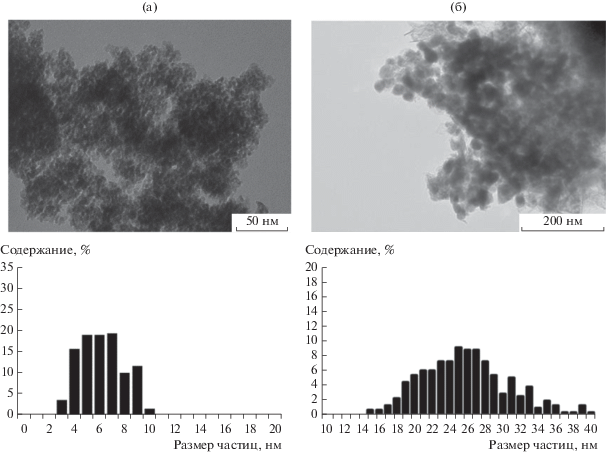
Для катализатора Co–Re/γ-Al2O3 после 300 ч работы наблюдается значительное увеличение среднего размера частиц Со (с 13.9 до 27.6 нм), которое является следствием агломерации частиц кобальта под влиянием реакционной среды.
ЗАКЛЮЧЕНИЕ
Изучена стабильность работы нанесенных кобальтовых катализаторов в синтезе углеводородов из СО и Н2 в непрерывных испытаниях. Определено, что наибольшей активностью обладает кобальтсиликагелевый катализатор, промотированный Al2O3. Введение Al2O3 в Co/γ-Al2O3 повышает степень превращения СО и селективность по углеводородам С5+, а также ингибирует агрегацию частиц Со в условиях синтеза ФТ под действием реакционной среды.
Добавление рения в катализатор Со/γ-Al2O3 приводит к значительному (в ~2 раза) росту конверсии СО в начальный период его работы (первые 50 ч). Однако скорость дезактивации на промотированном образце выше практически в три раза, что, вероятно, обусловлено двукратным увеличением размеров частиц кобальта под действием реакционной среды. Тем не менее даже после 200 ч синтеза в присутствии Co–Re/γ-Al2O3 конверсия СО была больше в 1.5 раза.
Для всех катализаторов в процессе синтеза наблюдается повышение селективности образования метана, что может быть следствием накопления в порах катализатора жидких углеводородов С5+, приводящего к возрастанию мольного отношения H2/CO внутри гранулы катализатора из-за разной скорости диффузии реагентов.
В результате работы определено, что скорость дезактивации катализаторов увеличивается в ряду: Co/γ-Al2O3 < Co–Al2O3/SiO2 < < Co/SiO2 < Co–Re/γ-Al2O3.
Список литературы
Prieto G., De Mello M.I.S., Concepcion P., Murciano R., Pergher S.B.C., Martınez A. // ACS Catalysis. 2015. V. 5. P. 3323.
Ellepola J., Thijssen N., Grievink J., Baak G., Avhale A., Schijndel J. // Comput. Chem. Eng. 2012. V. 42. P. 2.
Крылова А.Ю., Куликова М.В., Лапидус А.Л. // Химия твердого топлива. 2014. № 4. С. 18.
Крылова А.Ю. // Химия твердого топлива. 2014. № 1. С. 23.
Dry M.E. // Catal. Today. 2002. V. 71. P. 227.
Калечиц И.В. Химические вещества из угля. Москва: Химия, 1980. 616 с.
Lapidus A.L., Krylova A.Yu., Mikhailova Ya.V., Sineva L.V., Erofeev A.B. // Solid Fuel Chem. 2011. V. 45. P. 71.
Крылова А.Ю. // Кинетика и катализ. 2012. Т. 53. № 6. С. 790.
Bessell S. // Appl. Catal. A: Gen. 1993. V. 96. P. 253.
Jung J.S., Lee J.S., Choi G., Ramesh S., Moon D.J. // Fuel. 2015. V. 149. P. 118.
Ma W., Jacobs G., Keogh R.A., Bukur D.B., Davis B.H. // Appl. Catal. A: Gen. 2012. V. 437–438. P. 1.
Jacobs G., Ma W., Gao P., Todic B., Bhatelia T., Bukur D.B., Khalid S., Davis B.H. // Top. Catal. 2012. V. 55. P. 811.
Ghasvareh P., Smith K.J. // Energy Fuels. 2016. V. 30. P. 9721.
Merino D., Perez-Miqueo I., Sanz O., Montes M. // Top. Catal. 2015. V. 59. P. 207.
Bor Ø., Eri S., Blekkan E.A., Storsæter S., Wigum H., Rytter E., Holmen A. // J. Catal. 2007. V. 248. P. 89.
Xiong J., Borg O., Blekkan E.A., Holmen A. // Catal. Commun. 2008. V. 9. P. 2327.
Лапидус А.Л., Павлова В.А., Чинь Н.К., Елисеев О.Л., Гущин В.В., Давыдов П.Е. // Нефтехимия. 2009. Т. 49. № 4. С. 319.
Михайлова Я.В., Свидерский С.А., Крылова А.Ю., Лапидус А.Л. // Химия твердого топлива. 2010. № 5. С. 60.
Нарочный Г.Б., Яковенко Р.Е., Савостьянов А.П., Бакун В.Г. // Катализ в промышленности. 2016. № 1. С. 37.
Prieto G., Martínez A., Concepción P., Moreno-Tost R. // J. Catal. 2009. V. 266 P. 129.
Wang S., Yin Q., Guo J., Ru B., Zhu L. // Fuel. 2013. V. 108. P. 597.
Borg Ø., Dietzel P.D.C., Spjelkavik A.I., Tveten E.Z., Walmsley J.C., Diplas S., Eri S., Holmen A., Rytter E. // J. Catal. 2008. V. 259. P. 161.
Tavasoli A., Mortazavi Y., Khodadadi A.A., Mousavian M.A. // Catal. Iran. J. Chem. Chem. Eng. 2005. V. 24. P. 9.
Das T.K., Jacobs G., Patterson P.M., Conner W.A., Li J., Davis B.H. // Fuel. 2003. V. 82. P. 805.
Borg Ø., Hammer N., Eri S., Lindvag O.A., Myrstad R., Blekkan E.A., Rønning M., Rytter E., Holmen A. // Catal. Today. 2009. V. 142. P. 70.
Borg Ø., Eri S., Blekkan E.A., Storsæter S., Wigum H., Rytter E., Holmen A. // J. Catal. 2007. V. 248. P. 89.
Tavasoli A., Mortazavi Y., Ali K., Ali A.A., Ali M., Ali M. // Catalysts Iran. J. Chem. Chem. Eng. 2005. V. 24. P. 9.
Zhang Y., Nagamori S., Hinchiranan S., Vitidsant T., Tsubaki N. // Energy Fuels. 2006. V. 20. P 417.
Pardo-Tarifa F., Cabrera S., Sanchez-Dominguez M., Boutonnet M. // Int. J. Hydrogen energy. 2017. V. 42. P. 9754.
Fischer N., Steen E., Claeys M. // J. Catal. 2013. V. 299. P. 67.
Лапидус А.Л., Крылова А.Ю., Михайлова Я.В., Синева Л.В., Ерофеев А.Б. // Химия твердого топлива. 2011. № 2. С. 3.
Borg Ø., Yu Z., Chen D., Blekkan E.A., Rytter E., Holmen A. // Top. Catal. 2014. V. 57. P. 491.
Jacobs G., Patterson P.M., Zhang Y., Das T., Li J., Davis B.H. // Appl. Catal. A: Gen. 2002. V. 233. P. 215.
Dalai A.K., Davis B.H. // Appl. Catal. A: Gen. 2008. V. 348. P. 1.
Zhou W., Chen J., Fang K., Sun Y.H. // Fuel Proces. Technol. 2006. V. 87. P. 609.
Iglesia E., Soled S.L., Fiato R.A., Via G.H. // J. Catal. 1993. V. 143. P. 345.
Хасин А.А. // Газохимия. 2008. № 2. С. 28.
Botes F.G., Niemantsverdriet J.W., Loosdrecht J.A. // Catal. Today. 2013. V. 215. P. 112.
Vada S., Hoff A., Adnanes E., Schanke D., Holmen A. // Top. Catal. 1995. V. 2. P. 155.
Jacobs G., Das T.K., Patterson P.M., Li J., Sanchez L., Davis B.H. // Appl. Catal. A: Gen. 2003. V. 247. P. 335.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ