

Кинетика и катализ, 2020, T. 61, № 3, стр. 359-365
Влияние предварительной механохимической обработки на фотокаталитическую активность феррита висмута
Т. В. Краснякова a, b, С. А. Юрчило a, В. В. Моренко a, И. К. Носолев c, Е. В. Глазунова d, С. В. Хасбулатов d, И. А. Вербенко d, С. А. Митченко a, e, *
a Институт физико-органической химии и углехимии им. Л.М. Литвиненко
283114 Донецк, ул. Р. Люксембург, 70, Украина
b Луганский национальный университет им. Тараса Шевченко
91011 Луганск, ул. Оборонная, 2, Украина
c Донецкий физико-технический институт им. А.А. Галкина
283114 Донецк, ул. Р. Люксембург, 72, Украина
d Научно-исследовательский институт физики Южного федерального университета
344090 Ростов-на-Дону, просп. Стачки, 194, Россия
e ФГБОУ ВО Южно-Российский государственный политехнический университет (НПИ) им. М.И. Платова
346400 Новочеркасск, ул. Просвещения, 132, Россия
* E-mail: samit_RPt@mail.ru
Поступила в редакцию 23.08.2019
После доработки 19.11.2019
Принята к публикации 28.11.2019
Аннотация
Феррит висмута BiFeO3, полученный двухстадийным твердофазным синтезом, проявляет каталитическую активность в фотодеградации бромфенолового синего под действием пероксида водорода. Предварительная механоактивация BiFeO3 в атмосфере аргона приводит к увеличению активности фотокатализатора, обусловленному ростом его удельной поверхности. По достижении предельной удельной поверхности активацией порошка катализатора в аргоне продолжение обработки в атмосфере водорода ведет к дальнейшему повышению фотокаталитической активности за счет возрастания поверхностной концентрации кислородных вакансий.
ВВЕДЕНИЕ
Развитие химических технологий и их использование в современной промышленности приводят к значительному увеличению выбросов в окружающую среду разного рода отходов, в том числе токсичных и биологически не разлагаемых органических соединений. Очистка сточных вод от таких загрязнений в последнее время стала насущной проблемой современного высокотехнологичного общества [1]. Одним из путей решения этой проблемы может быть внедрение отвечающих принципам “зеленой химии” систем очистки воды, обеспечивающих глубокую минерализацию органических экополлютантов. Многообещающим здесь представляется фотокаталитическое разложение последних при участии порошковых или нанесенных катализаторов на основе оксидов переходных металлов, например, TiO2, СuO, Cr2O3, Co3O4, Fe2O3, MnO2, ZnO и др. [2–7]. Однако эти фотокатализаторы проявляют высокую активность при облучении светом ультрафиолетовой области спектра, поскольку обладают значительной шириной запрещенной зоны. Существенное ослабление интенсивности УФ-излучения в водной среде, особенно при ее загрязнении, заметно снижает эффективность процесса. Поэтому предпочтительней использовать фотокаталитические системы с меньшей шириной запрещенной зоны, отвечающей излучению видимого диапазона.
Одним из перспективных в этом плане фотокатализаторов является феррит висмута BiFeO3 (BFO). Это соединение со структурой типа перовскита обладает комплексом уникальных физико-химических свойств: при комнатной температуре ему присущи одновременно сегнетоэлектрическое и антиферромагнитное упорядочения [8–11], химическая устойчивость, относительно небольшая (~2.2 эВ) ширина запрещенной зоны [9–14]. Последний факт в сочетании с отсутствием токсичности открывает широкие возможности применения BFO для разложения разнообразных органических загрязнений окружающей среды с использованием практически неисчерпаемой и экологически чистой солнечной энергии.
В гетерогенных системах каталитическая активность определяется площадью поверхности катализаторов и поверхностной концентрацией активных центров. Так, наночастицы феррита висмута в виде нанопроволок и микрокубов, пористые тонкие пленки BFO, обладая большой удельной поверхностью, демонстрируют высокую фотокаталитическую активность [10, 15, 16]. Обеспечить развитую поверхность BFO можно, не только регулируя его морфологию процессами синтеза, но и, например, путем механохимического воздействия. Кроме увеличения поверхности механическая активация твердых веществ приводит также к возникновению чрезвычайно реакционно-способных приповерхностных состояний – точечных и протяженных дефектов кристаллической структуры, вакансий, свободных радикалов и т.п. [17–26], инициирующих протекание химических реакций. Активные центры фотокатализатора BFO ассоциируют с кислородными вакансиями [27, 28], которые в сверхравновесных количествах получают, например, путем отжига керамики в присутствии водорода при повышенном давлении [29]. Можно полагать, что механохимическая активация керамики BFO в атмосфере водорода также будет приводить к аналогичному эффекту, вследствие чего следует ожидать роста фотокаталитической активности механоактивированных образцов. Проверка этого предположения является целью настоящей работы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез катализаторов
Экспериментальные образцы катализаторов BFO синтезировали из оксидов Bi2O3 и Fe2O3 квалификации “ч. д. а.” в молярном соотношении 1 : 1 путем двукратных обжигов с промежуточным контролем фазового состава и измельчением материала шарами из ZrO2 с использованием планетарной мельницы-активатора АГО-2 (Россия) в дисперсионной среде этанола. На основе серии пробных образцов были выбраны оптимальные условия синтеза: первый обжиг проводили при Т1 = 850°С в течение τ1 = 10 ч, далее после размола и перемешивания материал отжигали второй раз при Т2 = 870°С в течение τ2 = 10 ч. Полноту синтеза контролировали методом порошковой дифрактометрии на рентгеновском дифрактометре ДРОН-3 (“Буревестник”, Россия).
Навеску синтезированного феррита висмута массой 1 г помещали в стеклянный виброреактор со стеклянными мелющими телами, продували аргоном или водородом и герметизировали, после чего осуществляли механоактивацию при помощи микровибромельницы ММVE-0.005 (“Гефест”, Россия) с рабочей частотой 50 Гц и амплитудой 5.5 мм, что отвечает удельной энергонапряженности I = 15 Вт/кг. Удельную дозу поглощенной порошком механической энергии определяли согласно соотношению [30] Dуд = I × t, где t – время механической обработки порошка.
Изучение поверхности катализаторов
Рентгенофазовый анализ катализатора проводили с помощью автоматизированного рентгеновского дифрактометра ДРОН-3 в CuKα-излучении с использованием никелевого фильтра в геометрии съемки по Брэггу–Брентано. Съемки вели при напряжении на рентгеновской трубке 30 кВ и силе тока 20 мА в режиме пошагового сканирования с шагом Δ(2θ) = 0.01°. Время экспозиции в каждой точке составляло 0.3 с, скорость счетчика – 2°/мин.
Удельную поверхность исходных и механоактивированных порошков определяли на приборе СОРБИ MS (“Meta”, Россия) динамическим методом БЭТ при четырех различных значениях относительного парциального давления смеси инертных газов азота и гелия, проходящей через исследуемый образец при температуре жидкого азота (77 К).
Морфологию поверхности изучали методом растровой электронной микроскопии при помощи сканирующего электронного микроскопа JSM-6490LV (“JEOL”, Япония). Съемку осуществляли в режиме вторичных электронов при ускоряющем напряжении 10 кВ и увеличении ×30 000. Микрорентгеноспектральный элементный анализ проводили с использованием энергодисперсионного спектрометра INCA Penta Fetх3 (“OXFORD Instruments”, Великобритания).
Фотокаталитическая активность
Фотокаталитическую активность BFO изучали по обесцвечиванию водного раствора бромфенолового синего (БФС). Перед началом реакции к 100 мл 1.46 × 10–5 М раствора БФС добавляли навеску 0.2 г катализатора и оставляли в темноте на 1 ч для достижения адсорбционного равновесия, затем в систему вводили 3.84 × 10–3 М перекиси водорода. После этого полученную взвесь облучали светом видимого диапазона при помощи LED-лампы (“Еpistar Leds”, Китай) с техническими характеристиками 6000 К, 4620 лм, 62 Вт при постоянном перемешивании с применением магнитной мешалки. Пробы объемом 10 мл отбирали через фиксированные промежутки времени, помещали в пробирку и отделяли катализатор при помощи центрифуги ОПн-3У4.2 (“Поликом”, Россия) в течение 5 мин на частоте 3000 об/мин. Надосадочную жидкость переносили в кювету и анализировали спектрофотометрически. Кинетику процесса контролировали по максимуму светопоглощения БФС на длине волны 591 ± 2 нм с использованием спектрофотометра SpectroVis®Plus (“Vernier Software&Technology”, США). По окончании анализа аликвоту тщательно встряхивали и возвращали в реакционную смесь.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристика катализатора
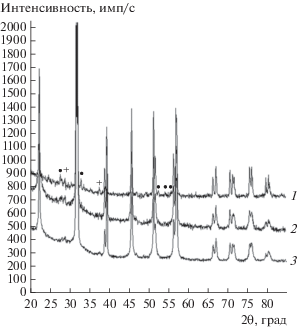
При исследовании кристаллической структуры исходного образца обнаружены все рентгеновские максимумы, соответствующие ферриту висмута со структурой типа перовскита [31] (рис. 1). Кроме того, наблюдаются слабые рефлексы, присущие примесным фазам силленита Bi25FeO40 [32] и муллита Bi2Fe4O9 [33], которые практически всегда имеют место при твердофазном синтезе. Анализ дифрактограмм свидетельствует, что механическая обработка не приводит к изменению фазового состава образцов и возникновению текстуры.
Рис. 1.
Дифрактограммы исходного (1) и механоактивированного в течение 30 (2) и 60 мин (3) BFO. Немаркированные рефлексы принадлежат фазе BiFeO3, ⚫ – Bi25FeO40, + – Bi2Fe4O9.
При увеличении времени механоактивации порошка BFO в среде аргона его удельная поверхность монотонно растет. Зависимость удельной поверхности S порошка от дозы D поглощенной порошком механической энергии имеет вид кривой с насыщением (рис. 2) и отвечает уравнению:
где S0 – удельная поверхность исходного порошка, (S0 + S*) – предельное значение удельной поверхности, ηsp – константа, характеризующая эффективность образования новой поверхности – величина, обратная удельной работе образования новой поверхности.Рис. 2.
Рост удельной поверхности катализатора в зависимости от дозы поглощенной порошком механической энергии при обработке BFO в атмосфере аргона. Точки на графике отвечают экспериментальным значениям S, огибающая кривая – расчет по уравнению (1) при S0 = (0.2 ± 0.1) м2/г, S* = (1.1 ± 0.1) м2/г, ηsp = (8 ± 2) × 10–2 г/Дж.
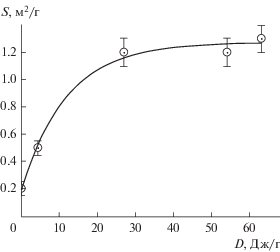
Из рис. 2 видно, что после поглощения катализатором дозы механической энергии ~25 Дж/г (в наших условиях это отвечает механообработке длительностью ~30 мин) достигается практически предельное значение S. Среда диспергирования не оказывает существенного влияния на удельную поверхность порошка: при обработке в атмосфере водорода в течение 60 мин значение удельной поверхности составляет 1.3 ± 0.1 м2/г и совпадает в пределах экспериментальных ошибок с полученным для BFO, активированного в течение того же времени в атмосфере аргона – 1.2 ± 0.1 м2/г.
Распределение всех элементов, входящих в состав BFO, для исходного и механоактивированных порошков по поверхности образцов равномерное (рис. 3).
Рис. 3.
Типичная микрофотография СЭМ поверхности BFO (а) и распределение элементов О (б), Fe (в), Bi (г) по поверхности порошка (по данным микрорентгеноспектрального анализа).

По данным микрорентгеноспектрального анализа атомное соотношение Fe/O в приповерхностных слоях экспериментальных образцов составляет 0.30, 0.31 и 0.34 для неактивированного и обработанного в атмосфере аргона и водорода феррита висмута соответственно.
Кинетика фотодеградации BFO
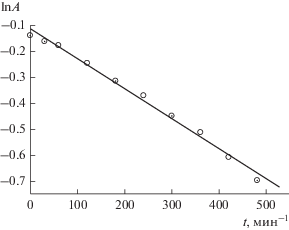
Для разложения БФС необходимо наличие в системе BFO–БФС–Н2О2 всех трех компонентов – при отсутствии феррита висмута или пероксида водорода деградация красителя с заметной скоростью не протекает; без облучения реакция идет также крайне медленно. Расходование красителя осуществляется по первому порядку, в присутствии необработанного механохимически катализатора (рис. 4) константа скорости составляет 1.2 × 10–3 мин–1.
Предварительная механическая активация феррита висмута в аргоне приводит к росту фотокаталитической активности ВFO (табл. 1). Зависимость константы скорости расходования БФС от времени предварительной активации катализатора имеет вид кривой с насыщением (рис. 5, кривая 1) и отвечает уравнению:
где k0 – константа скорости реакции на исходном образце, не подвергнутом предварительной механоактивации, (k0 + k*) – предельное значение константы скорости реакции, η – константа, характеризующая рост каталитической активности фотокатализатора.Таблица 1.
Предельные значения константы k скорости реакции окисления БФС и время его полупревращения t1/2 в системах с исходным ферритом висмута (BFO) и предварительно активированным в атмосфере аргона (BFO1) и водорода (BFO2) катализаторами
| Фотокатализатор | k × 103, мин–1 | t1/2, ч |
|---|---|---|
| BFO | 1.2 ± 0.1 | 9.6 |
| BFO1 | 4.6 ± 0.3 | 2.5 |
| BFO2 | 8.6 ± 0.4 | 1.3 |
Рис. 5.
Зависимость константы скорости расходования БФС от времени предварительной активации феррита висмута в атмосфере аргона (1) и водорода (2). Точки на графике отвечают экспериментальным значениям k, огибающая кривая – расчет по уравнению (2) при: k0 = (1.1 ± 0.1) × 10–3 мин–1, k* = (3.5 ± ± 0.2) × 10–3 мин–1, η = (10 ± 1) × 10–2 г/Дж (1); k0 = = (4.4 ± 0.2) × 10–3 мин–1, k* = (4.2 ± 0.3) × 10–3 мин–1, η = (4.0 ± 0.7) × 10–2 г/Дж (2).
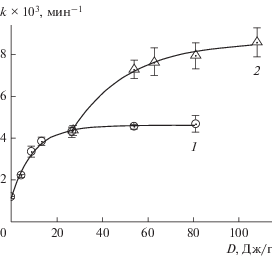
Поскольку среда диспергирования не влияет на развитие удельной поверхности порошков феррита висмута, для исключения вклада роста поверхности в каталитическую активность образцы катализатора сначала подвергали механоактивации в атмосфере аргона в течение 30 мин до достижения практически предельной удельной поверхности, а затем – в атмосфере водорода. Наблюдаемая константа скорости реакции в присутствии такого катализатора растет с увеличением времени его обработки (рис. 5, кривая 2), достигая предельного значения (8.6 ± 0.4) × 10–3 мин–1. Это в 7 раз больше, чем у исходного образца, и превышает активность катализатора, обработанного только в атмосфере аргона, примерно в 2 раза. Отметим, что в наших условиях практически полная фотодеградация БФС (степень превращения > 98%) в системе c наиболее активным катализатором BFO2 проходила примерно за 8 ч (табл. 1).
Увеличение активности BFO при его механоактивации обусловлено двумя факторами – ростом удельной поверхности катализатора и повышением поверхностной концентрации кислородных вакансий. При диспергировании BFO в атмосфере аргона подводимая механическая энергия расходуется, главным образом, на развитие поверхности, что обусловливает возрастание его фотокаталитической активности. Это подтверждается совпадением в пределах экспериментальных погрешностей величин ηsp и η, характеризующих развитие поверхности катализатора и увеличение его фотокаталитической активности соответственно.
Диспергирование в атмосфере водорода способствует связыванию кислорода с образованием кислородных вакансий в приповерхностных слоях BFO. Такое предположение подтверждается данными микрорентгеноспектрального анализа: атомное соотношение Fe/O в приповерхностных слоях катализатора, диспергированного в атмосфере водорода, выше, чем у BFO, активированного в аргоне. Это может служить причиной наблюдаемой в эксперименте большей активности катализатора, обработанного в водороде, по сравнению с диспергированным в аргоне.
ЗАКЛЮЧЕНИЕ
Таким образом, феррит висмута BiFeO3, полученный двухстадийным твердофазным синтезом, демонстрирует фотокаталитическую активность в разложении бромфенолового синего под действием пероксида водорода при облучении светом видимого диапазона. Предварительная механоактивация BiFeO3 сопровождается повышением его фотокаталитической активности. Для диспергированного в аргоне феррита висмута это обусловлено преимущественно ростом удельной поверхности катализатора, а для активированного в атмосфере водорода после 30-минутной обработки в аргоне – еще и увеличением поверхностной концентрации активных центров, которыми, вероятно, служат кислородные вакансии.
Список литературы
Stasinakis A.S. // Global NEST J. 2008. V. 10. № 3. P. 376. https://doi.org/10.30955/gnj.000598
Pare B., Jonnalagadda S.B., Tomar H., Singh P., Bhagwa V.W. // Desalination. 2008. V. 232. P. 80. https://doi.org/10.1016/j.desal.2008.01.007
Zhou B., Liu Z., Wang H., Yang N., Su W. // Catal. Lett. 2009. V. 132. P. 75. https://doi.org/10.1007/s10562-009-0063-3
Rauf M.A., Meetani M.A., Hisaindee S. // Desalination. 2011. V. 276. P. 13. https://doi.org/10.1016/j.desal.2011.03.071
Jing L., Zhou W., Tian G., Fu H. // Chem. Soc. Rev. 2013. V. 42. № 24. P. 9509. https://doi.org/10.1039/C3CS60176E
Hajjouji H.E., Barje F., Pinelli E., Bailly J.R., Richard C., Winterton P., Revel J.C., Hafidi M. // Bioresource Technol. 2009. V. 99. P. 7264. https://doi.org/10.1016/j.biortech.2007.12.054
Karunakaran C., Anilkumar P. // J. Mol. Catal. A: Chem. 2007. V. 265. P. 153. https://doi.org/10.1016/j.molcata.2006.10.016
Huang J., Tan G., Yang W., Zhang L., Ren H., Xia A. // Mater. Sci. Semicond. Process. 2014. V. 25. P. 84. https://doi.org/10.1016/j.mssp.2013.08.012
Wang X., Mao W., Zhang Q., Wang Q., Zhu Y., Zhang J., Yang T., Yang J., Li X., Huangac W. // J. Alloys Compd. 2016. V. 677. P. 288. https://doi.org/10.1016/j.jallcom.2016.02.246
Gao X., Da Y., Fu F., Hua X. // Solid State Sci. 2016. V. 62. P. 6. https://doi.org/10.1016/j.solidstatesciences.2016.10.014
Lingwei L., Meilin L., Gang L., Xiaoxiang X. // Appl. Surf. Sci. 2017. V. 391. P. 535. https://doi.org/10.1016/j.apsusc.2016.06.160
Tong T., Zhang H., Chen J., Jin D., Cheng J. // Catal. Commun. 2016. V. 87. P. 23. https://doi.org/10.1016/j.catcom.2016.08.030
Wang L., Niu C.-G., Wang Y., Wang Y., Zeng G.-M. // Ceram. Int. 2016. V. 42. № 16. P. 18605. https://doi.org/10.1016/j.ceramint.2016.08.204
Soltani T., Lee B.-K. // Chem. Eng. J. 2016. V. 306. P. 204. https://doi.org/10.1016/j.cej.2016.07.051
An J., Zhang G., Zheng R., Wang P. // J. Environ. Sci. 2016. V. 48. P. 218. https://doi.org/10.1016/j.jes.2016.01.024
Pattnaik S.P., Behera A., Martha S., Acharya R., Parida K. // J. Nanopart. Res. 2018. V. 20. № 1. P. 10. https://doi.org/10.1007/s11051-017-4110-5
Бутягин П.Ю. // Кинетика и катализ. 1987. Т. 28. № 1. С. 5.
Бутягин П.Ю. // Успехи химии. 1994. Т. 63. № 12. С. 1031.
Heinike G. Tribochemistry. Berlin: Akademie Verlag, 1984. 495 p.
Болдырев В.В. Экспериментальные методы в механохимии твердых неорганических веществ. Новосибирск: Наука, 1983. 65 с.
Авакумов Е.Г. Механические методы активации химических процессов. Новосибирск: Наука, 1986. 306 с.
Молчанов В.В., Буянов Р.А. // Успехи химии. 2000. Т. 69. № 5. С. 476.
Молчанов В.В., Буянов Р.А. // Кинетика и катализ. 2001. Т. 42. № 3. С. 406.
Boldyrev V.V., Tkačova K. // J. Mater. Synth. Process. 2000. V. 8. № 3/4. P. 121. https://doi.org/10.1023/A:1011347706721
Steinike U., Tkačova K. // J. Mater. Synth. Process. 2000. V. 8. № 3/4. P. 197. https://doi.org/10.1023/A:1011364110355
Митченко С.А. // Теоретическая и экспериментальная химия. 2007. Т. 43. № 4. С. 199.
Che D., Niu F., Qin L., Wang S., Zhang N., Huang Y. // Sol. Energy Mater. Sol Cells. 2017. V. 171. P. 24. https://doi.org/10.1016/j.solmat.2017.06.021
Di L., Yang H., Xian T., Chen X. // Materials. 2017. V. 10. I. 10. P. 1118. https://doi.org/10.3390/ma10101118
Wang S., Chen D., Niu F., Zhang N., Qin L., Huang Y. // J. Alloys Compd. 2016. V. 688. P. 399. https://doi.org/10.1016/j.jallcom.2016.07.076
Бутягин П.Ю. // Приборы и техника эксперимента. 1986. № 6. С. 201.
PDF-2: International Centre for Diffraction Data, № 73-548
PDF-2: International Centre for Diffraction Data, № 46-416
PDF-2: International Centre for Diffraction Data, № 72-1832
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ