

Кинетика и катализ, 2020, T. 61, № 3, стр. 312-333
Биоинспирированное окисление метана: от академических моделей метанмонооксигеназ к процессу прямого получения метанола
А. А. Штейнман *
ФГБУН Институт проблем химической физики РАН
142432 Черноголовка, просп. Акад. Семенова, 1, Россия
* E-mail: shteinman2002@mail.ru
Поступила в редакцию 08.11.2019
После доработки 31.01.2020
Принята к публикации 03.02.2020
Аннотация
Изучение механизма действия метанмонооксигеназ (ММО) в энзимологии и задача селективного окисления метана в химии являются двумя родственными, притягательными для многих ученых, параллельными фундаментальными направлениями, обогащающими и дополняющими одно другое. С прикладной точки зрения селективное окисление метана рассматривается как один из сильнейших вызовов для катализа. Несмотря на большие усилия, приемлемый для промышленности процесс прямого получения метанола, легко транспортируемого и универсального базового сырья, до сих пор не найден, и эта задача продолжает оставаться крайне актуальной. Большие надежды связаны с моделированием природных катализаторов ММО, способных при окружающих условиях превращать метан в метанол с селективностью до 100%. В обзоре представлены результаты длительных и драматических исследований, позволившие прояснить структуры активных центров и загадочные механизмы действия двух наиболее изученных ММО: железосодержащей растворимой (sMMO) и медьсодержащей мембраносвязанной (pMMO). Большое внимание в обзоре уделено различным биоинспирированным моделям этих ММО, как гомогенным, так и гетерогенным. Недавнее совершенствование гетерогенных химических аналогов ММО, эффективно катализирующих прямое окисления метана пероксидом водорода или О2 селективно до метанола, несомненно открывает новые горизонты для химической промышленности и активно обсуждается в последние годы на предмет адаптации к современным технологическим требованиям.
ВВЕДЕНИЕ
Аэробные метановые бактерии используют метан в качестве единственного источника строительного материала и энергии для своего существования. Первой, наиболее трудной стадией метаболизма метана с участием О2 является разрыв исключительно прочной связи С–Н метана (105 ккал/моль) с образованием метанола. Для этой цели метановые бактерии продуцируют два родственных биокатализатора: более активную, зависящую от ионов меди мембраносвязанную (particular) форму метанмонооксигеназы (рММО), а при дефиците меди – железосодержащую растворимую (цитоплазматическую) форму (sMMO). Обе оксигеназы содержат биядерные металлоцентры [1–3]. pMMO обладает высокой субстратной специфичностью, регио- и стереоселективностью. Гидроксилирование связи С–Н протекает с сохранением конфигурации при окисляемом центре.
В отличие от мембраносвязанной, растворимая форма изучена лучше, однако, несмотря на большие усилия многих исследователей в течение последних 25 лет, природа интермедиатов и интимный механизм действия sMMO до сих пор полностью не выяснены. Задачи полной расшифровки механизма биологического окисления метана с участием MMO, моделирования структуры ее активного центра (АЦ) и эффективного биоинспирированного окисления метана до метанола являются актуальным вызовом современной науки [4].
Оксигеназа sMMO была выделена и описана в 1970–1980 гг. Она сразу же привлекла большое внимание не только биохимиков, но и химиков благодаря уникальной способности катализировать селективное и специфичное окисление наиболее трудно окисляемого углеводорода метана до метанола в мягких, физиологических условиях, то есть при обычных температурах и давлениях кислородом воздуха в водной среде. Эта реакция была беспрецедентной для химиков, привыкших считать насыщенные углеводороды (алканы, парафины, от parum affinis – лишенные сродства) крайне инертными соединениями. Действительно, большинство реакций алканов требуют достаточно жестких условий (высокие температуры и давления, сильные реагенты) и, к тому же, дают большой набор продуктов, то есть являются неселективными.
Моделирование металлсодержащих ферментов – важная ветвь бионеорганической химии, представляющая интерес как для энзимологии, так и для металлокомплексного катализа. Информация, полученная при изучении простых химических моделей, позволяет подтвердить найденные при изучении самих ферментов характеристики их активных центров и выдвинутые гипотезы, а иногда и сформулировать новые идеи о структуре АЦ, строении интермедиатов и каталитическом цикле фермента. Действительно, химический механизм действия природного катализатора и структура интермедиатов не могут считаться надежно обоснованными, пока не будут подтверждены химическими моделями. С другой стороны, воспроизведение высокой активности и специфичности ферментов относительно простыми химическими аналогами позволяет расширить границы химического катализа. В качестве справедливости этих положений можно привести результаты, достигнутые в моделировании широко распространенной гемовой оксигеназы цитохрома Р-450, играющей исключительно важную роль в метаболизме животных и человека [5]. Изучение активации молекулярного кислорода простыми порфириновыми комплексами железа (гемами), спектральное наблюдение оксокомплексов высоковалентного железа (оксоферрильных комплексов) при взаимодействии гемов с донорами атома кислорода ХО (PhIO, NaOCl, RCO2OH, H2O2 и др.) при низких температурах и наблюдение селективного окисления алканов и олефинов при реакции их с оксоферрильными порфириновыми комплексами создали прочную основу для формулирования механизма гемовой монооксигеназы цитохрома Р-450. При этом постулируемый нативный феррильный интермедиат этой монооксигеназы удалось наблюдать только в самое последнее время вследствие его большой реакционоспособности и короткого времени жизни [5]. С другой стороны порфириновые комплексы металлов, биоинспирированные модели цитохрома Р-450, заняли в настоящее время достойное место в арсенале химических катализаторов для селективного окисления различных органических соединений, в том числе и в промышленности.
Давней и до сих пор нерешенной проблемой химии остается проблема прямого селективного окисления метана до метанола. Метанол является важным полупродуктом промышленного органического синтеза и может быть также использован как легко транспортируемый энергоноситель. Процесс производства метанола из метана – двустадийный, высокотемпературный и высокозатратный, сдерживает эффективное сырьевое использование метана. Eсли бы удалось сконструировать биомиметический индустриальный катализатор, воспроизводящий реакционную способность ММО, то крупномасштабное превращение метана в метанол могло бы существенно увеличить его использование как дешевого источника энергии и материала для разнообразных технологических процессов.
Несмотря на длительную историю исследования MMO и моделирования их АЦ, вопросы, почему они так активны в окислении метана в метанол, строение их активных интермедиатов и химический механизм долгое время оставались невыясненными. В этом отношении ситуация много лучше для моноядерных негемовых оксигеназ, для которых созданы довольно эффективные структурно-функциональные модели [6], и также для sMMO по сравнению с более сложной рММО; к тому же глубокое исследование последней началось сравнительно недавно.
Целью представленного обзора является обсуждение результатов, достигнутых к настоящему времени на пути изучения механизма действия MMO, их химического моделирования и рассмотрение перспектив дальнейшего развития этой области. Такое обсуждение кажется своевременным, хотя отдельные аспекты этой проблемы уже освещались в ряде предшествовавших обзоров [1–3, 7–9].
СТРУКТУРА АЦ sMMO И МЕХАНИЗМ АКТИВАЦИИ О2 И МЕТАНА
Оксигеназы
В ходе химической эволюции возникли уникальные биокатализаторы способные к селективному окислению углеводородов и других соединений молекулярным кислородом. Это гемовые оксигеназы высших организмов, такие как содержащий порфириновый комплекс железа цитохром Р-450 [10] и бактериальные негемовые оксигеназы, моно- и биядерные [11, 12]. Важное место среди последних занимает sMMO, принадлежащая к большому семейству многокомпонентных негемовых биядерных ферментов. Как и для других монооксигеназ, окисление трудно окисляемого субстрата с участием sMMO становится возможным благодаря сопряжению этой реакции с окислением физиологического восстановителя никотинадениндинуклеотида (NADH):
(I)
$\begin{gathered} {\text{RH}} + {{{\text{O}}}_{2}} + 2{{{\text{e}}}^{{\text{ - }}}} + 2{{{\text{H}}}^{ + }} = {\text{ROH}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}, \\ {\text{NADH}} = {\text{NA}}{{{\text{D}}}^{ + }} + 2{{{\text{e}}}^{{\text{ - }}}} + {{{\text{H}}}^{ + }}. \\ \end{gathered} $(II)
${\text{Fe}} + {{{\text{O}}}_{2}} + 2{{{\text{e}}}^{{\text{ - }}}} + 2{{{\text{H}}}^{ + }} = {\text{FeO}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}},$(III)
${\text{FeO}} + {\text{RH}} = \left\{ {{\text{FeOH}}\,\,\cdot\,\,{\text{R}}} \right\} = {\text{Fe}} + {\text{ROH}}.$Механизм оксигенирования связи С–Н по реакции (III) позволяет избежать неселективных радикально-цепных процессов и таким образом представляет важный элемент высокой организации фермента. Следствием быстрой рекомбинации углеводородного радикала с гидроксильной группой на железе является сохранение конфигурации (RC, retention of configuration) у атакуемого углеродного атома субстрата, то есть стереоспецифичность реакции, которая служит важным признаком участия в ней феррильного интермедиата. В случае выхода радикала из клетки происходит его рацемизация, и процесс становится нестереоспецифическим, что нередко наблюдается для модельных систем. Для проверки стереоспецифичности и природы АЦ используют различные субстратные пробы. Полное или значительное сохранение конфигурации указывает на двухэлектронный механизм и/или участие металлоцентра в отрыве атома Н от алкана (metal based mechanism).
sMMO
Оксигеназа sMMO состоит из трех компонентов, образующих белковый комплекс [3, 13]. Основной компонент, гидроксилаза (ММОН, 251 кДа), представляет собой димер, включающий три субъединицы (αβγ)2. Окисление метана происходит на биядерном металлоцентре, находящемся внутри белковой глобулы ММОН на расстоянии 12 Å от поверхности α-субьединицы. Второй компонент, редуктаза (MMOR, 38.5 кДа), является флавопротеином, который переносит электроны от NADH на ММОН. Восстановленная дитионитом гидроксилаза способна осуществлять стехиометрическое окисление метана кислородом (один цикл в отсутствие MMOR). Это доказывает, что функция последней состоит только в переносе электронов на гидроксилазу. Ферри-ММОН сама способна катализировать гидроксилирование метана пероксидом водорода вместо О2, но с меньшей эффективностью и иной региоселективностью в окислении н-алканов. Третий компонент MMOB (15.9 кДа) регулирует специфическую активность sMMO за счет индуцирования конформационных изменений в ММОН, в результате которых облегчается доступ О2 и метана к АЦ и ограничивается доступ протонов. Этот небольшой белок В, не содержащий какого-либо кофактора, связываясь с ММОН, увеличивает ее активность в 150 раз, ускоряет в 1000 раз восстановление О2, изменяет координационное окружение и спектроскопические характеристики Fe2-центра и влияет на регио- и стереоспецифичность реакций sMMO [3, 13]. Связывание белка В к гидроксилазе изменяет координационное окружение только у одного атома железа, помогая созданию свободного координационного места для взаимодействия с кислородом. В экспериментах было показано, что sMMO способна гидроксилировать гомологи метана, причем белок В сдвигает региоселективность в сторону первичных спиртов. Таким образом, взаимодействие между компонентами играет решающую роль в согласованном переносе электронов и протонов для активации О2 и метана и является биохимическим механизмом действия sMMO [1–3].
Строение активного центра sMMO
Строение АЦ sMMO было окончательно установлено в результате рентгеноструктурного анализа (РСА) ее ферри-формы в 1993 г. и восстановленной, функционально активной ферро-формы в 1995 г. [2], что, вместе с предшествовавшим десятилетним периодом интенсивных спектральных исследований [12], положило начало изучению химического механизма действия sMMO. Согласно РСА, Fe2-центр sMMO связан с белком посредством двух гистидинов и четырех глутаматов (рис. 1). В ферри-форме два атома железа связаны двумя группами ОН и карбоксилатным мостиком одного из глутаматов (рис. 1а). При восстановлении оба мостиковых гидроксида протонируются, давая две молекулы воды, одна из которых заменяется кислородным атомом глутамата в результате карбоксилатного сдвига, и расстояние между атомами железа изменяется от 3.1 до 3.3 Å (рис. 1б). Атомы железа в обеих формах являются неравноценными вследствие различного координационного окружения. Кроме того при восстановлении Fe(2) становится пятикоординированным со свободным координационным местом для связывания О2. Таким образом, в АЦ sMMO атомы железа с координированными аминокислотными остатками образуют биядерный биокомплекс железа, который можно рассматривать аналогично обычным химическим комплексам.
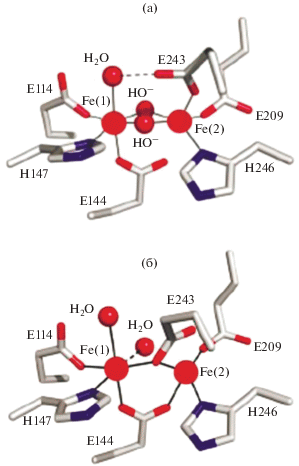
Активные интермедиаты и гипотеза мостикового механизма их образования
В 1993–1994 гг. с использованием оптической и мессбауэровской спектроскопии были охарактеризованы активные интермедиаты sMMO: пероксидный P и феррильный Q [3]. Бесцветный интермедиат Р быстро образуется после смешивания в проточной установке потоков ферро-ММОН и О2 и затем медленно превращается в желтый интермедиат Q. Интермедиат Р имеет оптический спектр с λмакс = 420, 720–725 нм. Его спектр Мессбауэра (δ = 0.66 мм/с, ΔEQ = 1.51 мм/с) характерен для высокоспинового Fe(III). На основании этих данных и теоретических расчетов для интермедиата Р предполагались структуры μ-1,2-пероксо, линейная (a) и зигзагообразная (gauche, b), а также структура μ-η2,η2-пероксида (c) (схема 1 ):
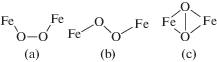
Схема 1 . Предполагаемые структуры интермедиата Р: μ-1,2-пероксо линейная (a) и зигзагообразная (gauche, b) и структура μ-η2,η2-пероксида (c).
Сравнение с модельными комплексами и пероксо-интермедиатами родственных Fe-оксигеназ предполагает для Р наиболее вероятной структуру (а). Интермедиат Р самопроизвольно превращается в интермедиат Q, ответственный за активацию связи С–Н в метане. Дальнейшие кинетические исследования каталитического цикла sMMO позволили идентифицировать еще четыре интермедиата, включая Т, который является продуктным комплексом. В отсутствие метана интермедиат Q накапливается в заметной концентрации и может быть сохранен методом быстрого замораживания (rapid freeze-quench, RFQ). Мессбауэровский спектр замороженных растворов Q указывает, что железо в нем находится в высоковалентном и высокоспиновом состоянии FeIV (S = 2). Интермедиат Q обладает двумя антиферромагнитно связанными атомами FeIV и имеет поглощение в области 420–430 нм. Будучи антиферромагнитно связанными, атомы железа образуют диамагнитный кластер FeIV–О–FeIV. Интермедиат Q реагирует с метаном с константой скорости 3–4 с–1 при 4°С, а в отсутствие субстратов превращается в неактивный интермедиат Q*, структура которого близка к структуре Т. Важным отличием реакций Р и Q с углеводородными субстратами является туннелирование атома Н в случае Q и нормальный кинетический изотопный эффект (КИЭ) для Р. Несмотря на большие усилия в исследовании механизма действия sMMO, точная структура интермедиатов Р и Q не доказана до последнего времени. В ряде ранних работ предполагалось, что каталитический цикл sMMO может быть подобен хорошо установленному каталитическому циклу гемовой оксигеназы цитохрома Р-450, содержащей протопорфириновый комплекс железа с аксиальным тиолатным лигандом. Отличительным элементом химического механизма цитохрома Р-450 является участие протонов в активации О2 c гетеролитическим разрывом связи О–О.
С учетом биядерной структуры АЦ sMMO и некоторых спектральных исследований в 1995 г. был предложен альтернативный механизм sMMO [14] (схема 1 ), названный мостиковым для различения с терминальным механизмом цитохрома Р-450.
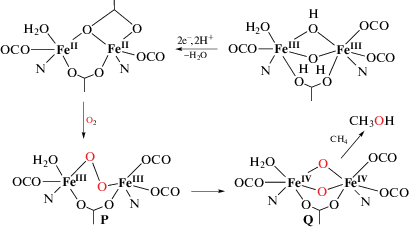
Схема 2 . Мостиковый механизм активации О2 и метана в АЦ sMMO [14].
Сущность нового механизма составляют две принципиальные особенности. Во-первых, для интермедиата Q в мостиковом механизме была предположена структура бис-µ-оксокомплекса высоковалентного железа ${\text{Fe}}_{{\text{2}}}^{{{\text{IV}}}}$(μ-O)2 на основании имевшихся данных мессбауэровской спектроскопии и некоторых химических аналогий. Действительно, подобный Mn2(μ-O)2-комплекс является сильным окислителем, способным окислять воду до О2. Во-вторых, предполагалось, что активация О2 начинается с образования мостикового пероксидного интермедиата, и последующий гомолитический разрыв связи О–О не требует участия протонов в этом механизме (схема 2 ). Необходимый для завершения каталитического цикла перенос на железо 2e- и 2H+ с выделением молекулы воды в мостиковом механизме осуществляется после завершения реакции интермедиата Q с алканом, а не до, как в случае цитохрома Р-450. Это обстоятельство сохраняет гидрофобную полость sMMO до и во время реакции оксигенирования алкана более сухой, что может иметь преимущества для связывания гидрофобного субстрата, такого как метан, контроля подачи протонов в АЦ и поддержания высокой реакционной способности феррильного интермедиата в этих условиях, а также для диффузии образовавшегося метанола в водную среду. Таким образом, можно отметить различную последовательность отщепления фрагментов молекулы О2 при ее активации в АЦ монооксигеназ: О2–/О – в случае моноядерных комплексов, О/О2– – в случае биядерных комплексов железа.
В последующие годы мостиковый механизм получил ряд экспериментальных и теоретических подтверждений, был уточнен и модифицирован. Подтверждение циклической структуры интермедиата Q было достигнуто методом рентгено-абсорбционной спектроскопии EXAFS [15]. Получены модельные комплексы с ядром Fe2(μ-O)2 [16], и такая структура для интермедиата Q sMMO согласуется с теоретическими расчетами [17, 18]. Несмотря на это, структура интермедиата Q вызывала большие сомнения и споры [18, 19]. Квантово-химические расчеты не cмогли воспроизвести расстояние r(Fe⋅⋅⋅Fe) = 2.46 Å между атомами железа, предсказанное из EXAFS-измерений. К тому же первый синтетический комплекс с ядром [${\text{Fe}}_{{\text{2}}}^{{{\text{IV}}}}$(μ-O)2] имел значительно большее r(Fe⋅⋅⋅Fe) = 2.73 Å и был малоактивным [20]. Впрочем в качестве более активной формы интермедиата Q может выступать один из электромеров [${\text{Fe}}_{{\text{2}}}^{{{\text{IV}}}}$(μ-O)2] [14], который бы образовался при связывании белка В или метана с ММОН. Структура интермедиата Q могла бы проясниться с помощью резонансной рамановской спектроскопии (RRS), однако препятствием для использования этого метода были низкая достижимая концентрация Q и, возможно, его разрушение лазером в результате облучения; до последнего времени этот эксперимент не был успешным.
Была получена также информация, которая не согласовывалась с первоначальной версией мостикового механизма для sMMO. Оказалось, что на стадии активации О2 необходимо участие двух протонов [21]. В связи с этим многие авторы предпочитали для sMMO модифицированный мостиковый механизм с участием протона в разрыве связи О–О, напоминающий гетеролитический механизм действия цитохрома Р-450 (см. однако [22]). Стоит еще раз подчеркнуть, что принципиальным отличием мостикового механизма является участие обоих атомов О2 в образовании интермедиата Q и отсутствие необходимости Н+ для разрыва связи О–О.
Важным подтверждением мостикового механизма для sMMO и, в частности, возможности активации О2 без участия протонов было наблюдение обратимого превращения полученного с использованием лиганда 2 (схема 3 ) пероксо-производного комплекса 7 (схема 4 ) в диферрил (рис. 2) [23, 24].
Рис. 2.
Обратимое превращение пероксо-${\text{Fe}}_{{\text{2}}}^{{{\text{III}}}}$-комплекса в диферрил [23].
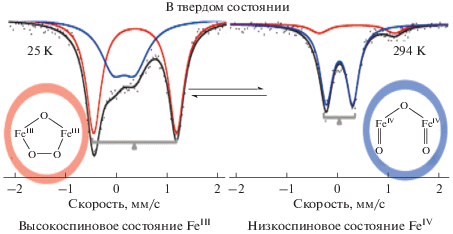
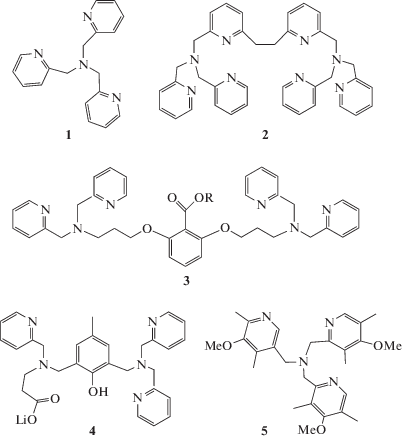
Схема 3 . Лиганды, моделирующие координационное окружение АЦ sMMO.
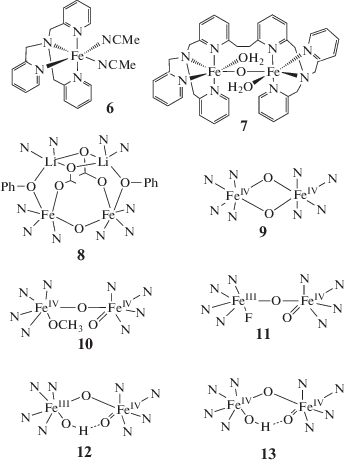
Схема 4 . Модельные комплексы для АЦ sMMO (6–8) и интермедиата Q (9–13 на базе лиганда 5 (схема 2 )). В комплексах 8–13 пиридины представлены только N-донором.
Пероксидный комплекс был приготовлен реакцией пероксида водорода с комплексом 7 в растворе ацетонитрила при –40°С. Поскольку этот пероксидный комплекс неустойчив в растворе даже при такой низкой температуре, он был высажен добавлением эфира и подвергнут изучению в сухом твердом состоянии методом температурноварьируемой мессбауэровской спектроскопии в интервале 25–294 К (рис. 2).
В спектрах на рис. 2 наблюдаются два квадрупольных дублета: в то время как один имеет параметры типичные для (μ-оксо)(μ-1,2-пероксо)комплексов железа (моделей P), другой напоминает параметры комплексов FeIV=O (S = 2) (моделей Q). Равновесие между пероксидным интермедиатом и диферрилом зависит от температуры, сдвигаясь в сторону пероксида при низких температурах и в сторону диферрила при повышении температуры.
БИЯДЕРНЫЕ КОМПЛЕКСЫ ЖЕЛЕЗА КАК СТРУКТУРНЫЕ И ФУНКЦИОНАЛЬНЫЕ МОДЕЛИ АЦ sMMO
Биядерные комплексы железа исследовали главным образом как структурные или спектроскопические модели АЦ sMMO [26, 27]. Значительно меньше изучено их участие в каталитическом окисления углеводородов. В большинстве работ использовали системы FeIII-комплекс–Н2О2–MeCN–циклогексан, и сравнительно мало исследований посвящено непосредственно окислению метана. Данные о механизме гидроксилирования алканов c участием этих комплексов представляют большой интерес в свете проблемы моделирования sMMO, однако они редки и не всегда обстоятельны. В ряде случаев реальным катализатором мог быть моноядерный комплекс железа, образовавшийся в результате расщепления биядерного центра [28]. В других высокий выход продуктов окисления, скорее всего, связан с развитием радикально-цепных реакций, инициированных металлокомплексом. Решающим фактором в создании биомиметического катализатора является рациональный дизайн лиганда.
В то время как монооксигеназы более богаты кислородными донорами у металла, в химических моделях обычно используют полидентатные N-донорные лиганды (схема 3 ), поскольку они образуют более стабильные комплексы. Например, биядерные комплексы железа с лигандом 2 являются эффективными катализаторами гидроксилирования алканов с участием мета-хлорпербензойной кислоты как окислителя [29, 30]. В качестве кислородного лиганда предпочтителен карбоксилат, обладающий с точки зрения возможных стерических затруднений минимальным объемом и имеющий высокую устойчивость в агрессивных окислительных системах. Было показано [29] эффективное каталитическое эпоксидирование олефинов пероксидом водорода с использованием биядерного комплекса железа 7 (схема 4 ) с гексапиридиламиновым динуклеирующим лигандом 2 (схема 3 ). Однако замещение двух пиридильных доноров в этом лиганде на карбоксилатные группы хотя и привело к существенному ускорению расщепления связи О–О в пероксидном интермедиате, но одновременно снизило каталитическую эффективность этого катализатора в 3000 раз [30].
Ранние химические модели биядерных оксигеназ
Прорыв в функциональном моделировании негемовых оксигеназ был достигнут в 1996 г., когда на ранних химических моделях биядерных оксигеназ была продемонстрирована возможность переноса атома кислорода в связь С–Н алканов по скрыто-радикальному механизму отщепления–рекомбинации, подобному механизму гемовых оксигеназ (III). В качестве моделей АЦ ферри-sMMO были использованы биядерные комплексы железа с бипиридильными (bpy) и фенантролиновыми (phen) лигандами – [Fe2O(bpy)2(RCOO)2(H2O)2](ClO4)2 и [Fe2O(phen)4(H2O)2](ClO4)4 [31]. Оба комплекса содержат молекулы воды, легко замещаемые пероксидом водорода, которые маркируют так называемые лабильные координационные места каталитического металлоцентра, необходимые для связывания субстрата, и катализируют при комнатной температуре окисление циклогексана пероксидом водорода в ацетонитриле до спирта и кетона. Реакция протекает с заметным кинетическим изотопным эффектом, указывающим, что разрыв связи С–Н происходит в лимитирующей стадии. Число оборотов катализатора TON (turnover number) доходит до 30. Этот параметр обычно используется как характеристика жизнеспособности катализатора или каталитической системы. Выход продуктов окисления в расчете на пероксид водорода достигает 25% за 2 ч. Две группы наблюдений дают основания рассматривать эти комплексы как простейшие модели АЦ sMMO. При окислении метана (40–50 ат, 20°С) в этих системах происходит образование метанола с выходом около 1–2% на пероксид:
Более важно, что на примере циклогексана здесь впервые для негемовых комплексов была показана возможность двухэлектронного переноса атома О на связь С–Н, ранее известная только для порфириновых комплексов железа, моделей гемовой оксигеназы цитохрома Р-450.
Действительно (схема 5 ), основным продуктом окисления олефинов в этих системах является эпоксид, что не характерно для свободно-радикальных реакций, для которых типично аллильное окисление олефинов. При гидроксилировании третичной связи С–Н в цис- или транс-1,2-диметилциклогексане происходит заметное сохранение конфигурации у атакуемого третичного углеродного атома (RC = 35–70%), т.е. только часть образующихся радикалов R· выходит из сферы железа в раствор и рацемизуется. В случае свободно-радикального окисления RC как правило близко к 0. Позднее для моноядерных комплексов железа типа [Fe(TPA)(H2O)2](ClO4)2 (6, схема 4 ) с трипиколиламинным (TPA) лигандом (1, схема 3 ), изучаемых в качестве моделей моноядерных негемовых оксигеназ, удалось наблюдать почти полное сохранение конфигурации (RC ≈ 100%) в системах с пероксидом водорода [32]. Для этих комплексов также отмечалось вхождение 18О в образующийся спирт при проведении оксигенирования в присутствии меченой по кислороду воды Н218О, которое по аналогии с гемовыми системами обычно рассматривается как доказательство участия феррильного интермедиата в реакции. При гидроксилировании метильной группы транс-1,2-метилфенилциклопропана, катализируемом комплексом [Fe2O(phen)4(H2O)2](ClO4)4, наблюдается заметное сохранение целостности циклопропанового кольца в образующемся спирте (схема 5 ); в случае же появления на углероде, соседнем к циклопроановому кольцу, долгоживущего радикала должна происходить перегруппировка с раскрытием цикла.
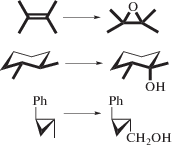
Схема 5 . Тесты на скрыто-радикальный двух-электронный механизм переноса атома О на связь С–Н. Сверху вниз: эпоксидирование, сохранение конфигурации, сохранение циклопропанового кольца.
Продвинутые модели с каркасными лигандами
Уникальная активность ферментов обусловлена высокой степенью организации их АЦ, которая достигается посредством относительно жесткого полипептидного каркаса. Очевидно, в ходе эволюции ММО сложился оптимальный баланс между жесткостью и гибкостью такого каркаса. В случае АЦ sMMO остатки аминокислот, связанные с этим каркасом, образуют пространственно организованную координационную сферу комплекса, которая комплементарна с биядерным центром и обеспечивает его целостность в течение всего каталитического цикла. Индивидуальный состав доноров и стереохимия этой координационной сферы, включая наличие и положение лабильных координационных мест, задается полипептидной структурой. С другой стороны, этот каркас должен адаптироваться к переходным состояниям и интермедиатам в ходе каталитического цикла.
Недостаток простейших комплексов в качестве функциональных моделей АЦ sMMO заключался в потере их индивидуальности при катализе вследствие диссоциации монодентатных лигандов в растворе. Дальнейшая эволюция моделей требовала закрепления лигандов на некотором каркасе подобно опыту Природы. Хотя подобные лиганды в химии обычно называют полидентатными, наименование “каркасные” более уместно в контексте данного обзора и, как будет показано далее, оно гораздо шире, так как включает и другие типы лигандов.
В соответствии со структурой АЦ sMMO дизайн и синтез первого каркасного лиганда (3, схема 3 ) предусматривал включение центрального бидентатного карбоксилата и наличие в модельном комплексе необходимых для катализа лабильных координационных мест. При взаимодействии лиганда 3 с перхлоратом железа(III) получен комплекс железа(III) состава [Fe2O(3)(H2O)2](ClO4)3 (3а), который при реакции с бензойной кислотой дает другой комплекс [Fe2O(3)(PhCOO)](ClO4)2 (3b). Монокристаллы комплексов были охарактеризованы с помощью рентгеноструктурного анализа (РСА), а их растворы – методом электроспрей-ионизационной масс-спектрометрии (ESI-MS), и показали высокую каталитическую активность в окислении алканов Н2О2 в ацетонитриле при 20°С [33–35]. Так, в окислении циклогексана при катализе комплексом 3а с выходом 50% на пероксид получены спирт (А) и кетон (К) (А/К = 4). При проведении реакции в присутствии меченой по кислороду воды ${\text{Н}}_{{\text{2}}}^{{{\text{18}}}}{\text{О}}$ около 13% образующегося спирта содержат меченый кислород в гидроксильной группе, указывая на участие в реакции окисления феррильного интермедиата. Этот вывод подтверждается высокой стереоспецифичностью в окислении 1,2-цис-диметилциклогексана (RC = 94%). В окислении метана ТON = 2 при 20°С и увеличивается до 7 при 60°С.
Согласно данным РСА комплексы 3а и 3б являются димерами ожидаемых биядерных комплексов и имеют формально тетраядерную структуру [33]. С другой стороны, согласно ЭСМС в растворе ацетонитрила (MeCN) имеется равновесие между биядерными комплексами и их димерами, зависимое от концентрации комплексов в растворе. На рис. 3 представлено строение биядерного фрагмента 3а. Кислородные атомы О1, О6 и О7 маркируют четыре лабильных координационных места, по два у каждого атома железа. Существенно, что эти лабильные координационные места находятся в благоприятной конфигурации для связывания и активации О2 в соответствии с мостиковым механизмом. Расстояние между атомами железа Fe(1) и Fe(2) (3.1Å) также хорошо подходит для образования пероксидного комплекса. Поскольку в димере биядерные центры достаточно отдалены друг от друга, они должны работать независимо. Действительно, никаких экспериментальных подтверждений обратного не найдено.
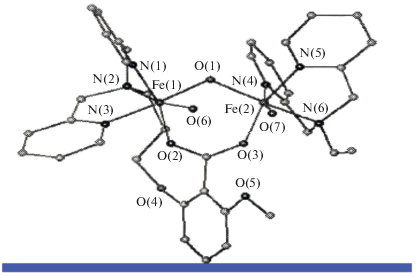
Так как функционально активной является восстановленная форма фермента ферро-sMMO, была предпринята попытка приготовить модель этой формы с лигандом 3 (схема 3 ). Соответствующий комплекс железа(II) синтезирован реакцией натриевой соли лиганда с трифлатом железа FeII(OTf)2(MeCN)2 в растворе MeCN в отсутствие О2 [36]. Ожидалось, что реакция этого комплекса с О2 будет моделировать активацию последнего и, возможно, позволит получить прямые доказательства мостикового механизма. Однако данные о кристаллической структуре комплекса показали, что за счет вовлечения в координацию с железом(II) эфирных кислородных атомов лиганда 3 иммобилизованный карбоксилат в этом комплексе принял довольно редкую анти,анти-конфигурацию в отличие от обычной син,син-конфигурации, такой как в АЦ sMMO. Вследствие этого расстояние Fe–Fe увеличилось до 6.23 Å и стало слишком большим для мостикового связывания О2 с образованием пероксидного интермедиата. Терминальный пероксидный интермедиат также не мог образоваться вследствие отсутствия лабильных координационных мест. Таким образом, при переходе к модели АЦ ферро-sMMO обнаружился главный недостаток первого каркасного лиганда – нарушение баланса между жесткостью и гибкостью.
Многочисленные биядерные комплексы железа с каркасными лигандами, которые моделируют структурные и, в меньшей степени, функциональные черты АЦ sMMO, включая его полипептидное окружение, описаны в прекрасном обзоре [25]. Важным достижением авторов был синтез модели ферро-формы АЦ sMMO с ранее недоступной конфигурацией двух N-доноров в син-ориентации по отношению к вектору Fe–Fe, как в АЦ sMMO. При взаимодействии некоторых из этих комплексов с О2 удается наблюдать образование пероксидных интермедиатов.
Необычный комплекс [${\text{Fe}}_{{\text{2}}}^{{{\text{III}}}}{\text{O}}$(4)]2+, полученный с лигандом 4 (схема 3 ), согласно РСА имеет строение тетраядерного гетерокластера Fe2Li2 (8, схема 4 ) и содержит встроенный биядерный ферри-центр, обладающий интересными свойствами [37]. В этом центре связи карбоксилатов с атомами железа являются наиболее слабыми и потому лабильными, то есть легко замещаются на молекулы растворителя. В окислении насыщенных углеводородов пероксидом водорода с комплексом 8 в качестве катализатора характер образующихся продуктов и тесты на механизм доказывают участие в реакции феррильных интермедиатов. С другой стороны, низкотемпературная УФ-спектроскопия и ESI-MS позволяют обнаружить и охарактеризовать короткоживущий пероксидный интермедиат [Fe2O(4а)(O2)], очевидно находящийся в равновесии с диферрильным интермедиатом [(Fe=O)2(O)(4а)], который и является ключевым окислителем в этой реакции. Это первая каталитическая окислительная система с биядерным ферри-центром, которая предположительно моделирует образование и взаимопревращение P- и Q-подобных интермедиатов в окислительном процессе.
МОДЕЛИ ИНТЕРМЕДИАТА Q И ИХ РЕАКЦИОННАЯ СПОСОБНОСТЬ
Наблюдение интермедиатов при взаимодействии биядерных карбоксилатных комплексов FeII с молекулярным кислородом
Одним из главных вопросов в изучении химического механизма sMMO является уточнение структуры интермедиата Q и выяснение его реакционной способности. В связи с этим большой интерес приобретает изучение биядерных оксокомплексов высоковалентного железа. Имеется много литературных данных о реакционной способности моноядерных оксоферрильных комплексов, как гемовых, так и негемовых [38, 39]. В связи с тем, что феррильные интермедиаты наиболее активных каталитических систем, как правило, неустойчивы даже в условиях очень низких температур, обычно синтезируют и исследуют менее активные, но более устойчивые феррильные комплексы азотсодержащих лигандов. Особый интерес представляет изучение высокоспиновых оксоферрильных комплексов железа и комплексов, содержащих кислородные доноры, так как эти факторы считаются решающими для высокой реакционной способности природных оксигеназ. Синтез комплексов FeII общей формулы [Fe2(O2CR)4L2] (схема 6 ), содержащих в качестве лигандов L объемистые мета-терфенилкарбоксилаты, позволяет избежать обычного для карбоксилатов железа образования олигомеров и является важным прорывом в моделировании богатой кислородными донорами структуры биядерного центра sММО [40].
Схема 6 . Карбоксилатные модели с объемистыми лигандами: L = 2,6-ди(р-мезитил)бензоат (–O2CArMes) или 2,6-ди(р-толил)бензоат (–O2CArTol).
Эти комплексы могут присоединять и активировать О2 с образованием при низких температурах различных кислородных интермедиатов и дают возможность исследовать реакции последних с углеводородами. Так, взаимодействие комплекса [Fe2(O2CR)4L2] (L = 4-tBuPy и Py) с О2 при ‒78°С в дихлорметане или толуоле приводит к смеси комплексов FeIIFeIII и FeIIIFeIV (1 : 1), вероятно вследствие реакции образующегося аналога интермедиата Q с исходным комплексом:
(IV)
$\begin{gathered} {\text{F}}{{{\text{e}}}^{{{\text{II}}}}}{\text{F}}{{{\text{e}}}^{{{\text{II}}}}} + {{{\text{О}}}_{2}} \to {\text{F}}{{{\text{e}}}^{{{\text{III}}}}}{\kern 1pt} --{\kern 1pt} {\text{O}}{\kern 1pt} --{\kern 1pt} {\text{O}}{\kern 1pt} --{\kern 1pt} {\text{F}}{{{\text{e}}}^{{{\text{III}}}}} \to \\ \to {\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{{\left( {\text{O}} \right)}_{2}}{\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}, \\ \end{gathered} $(V)
$\begin{gathered} {\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{{\left( {\text{O}} \right)}_{2}}{\text{F}}{{{\text{e}}}^{{{\text{IV}}}}} + {\text{F}}{{{\text{e}}}^{{{\text{II}}}}}{\text{F}}{{{\text{e}}}^{{{\text{II}}}}} \to \\ \to {\text{F}}{{{\text{e}}}^{{{\text{II}}}}}{{\left( {\text{O}} \right)}_{2}}{\text{F}}{{{\text{e}}}^{{{\text{III}}}}} + {\text{F}}{{{\text{e}}}^{{{\text{III}}}}}{{\left( {\text{O}} \right)}_{2}}{\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}. \\ \end{gathered} $Феррильные биядерные комплексы железа
Большая работа была выполнена по синтезу биядерных комплексов железа, содержащих один или два феррильных центра, и исследованию их реакционной способности. К настоящему времени синтезированы многочисленные циклические и открытые (нециклические) биядерные Fe2O2-комплексы высоковалентного железа, где в качестве лиганда часто используется тетрадентатный лиганд трипиколиламин (tpa) (1, схема 3 ) или его алкоксильные и/или алкильные замещенные, такие как tpa* (5, схема 3 ) [41]. Электронодонорные заместители на каждом из пиридинов лиганда 5 (схема 3 ) позволили увеличить термическую стабильность новых комплексов железа. В этих комплексах один или оба атома железа находятся в состоянии FeIV. Некоторые из вышеуказанных комплексов воспроизводят предполагаемую бис-μ-оксо-мостиковую структуру интермедиата Q и моделируют отдельные окислительные реакции монооксигеназ, хотя гидроксилирование метана не изучалось ни в одном случае. Первый синтетический комплекс, частично воспроизводящий предполагаемую бис-μ-оксо-${\text{Fe}}_{{\text{2}}}^{{{\text{IV}}}}$-структуру Q, комплекс 9 (схема 4 ), был получен прямым взаимодействием µ-оксокомплекса FeIII с пероксидом водорода в присутствии хлорной кислоты [19] или электрохимическим окислением его FeIIIFeIV-предшественника [20]. Хотя новый комплекс формально моделирует предполагаемую циклическую структуру ядра интермедиата Q, правда без мостикового карбоксилата, его активность много ниже. Это подтверждает, что беспрецедентная активность интермедиата Q в реакции с метаном обусловлена множеством тонких структурных факторов, которые еще предстоит понять.
В последнее десятилетие были получены модели интермедиата Q с открытым Fe2O2-ядром типа 10 и 11 (схема 4 ) [42, 43]. Комплекс 10 имеет мессбауэровские параметры близкие к параметрам интермедиата Q [41]. Комплекс 11 оказался наиболее реакционноспособным из всех модельных комплексов интермедиата Q – его отличие в реактивности от других моделей достигает 7.5 порядков величины [41]. Образование водородной связи между феррильным О и ОН-группой превращает линейные комплексы в псевдоциклические 12 и 13 [41, 42]. Синтез и изучение комплексов 9–13 позволяют сделать выводы о влиянии структурных факторов на спектральные свойства и реакционную способность этих моделей и продвинуться в понимании природы интермедиата Q. В работе [41] на основании исследования многочисленных химических моделей авторы заключают, что “два главных фактора контролируют скорость разрыва связи С–Н: будет ли оксо-группа мостиковой или терминальной и будет ли спиновое состояние Fe=O-центра 1 или 2”. Авторы [41] показали, что терминальный О реагирует со связью С–Н в 100 раз быстрее мостикового, и FeIV=O (S = 2) оказался в 103–104 раз более реактивным, чем FeIV=O (S = 1).
Поскольку моноядерные негемовые оксигеназы не способны окислять метан, возникает вопрос о роли второго атома железа в формировании уникальной активности биядерного центра sММО. Недавно [44] была показана активация негемового пероксидного интермедиата Fe–OOH в моноядерном модельном комплексе [FeII(β-BPMCN)(CH3CN)2]2+ (где BPMCN = = N,N'-бис(пиридил-2-метил)-N,N'-диметил-транс-1,2-диаминоциклогексан) с помощью FeIII(OTf)3 с образованием сильного окислителя, способного быстро окислять циклогексан при ‒40°С. Величина КИЭ, 100% включение 18О из Н218О2, высокое отношение А/К – все указывает на возможное участие в реакции окислении перферрильного интермедиата FeV=O. Авторы [44] предполагают, что Люисовская кислота FeIII(OTf)3 атакует терминальный атом кислорода гидропероксо-лиганда, облегчая гетеролиз связи О–О. По сравнению с известными моноядерными негемовыми окислителями, новый высоковалентный интермедиат является не только наиболее активным, но также и наиболее селективным в гидроксилировании циклогексана до циклогексанола. На основании полученных результатов авторы [44] предполагают, что второй атом железа в АЦ sMMO действует не только как редокс-центр, но также как сильная Люисовская кислота, чтобы генерировать мощный окислитель, способный гидроксилировать метан.
НЕДАВНИЙ ВКЛАД НОВЕЙШИХ СПЕКТРОСКОПИЧЕСКИХ МЕТОДОВ В ИЗУЧЕНИЕ ХИМИЧЕСКОГО МЕХАНИЗМА ДЕЙСТВИЯ sMMO И ПРИРОДЫ КЛЮЧЕВОГО АКТИВНОГО ИНТЕРМЕДИАТА Q
Строение биядерного феррильного интермедиата Q окончательно не выяснено. Наиболее вероятными (без учета изменений в координационном окружении) являются равновесные структуры FeIV(µ‑O)2FeIV$ \rightleftarrows $ O=FeIV⋅⋅⋅O=FeIV$ \rightleftarrows $ $ \rightleftarrows $ FeIII–O–FeV=O, причем первая, возможно, соответствует структуре Q в отсутствие субстрата (метана), а две последние могут быть его активной формой, образующейся при вхождении метана в активный центр [14]. Квантово-химические расчеты модельного диферрильного интермедиата [(EDTAH)FeIV=O]2, где EDTAH – монопротонированный этилендиамитетраацетатный лиганд, показали [45], что в водном окружении он связан с одной молекулой воды и находится в высокоспиновом основном состоянии S = 4. Включение молекулы Н2О и одновременное вытеснение карбоксилатного донора из внутренней координационной сферы превращает один из атомов железа в пятикоординированный и изменяет энергию и форму орбиталей оксо-групп относительно газовой фазы. Это приводит к относительной стабилизации орбитали 3σ* на одной из FeIV=О-групп и существенному увеличению акцепторной способности диферрильного интермедиата к отрыву Н от метана. Полученные результаты хорошо дополняют и развивают выводы, сделанные ранее при квантово-химическом моделировании интермедиата Q [17]
Не вызывает сомнения, что детали геометрической и электронной структуры крайне реактивного интермедиата Q представляют большой интерес для понимания удивительной способности sMMO окислять метан в физиологических условиях. Несмотря на многолетние интенсивные исследования химиков и биологов, вопросы об интимном механизме sMMO и структуре ключевого интермедиата Q остаются спорными. Особенное недоумение вызывают не согласующиеся между собой выводы разных работ о структуре ядра Fe2O2 интермедиата Q: замкнутой циклической, то есть бис-μ-оксо или открытой линейной с терминальным оксоферрилом FeIV=O. Дальнейший прогресс в понимании структуры Q и его уникальной реакционной способности связан со стремительным развитием спектроскопических методов исследования металлсодержащих ферментов. Успех этих методов в случае sMMO также обусловлен возможностью сравнения полученных спектров со спектрами ранее синтезированных и хорошо охарактеризованных биядерных высоковалентных модельных комплексов железа.
Резонансная рамановская спектроскопия
Препятствия для использования RRС в исследовании Q удалось недавно преодолеть авторам [46] с помощью специально сконструированного и оптимизированного инструмента [47] и применения оригинального подхода. Был использован метод RR-спектроскопии с разрешением во времени (TRRR – time-resolved resonance Raman), измерения проводили в потоке быстро смешивающихся струй реагентов в точке максимальной концентрации Q, и сигнал накапливался в течение многих часов. В дифференциальном (16О2–18О2) RRС в системе “восстановленная MMOH–белок B–О2” были обнаружены слабые сигналы колебаний атомов кислорода, связанных с железом, при 690 и 654 см–1. Добавление в систему метана позволило приписать исчезающий сигнал при 690 см–1 интермедиату Q, а появляющийся сигнал при 556 см–1 – продуктному интермедиату Т. Заключения о структуре Q были сделаны на основании сравнения его RRС с соответствующими спектрами синтетических модельных комплексов. Частота колебания 690 см–1 и изотопный сдвиг Δ18О равный 36 см–1 были близки только к соответствующим значениям в RR-спектрах комплекса 9 (схема 4 ) с ядром [${\text{Fe}}_{{\text{2}}}^{{{\text{IV}}}}$(μ-O)2], что убедительно подтверждало для интермедиата Q структуру бис-µ-оксокомплекса железа. Характерной спектральной чертой такой структуры является присутствие двух колебательно связанных μ-O-мостиков, приводящее к тетраатомным колебаниям с уникальной спектральной частотой (673 см–1) [48]. Появление новой частоты 673 см–1 в спектре Q при использовании смешанного изотопомера кислорода (16О18О) стало дополнительным доказательством такой структуры. Отсутствие этого колебания в дифференциальном (16О2–18О2) спектре, как и равная интенсивность характеристических частот (690 и 654 см–1) в RRС Q (16О2) и Q (18О2) доказывало отсутствие заметного обмена между μ-O и водой. Важным выводом из этих результатов было заключение, что интермедиат Q содержит оба атома исходной молекулы О2. Подобным образом было найдено, что Т включает только один атом О из О2 и имеет структуру Fe–O–Fe, поскольку неизменность спектров при замене Н2О на D2O указывала на μ-O, а не μ-ОН. Вместе с предыдущей информацией о вхождении одного атома из молекулы О2 в образующийся спирт эти данные позволяли проследить путь каждого атома О на протяжении полного каталитического цикла. Наблюдение, что оба атома О в интермедиате Q происходят из одной и той же молекулы О2 свидетельствует: разрыв связи О–О в интермедиате Р происходит гомолитически без непосредственного участия протона и, следовательно, механизмы активации О2 различны для sMMO и цитохрома Р-450, как и предполагалось ранее [49], а именно, мостиковый гомолитический – в первом случае и терминальный гетеролитический – во втором. Таким образом, казалось, что в многолетней дискуссии о механизме sMMO наконец поставлена последняя точка.
Рентгеновская спектроскопия
Использование несколькими годами позже продвинутых методов рентгено-абсорбционной спектроскопии (XAS) для более глубокого исследования природы интермедиата Q вновь добавило разногласия в результаты разных работ. Новейшие методы флюоресцентного детектирования сигнала с высокоэнергетическим разрешением HERFD (High-Energy Resolution Fluorescence Detected) XAS и HERFD EXAFS [50, 51] позволили преодолеть главные ограничения стандартных методов XAS и получить беспрецедентные детали электронного и геометрического строения металлоцентра интермедиата Q.
Сравнение спектров HERFD XAS интермедиата Q с соответствующими спектрами родственных модельных комплексов железа в сочетании с квантово-химическими расчетами методом TDDFT (time dependent density functional theory) привело авторов работы [52] к заключению, что полученные ими результаты лучше согласуются с открытой структурой ядра Fe2O2 интермедиата Q. Действительно, наблюдалось сильное различие в энергии и интенсивности сигналов в прикраевой области (pre-edge) спектра для открытого и замкнутого ядра Fe2O2 в модельных комплексах 9 и 13 (схема 4 ). Эти различия хорошо воспроизводились расчетами методом TDDFT и открывали возможность прямой корреляции найденных спектральных характеристик с геометрией и электронной структурой локального ядра Fe2O2 интермедиата Q.
Ранее на основании анализа кристаллических структур модельных комплексов и теоретических расчетов интермедиата Q было сделано заключение, что ключевым параметром для различения структур ядра Fe2O2 может быть расстояние Fe–Fe: ~3.4 Å для открытой структуры и 2.7–2.8 Å для замкнутой [53]. Недавно метод HERFD EXAFS, более точный относительно стандартного EXAFS, позволил получить дальнейшие подтверждения открытой структуры ядра Fe2O2 интермедиата Q [54] благодаря уточненным спектральным доказательствам более длинного расстояния между атомами железа (~3.4 Å) как в модельных комплексах 10–13 (схема 4 ) [41, 42] по сравнению с коротким расстоянием (~2.5 Å), измеренным ранее стандартным методом EXAFS. В то же время авторы работы [54] не считают этот результат приемлемым для окончательного установления структуры Q вследствие недостаточного разрешения полученных спектров и остающихся противоречий с другими методами. В связи с этим рассматриваются другие возможные структуры, в том числе квазициклическая, типа 13 (схема 4 ), в которой терминальный оксоферрил связан сильной Н-связью с ОН-группой при соседнем атоме железа. Нерешенные противоречия указывают на необходимость дополнительных синтетических моделей и продвинутых QM/MM расчетов.
АЦ рММО И ХИМИЧЕСКИЕ МОДЕЛИ
Структура АЦ и механизм действия pMMO
В то время как многие черты строения и функционирования sMMO были раскрыты еще в прошлом столетии, понимание структуры и механизма pMMO приходит только в самые последние годы [55, 56].
Согласно рентгеноструктурным данным трансмембранный протеин pMMO из Methylococcus capsulatus (Bath) состоит из трех субъединиц: pmoB или α (47 кДа), pmoA или β (24 кДа), pmoC или γ (22 кДа), сгруппированных в трехмерный комплекс (αβγ)3. С этим комплексом может связываться молекула метанобактина, содержащая 6–8 ионов меди и поставляющая ее в активный центр pMMO.
История исследования АЦ и механизма действия pMMO полна противоречий и заблуждений. В литературе рассматривались три основных представления о природе АЦ pMMO: 1) трехядерный кластер меди (Cu3) в функции как катализатора гидроксилирования метана, так и переносчика электронов [57]; 2) биядерный кластер железа, служащий каталитическим центром как и в sMMO [58]; 3) биядерный кластер меди как каталитический центр [59]. Позднейшие структурные исследования [55] согласовывались с последней точкой зрения, поскольку показали, что активность pMMO не зависит от железа, медный активный центр локализован в растворимом домене субъединицы pmoB, и, наконец, это – биядерный центр. Недавние спектральные исследования с использованием продвинутых методов электронного парамагнитного резонанса (ЭПР) [56, 60] дали убедительные доказательства, что АЦ pMMO содержит два моноядерных Cu-центра: один в растворимой pmoB-субъединице, а другой в мембраносвязанной pmoC-субъединице. Согласно РСА и спектроскопическим данным (CuII)В-центр имеет насыщенную экваториальную координацию прочно связанных лигандов: аминогруппы и трех имидазолов гистидина, а (CuII)С содержит 3 имидазола гистидина и связанную аксиально молекулу воды. Расстояние между этими центрами (около 2 нм) достаточно для прямого переноса электрона. Ранние исследования показали способность Cu-центров подвергаться редокс-превращениям при обработке pMMO пероксидом водорода или дитионитом. Хотя работы последних лет несколько прояснили структуру АЦ pMMO, механизм окисления метана этой монооксигеназой до сих пор не установлен [61].
Комплексы меди как структурные и функциональные модели pММО
Известно множество биядерных µ-η2: η2-пероксокомплексов меди(II), некоторые из которых могут обратимо давать комплексы бис-µ-оксо-${\text{Cu}}_{{\text{2}}}^{{{\text{III}}}},$ подобные интермедиату Q sMMO и соответствующим биядерным комплексам железа. Однако структура бис-µ-оксо-${\text{Cu}}_{{\text{2}}}^{{{\text{III}}}}$ никогда не наблюдалась в биологических системах. Описано гидроксилирование алканов пероксидом водорода при катализе биядерным β-дикетиминатным комплексом меди(II) [62]. При окислении циклогексана с участием этого комплекса образуются циклогексанол и циклогексанон (А/К = 3.7, TON = 26). Наблюдаемый КИЭ (kH/kD = 2.4), высокая региоселективность атаки на третичную связь адамантана (3°/2° = 5) и преобладание спирта в продуктах согласуются, по мнению авторов [62], с механизмом отщепления Н оксо-группой.
В работе [63] дан обзор методов синтеза и свойств CuII–O–CuII-комплексов, рассматриваемых в качестве моделей АЦ pMMO. Эти комплексы оказались довольно реакционноспособными, но неустойчивыми в гомогенных условиях, и потому с ними не удавалось наблюдать гидроксилирование алканов. Напротив, будучи иммобилизованными в наночастицах мезопористого силикагеля, они приобрели стабильность и способность катализировать при комнатной температуре аэробное окисление связей С–Н в толуоле с образованием бензилового спирта [64]. Трехядерные комплексы меди широко исследовались в качестве моделей АЦ pMMO [65]. Следуя теоретическим расчетам и основываясь на гипотезе о механизме гидроксилирования метана на трехядерном медном центре в работе [66], были сконструированы каркасные лиганды (L) и синтезированы неустойчивые ${\text{Cu}}_{{\text{3}}}^{{\text{I}}}{\text{L}}$-комплексы, которые реагируют с O2 с образованием предполагаемых на основании ESI-MS активных интермедиатов бис-µ3-оксо-CuIICuIICuIII. Эти комплексы в отсутствие субстрата гидроксилируют очень прочную связь С–Н в метильной группе ацетонитрила, используемого в качестве растворителя. В 2013 г. было показано [67], что подобные комплексы активируют О2 для окисления метана в метанол при комнатной температуре с небольшим TON, увеличивающимся при использовании пероксида водорода. Оказалось, что иммобилизация подобного комплекса в мезопористом силикагеле, моделирующим некоторые эффекты белковой глобулы фермента, позволяет осуществить селективное превращение метана в метанол при участии пероксида водорода с конверсией 17.4% и TON = 170 [68]. Авторы полагают, что такой эффект связан с включением комплекса в гидрофобную полость силикагеля.
ГЕТЕРОГЕННЫЕ АНАЛОГИ ММО, МОДЕЛИРУЮЩИЕ НЕКОТОРЫЕ ЭФФЕКТЫ БЕЛКОВОЙ ГЛОБУЛЫ
В то время как исключительная селективность MMO, по-видимому, обусловлена изоляцией их АЦ полипептидным каркасом (scaffolds) и гетерогенностью, попытки смоделировать эффекты белковой глобулы на гомогенных системах с помощью каркасных лигандов не были реализованы. Впрочем, недавно была опубликована работа, которая моделирует важную функцию белковой глобулы, а именно организацию гидрофобной полости вблизи АЦ. При связывании трис(2-пиридилметил)амина (TPA) с циклотривератриленом получен гемикриптофановый комплекс (железокавитин) [69], который представляет собой FeTPA, встроенный в каркасный лиганд (рис. 4) [70].
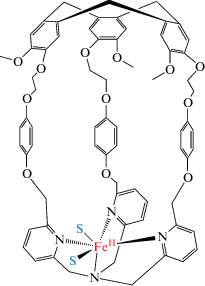
В отличие от комплекса FeTPA, не способного окислить метан, вышеуказанный модельный комплекс катализирует окисление метана пероксидом водорода до метанола и метилгидропероксида при 60°С в воде. Получены 1Н ЯМР доказательства включения метана в полость кавитина. Эту модель можно рассматривать как переходную от гомогенных систем к гетерогенным.
Проблема моделирования белковой глобулы фермента частично решается использованием гетерогенных систем типа цеолитов, обладающих микропористой структурой [71], или пористых координационных полимеров, более известных как металл-органические каркасы (MOF, МОФ) [72]. Они моделируют протеиновый каркас, помещая АЦ в жесткие поры и тем самым защищая его от нежелательных бимолекулярных реакций, однако при этом, к сожалению, ограничивают доступ к нему метана и кислорода, и потому часто лимитирующим становится транспорт реагентов и/или продуктов. В связи с этим стоит напомнить, что в случае sMMO ее исключительная селективность контролируется специальным регуляторным механизмом, учитывающим геометрические размеры субстратов и облегчающим доступ к АЦ наименьшей молекуле, и квантовым туннелированием в случае метана, которые совместно существенно ускоряют окисление метана по сравнению с другими субстратами [3].
Железосодержащие цеолиты
Некоторые металлсодержащие цеолиты, включая модифицированные введением катиона металла цеолиты MFI, MOR, FER и другие, образуют с окислителями оксокомплексы, которые при комнатной или не очень высокой (<150°С) температуре селективно гидроксилируют метан, что, по-видимому, в первую очередь обусловлено пористой структурой этого типа цеолитов.
Сравнительно давно обнаружено высокоселективное (94%) окисление метана до метанола закисью азота (N2O) при комнатной температуре на цеолите FeZSM-5 [73]. Активными центрами являются поверхностные комплексы железа (FeII), формирующиеся на внутренней поверхности пор при высокотемпературной обработке FeZSM-5 в результате термического восстановления FeIII с выделением О2. При 200–250°С эти комплексы образуют с N2O долгоживущие активные кислородные интермедиаты (FeО), способные переносить свой атом кислорода на связи С–Н алканов при комнатной и более низкой температурах. Процессы переноса атома О от N2O на алканы являются двухэлектронными и формально напоминают образование феррильных интермедиатов путем переноса атома О от доноров ХО к FeII-центрам железосодержащих оксигеназ или их моделей и реакции образующихся феррильных интермедиатов с RH. Данное обстоятельство, а также кинетические характеристики этих реакций и, наконец, способность селективно окислять метан в метанол при комнатной температуре позволили рассматривать FeII-комплексы на поверхности цеолита FeZSM-5 как возможную модель активного центра ММО [74]. Большой кинетический изотопный эффект (kH/kD) при окислении метана, равный 5.5–1.9 при температурах от –50 до +100°С, указывает на то, что лимитирующей стадией является разрыв С–Н-связи [75]. До сих пор не достигнуто полного согласия относительно геометрической и электронной структур активных поверхностных комплексов и механизма оксигенирования метана на цеолите FeZSM-5. Одним из спорных вопросов является ядерность этих поверхностных комплексов железа [76]. Разные авторы предполагают биядерные и/или моноядерные центры. В ранних наших исследованиях на основе мессбауэровских исследований было сделано предположение о биядерной природе последних [77]. При окислении метана FeII-центры превращаются в биядерные FeIII(µ‑O)FeIII–центры обратимо. Учитывая обратимость редокс-переходов поверхностных комплексов железа, можно заключить, что биядерная структура сохраняется для всего каталитического цикла. Поэтому вывод о биядерности кажется более вероятным, особенно принимая во внимание, что MS-спектры гидратированных образцов замечательным образом напоминают эти характеристики для АЦ sMMO. Мессбауэровские исследования обнаружили два типа долгоживущих активных интермедиатов (FeО) на поверхности FeZSM-5, оба формально имеют изомерный сдвиг, характерный для FeIII-состояния железа [78]. Один из этих центров (FeО)1, согласно изомерному сдвигу δ = 0.58, мог быть пероксидным интермедиатом, в то время как другой (FeО)2, согласно изомерному сдвигу δ = 0.38, с учетом координационного окружения железа мог бы быть феррилом или иметь строение FeIII–O•, изоэлектронное феррилу FeIV=O. Действительно, оксоферрильный комплекс [(Н2О)5FeIV=O] имеет изомерный сдвиг δ = 0.38. В термически активированном FeZSM-5 также идентифицированы два типа поверхностных комплексов (FeII), которые при реакции с N2O превращаются в два типа кислородных интермедиатов (FeО) [78]. Последние исчезают при реакции с метаном и, возможно, их различие обусловлено разным положением FeII-центров в порах цеолитов. Кинетические результаты указывают, что оба типа (FeО) обладают одинаковой реакционной способностью или способностью быстрого взаимопревращения. Активный окислительный интермедиат цеолита FeZSM-5 был также охарактеризован с помошью УФ-видимой и рамановской спектроскопии [79], причем было показано, что при обработке бензолом он исчезает, а в спектре появляются полосы, соответствующие фенолу, координированному к железу, то есть происходит перенос атома О на бензол. Недавно с использованием спектров магнитного кругового дихроизма (МКД) и других спектроскопических методов АЦ цеолита FeВЕА были идентифицированы как моноядерные высокоспиновые феррильные комплексы [80]. Такой неожиданный результат был объяснен эффектом топологии жесткой цеолитной матрицы, которая обеспечивает соответствующие геометрические напряжения вокруг железа. Как полагают авторы [80], в результате этих напряжений транс-положение к альфа-кислороду в координационной сфере железа сохраняется вакантным, и поддерживается энтатическое состояние всего активного центра, которое делает моноядерный центр способным к реакции с метаном.
Предложен каталитический (более 3000 оборотов катализатора) процесс гидроксилирования метана пероксидом водорода в присутствии воды на модифицированных солями меди FeZSM-5 цеолитах с селективностью более 85% [81]. Другие цеолиты на порядок менее активны, но более селективны. Так метанол может быть получен с селективностью 93% при конверсии 10% на Fe/Cu-силикалите-1 при 70°С и давлении метана около 3 атм. Скорость этих биомиметических процессов приближается к ферментативному окислению метана. Согласно данным EXAFS и RRС активными центрами являются поверхностные биядерные комплексы железа, которые при реакции с пероксидом водорода образуют Fe–OOH-интермедиаты [82]. Предполагается, что роль ионов меди состоит в ингибировании образования НО-радикалов, которые приводят к побочным продуктам и снижают селективность. Кроме того гидрофобный активный центр этих цеолитов имеет предпочтение к неполярному метану перед полярным метанолом подобно активному центру sММО. Хотя биядерный центр, водная среда, селективность и активность этой системы сближают ее с растворимой ММО, предложенный механизм отличается от ферментативного.
Показано низкотемпературное селективное окисление метана закисью азота на биядерных катионных центрах (Co, Ni, Fe) в ферриерите [83]. Недавно [84] был предложен эффективный жидкофазный метод окисления метана в метанол пероксидом водорода на цеолите Fe-MFI в сульфолан-водном растворителе при 50°С c селективноcтью 85%. Достижения и перспективы прямого окисления метана в метанол на цеолитах, содержащих поверхностные (extra-framework) d-металлы, проанализированы в недавних обзорах [84, 85]. В табл. 1 для сравнения приведены разрабатываемые в настоящее время инновационные процессы окисления метана: автотермический (№ 1) и гетерогеннокаталитические – на Fe-цеолитах (№ 2 и № 3), на Cu-цеолитах (№№ 4–9) и с участием пористых координационных полимеров.
Таблица 1.
Некоторые инновационные процессы прямого окисления метана в метанол*
| № | Катализатор/ фаза |
Окислитель | Р, бар | Т, °С | S, % | Y, мкмоль/г | А, моль/моль | Е, мкмоль г–1 ч–1 | Характер процесса | Ссылка |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | –**/г | Воздух | 60 | 450–550 | 50 | – | – | – | |Автотермический | 86 |
| 2 | FeZSM-5/г | N2O | 1 | 275(550) | 62 | – | – | – | В потоке, TON = 7 | 87 |
| 3 | FeMFI/ж | H2O2 | 30 | 50 | 85 | 950 | – | – | В автоклаве, 50% водный сульфолан | 83 |
| 4 | CuMOR/г | O2 | 1 | 200(450) | 90 | 90 | 0.32 | – | Стадийный | 88 |
| 5 | CuMOR/г | O2 | 37 | 200 | – | 56 | – | – | Изотермический | 89 |
| 6 | CuCHA/г | O2 | 1 | 200(500) | 90 | 125 | 0.20 | 8.9 | Стадийный | 90 |
| 7 | CuMOR/г | O2 | 1 | 200(500) | 90 | 170 | 0.47 | 12.1 | Стадийный | 91 |
| 8 | CuMAZ/ж | O2 | 30 | 200(450) | 95 | 200 | – | – | В автоклаве | 92 |
| 9 | CuFAU/г | O2 | 15 | 360 | 93 | 360 | 0.25 | 180 | Изотермический, стадийный | 93 |
| 10 | FeMIL-53/ж | H2O2 | 30 | 50 | 85 | – | 40 | – | В автоклаве, TOF = 90 ч–1 | 94 |
| 11 | CuMOF-808/г | N2O | 1 | 150 | 90 | 72 | – | – | В потоке, изотермический | 95 |
| 12 | CuNU-1000/г | O2 | 40 | 200 | 90 | 4.1 | 0.04 | – | Изотермический, стадийный | 96 |
* Катализаторы: цеолиты (№ 2–9), MOF (№ 10–12). Р – давление, Т – температура реакции (активации), S – селективность, Y – выход на 1 г катализатора, продуктивность, А – активность, Е – эффективность. ** Автотермический процесс. Примечание. Для опытов 4, 6, 7 и 9 результаты даны для одного цикла, активность катализатора сохраняется до 4–8 циклов [93]. Прочерки означают отсутствие данных.
Окисление метана молекулярным кислородом на Cu-содержащих цеолитах
Подобно поверхностным (extra-framework) α‑центрам в порах FeZSM-5 аналогичные α-центры были обнаружены для CuZSM-5. Они генерируются сходной высокотемпературной обработкой металлсодержащего цеолита, приводящей к образованию АЦ (CuI)α, и связывают атом О из закиси азота, образуя α-ОCu, способный селективно окислять метан до метанола в мягких условиях [97]. Также как и в случае FeZSM-5 в качестве модели АЦ sMMO, α-центры на CuZSM-5 и α-ОCu можно рассматривать как модель АЦ и активного окислителя pMMO. В отличие от FeZSM-5, Cu-содержащие цеолиты катализируют окисление метана до спиртов молекулярным кислородом при 125°С, однако активность их ниже чем у α-ОFe, окисляющего метан уже при минусовых температурах. В последние годы усилился интерес к природе α-ОCu, поскольку, во-первых, он образуется не только из N2O, но также из О2, во-вторых, поддерживает каталитическое окисление метана и, в-третьих, дает надежду понять природу активного окислителя pMMO, не поддающегося расшифровке уже более 20 лет. В ранних работах для активного окислителя на CuZSM-5 принималась структура бис-µ-оксо-${\text{Cu}}_{{\text{2}}}^{{{\text{II}}}}$ [97]. Позднее на основе всесторонних спектральных исследований α-О на CuZSM-5 была однозначно приписана структура изогнутого [Cu–O–Cu]2+-ядра, которая никогда ранее не наблюдалась и не предполагалась как каталитически активная частица [98, 99]. Как показали дальнейшие исследования [100], предшественником активного интермедиата в синтезе метанола над CuZSM-5 с участием кислорода является µ-η2:η2-пероксо-${\text{Cu}}_{{\text{2}}}^{{{\text{II}}}}$-интермедиат. Два электрона, необходимые для разрыва пероксидной связи, поставляются окружающими ионами меди, не участвующими в образовании α-центров. Опыты с 18О2 продемонстрировали, что один атом кислорода в этом процессе идет на образование ${\text{LCu}}_{{\text{2}}}^{{{\text{II}}}}{\text{O,}}$ тогда как другой связывается с решеткой цеолита, возможно, с ионами меди, не участвующими в образовании α-центров. Величина кинетического изотопного эффекта (КИЭ = 4) говорит о том, что лимитирующей стадией окисления метана на Cu ZSM-5 является отрыв атома Н. Расчеты методом функционала плотности подтверждают образование [Cu–OH–Cu]2+-интермедиата, причем вычисленные значения энергии активации и КИЭ находятся в хорошем согласии с экспериментальными величинами. Прямое превращение метана в метанол над Cu-цеолитами в постадийном циклическим процессе включает три последовательные стадии: высокотемпературную активацию в атмосфере кислорода при 400–500°С, реакцию с метаном при 200°С и выделение метанола парами воды [101, 102]. Больший интерес для промышленности представляет непрерывный изотермический процесс при 200°С, несмотря на меньший выход и селективность образования метанола [89]. В этом случае стадии активации катализатора и реакцию с метаном проводят при одной температуре и для увеличения выхода и селективности используют более высокое парциальное давление метана и возможно низкое парциальное давление кислорода (табл. 1, № 5). Дальнейшим развитием изотермического процесса была разработка непрерывной струевой технологии (в потоке) с online введением паров воды и отбором метанола без остановки процесса [103].
Природа АЦ и продуктивность Cu-цеолитных катализаторов зависит от типа исходного цеолита, отношений Cu/Al и Si/Al и условий синтеза, а также от условий проведения процесса прямого окисления метана. Редокс-свойства Cu-цеолита сильно влияют на активность и селективность [89]. Продемонстрировано [93], что при реакции CuFAU(3) с метаном в изотермическом режиме при 400°С и атмосферном давлении удается сохранять высокую производительность на уровне 88 мкмоль/г в течение многих циклов при неизменности температуры и давления. Повышение давления метана до 15 бар приводит к увеличению выхода метанола до 360 мкмоль/г – максимально достигнутой продуктивности Cu-цеолитов за один цикл (табл. 1, № 9). Этот результат представляет важный шаг в упрощении процесса и открывает новые возможности для разработки крупномасштабного производства.
Как было показано многочисленными исследованиями, ядерность центров, формирующихся при термической активации катализатора, меняется от моно- до полиядерных с преобладанием двуядерных [91]. Точная структура АЦ, образующихся при высокотемпературной обработке медьсодержащих цеолитов, все еще дебатируется. Получены убедительные доказательства, что наиболее активными являются биядерные комплексы меди [91], причем с использованием продвинутой спектральной техники точно установлена их структура: это биядерные комплексы меди транс-μ-1,2-пероксо-([${\text{Cu}}_{{\text{2}}}^{{{\text{II}}}}{{{\text{O}}}_{2}}$]2+) и моно-μ-оксо-([${\text{Cu}}_{{\text{2}}}^{{{\text{II}}}}{\text{O}}$]2+). В то же время анализ большого объема работ по Cu-цеолитам указывает, что, в зависимости от топологии цеолита, состава и условий активации, может образоваться разный набор Cu-центров [104]. Повидимому это положение справедливо и для Fe-цеолитов.
В связи со значительно меньшей прочностью С–Н-связей в метаноле по сравнению с метаном, для достижения высокой селективности необходимо ограничиваться низкой конверсией метана или стабилизировать метанол либо предшествующие ему металл-метоксильные интермедиаты от переокисления. С этой целью в стадийном процессе после высокотемпературной активации катализатора молекулярным кислородом катализатор освобождается от О2 перед контактом с метаном, а в непрерывном процессе отношение О2/СН4 поддерживается на минимальном уровне, и изыскиваются способы скорейшего удаления метанола из зоны реакции.
Пористые координационные полимеры (MOF)
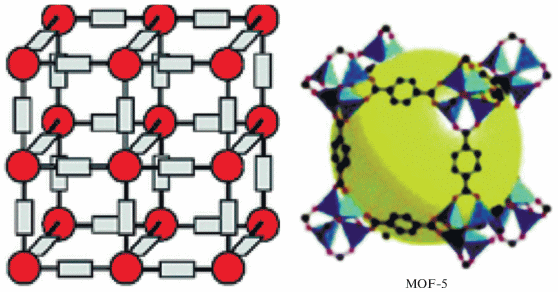
Металл-органические каркасные катализаторы [105] или просто металл-органические каркасы (metal organic frameworks, MOF) позволяют довольно высокую степень синтетического контроля и поэтому являются идеальными материалами для моделирования АЦ ММО [106]. Подобно цеолитам, MOF – это синтетические пористые материалы, представляющие собой объемную сетку, в узлах (node) которой находятся катионы или кластеры металлов, связанные друг с другом многофункциональными органическими молекулами (linker) (рис. 5).
Рис. 5.
Структура MOF. Каждая пора характеризуется размером транспортного окна и объемом полости, соответствующей Ван-дер-ваальсовой сфере (справа).
Преимущества гибридных (органических/неорганических) MOF над чисто неорганическими цеолитами состоят в том, что они обеспечивают более определенное координационное окружение и электронную структуру комплекса металла и, таким образом, являются более перспективными объектами для моделирования АЦ ферментов, изучения механизма и достижения высокой активности в окислении метана [106]. Большинство MOF – это кристаллические твердые вещества с упорядоченной структурой, что позволяет характеризовать их методом РСА (изолированные кристаллы) или порошковой дифракционной техникой. MOF аккумулируют лучшие черты гомогенного и гетерогенного катализа. Периодическая структура пор и возможность контролировать их форму и размер, также как и индивидуальность функциональных групп в них, открывают возможность конструирования АЦ с характеристиками ферментов [105].
В ранних работах было продемонстрировано, что FeMOF-74, используемый для разделения легких углеводородов, содержит равномерно распределенные изолированные координационно ненасыщенные FeII-центры, которые катализируют окисление этана закисью азота при 75°С до этанола и ацетальдегида в соотношении 10 : 1 [107]. На основании спектроскопических исследований и DFT-расчетов был предложен механизм, включающий высокоспиновые интермедиаты FeII и FeIV=O. Описан первый пример изолированных димеров железа в MIL-53(Fe,Al), которые при реакции с пероксидом водорода образуют высокоспиновые FeIV=O-интермедиаты, способные гидроксилировать метан [94]. В работах [108, 109] проанализированы возможные механизмы окисления метана пероксидом водорода на MIL-53(Al,Fe) и MIL-100(Fe). Моноядерные центры в узлах MIL-100(Fe) превращают пропан в пропанол в реакции с N2O при 150°С. Получены доказательства, что активация легких алканов с участием этого катализатора протекает на высокоспиновых квадратно-пирамидальных центрах FeII (S = 2), расположенных в узлах MIL-100(Fe) [108].
Выбор MOF-808 с подходящей структурой и топологией и постсинтетическое введение в него имидазольных лигандов и затем CuI в присутствии O2 позволили получить биомиметический катализатор, который показал высокую селективность в окислении метана в метанол при 150°С с использованием закиси азота (табл. 1, № 11). На основании спектроскопии и расчетов DFT предложен бис-μ-оксо-Cu2-центр в качестве наиболее вероятного АЦ катализатора [95].
Методом жидкофазного постсинтетического введения Cu в Zr6-узлы MOF NU-1000 приготовлен эффективный катализатор прямого окисления метана кислородом в метанол, содержащий биядерные комплексы меди [110]. При повышении давления метана от 1 до 40 бар выход метанола увеличивается в 4 раза, а селективность возрастает до 90%. Оценки показывают, что при этом около 11.2% Cu в катализаторе участвуют в активации метана (табл. 1, № 12).
Хотя изучение гетерогенного катализа с участием MOF, в том числе селективного окисления метана, находятся еще на очень ранней стадии развития, можно отметить быстрый рост числа работ в этом направлении. Это обусловлено, с одной стороны, многими замечательными характеристиками этих инновационных материалов, открывающими новые возможности для химического моделирования энзимов, с другой – перспективами применения в промышленности катализаторов на их основе [111].
ЗАКЛЮЧЕНИЕ
В последние годы достигнут большой прогресс в понимании биологического окисления метана и создании биоинспирированных каталитических процессов прямого получения метанола. Эти две области развивались параллельно, в симбиозе одна с другой. В химическом моделировании ММО можно отметить ряд этапов, начиная от гомогенных систем, включающих: а) простые металлокомплексные модели АЦ, затем б) металлокомплексы с каркасными лигандами (scaffolds) и с) металлокавитины, до гетерогенных, таких как металлсодержащие цеолиты и металл-органические каркасы (MOF).
Хотя двадцатипятилетний марафон в изучении механизма MMO пока не дал исчерпывающего ответа на главный вопрос: почему MMO так активны и селективны в окислении метана, однако он позволил накопить ценную информацию для выяснения природы активных интермедиатов и многих деталей механизма функционирования MMO. Поиски химической модели АЦ MMO стимулировали неорганический синтез: получены новые типы комплексов железа и меди, такие как моно- и бис-µ-оксо-мостиковые комплексы, комплексы с каркасными ди- и тринуклеирующими полидентатными лигандами, с объемистыми карбоксилатными донорами, и, наконец, комплексы с атомами железа или меди в высокоокисленном состоянии (high-valent methals). В будущем можно ожидать новых успехов на этом пути, особенно принимая во внимание перспективы использования для дизайна моделей инновационных областей химии, таких как наноструктуры, молекулярные каркасы, супрамолекулярные комплексы и прочее.
Академические исследования ММО и моделирования их АЦ сохраняют свою актуальность. Успехи последних лет на этом пути выдвинули вопрос об адаптации лабораторных достижений к особенностям и требованиям химической технологии. Хотя достигнута высокая селективность окисления метана в метанол, скорость и продуктивность этих процессов еще очень мала. Очевидно, что между лабораторными наработками и ожиданиями промышленности существует огромная пропасть, которую еще предстоит преодолеть. Усилия многих ученых привели к созданию ряда высоко селективных функциональных моделей MMO, особенно на основе каркасных комплексов меди и медьсодержащих цеолитов, и очевидно назрел вопрос о переходе к практической фазе исследований. Об этом свидетельствуют обсуждение в последних работах [93, 101, 104] путей и возможностей создания промышленного процесса прямого селективного получения метанола, включая экономический анализ [112], и появление ряда патентов по этому направлению (например, “Молекулярные катализаторы и методы окисления углеводородов”, патент США US 10005746 B2 Junе 26, 2018). Таким образом, в недалеком будущем, вероятно, можно ожидать впечатляющего прорыва на этом пути.
Список литературы
Sazinsky M.H., Lippard S.J. // In: Metal ions in life sciences. Springer, 2015. V. 15. P. 205.
Sirajuddin S., Rosenzweig A.C. // Biochem. 2015. V. 54. № 14. P. 2283.
Banerjee R., Jones J.C., Lipscomb J.D. // Annu. Rev. Biochem. 2019. V. 88. P. 409.
Ward T.R. // ACS Cent. Sci. 2019. V. 5. P. 1732.
Rittle J., Green M.T. // Science. V. 330. P. 933.
Puri M., Que L., Jr. // Acc. Chem. Res. 2015. V. 48. P. 2443.
Friedle S., Reisner E., Lippard S.J. // Chem. Soc. Rev. 2010. V. 39. P. 2768.
Westerheide L., Pascaly M., Krebs B. // Curr. Opin. Chem. Biol. 2000. V. 4. P. 235.
Que L., Tolman W.B. // Nature. 2008. V. 455. P. 333.
Poulos T.L. // Chem. Rev. 2014. V. 114. P. 3919.
Costas M., Mehn M.P., Jensen M.P., Que L., Jr. // Chem. Rev. 2004. V. 104. P. 939.
Solomon E.I., Brunold T.C., Davis M.I., Kemsley J.N., Lee S.-K., Lehnert N., Neese F., Skulan A.J., Yang Y.-S., Zhou J. // Chem. Rev. 2000. V. 100. P. 235.
Lee S.J. // J. Microbiol. 2016. V. 54. P. 277.
Shteinman A.A. // FEBS Lett. 1995. V. 362. P. 5.
Shu L., Nesheim J.C., Kauffmann K., Münck E., Lipscomb J.D., Que L., Jr. // Science. 1997. V. 275. P. 515.
Shan X., Que L., Jr. // J. Inorg. Biochem. 2006. V. 100. P. 421.
Gherman B.F., Baik M.-H., Lippard S.J., Friesner R.A. // J. Am. Chem. Soc. 2004. V. 126. P. 2978.
Mai B.K., Kim Y. // Chem. Eur. J. 2014. V. 20. P. 6532.
Xue G., DeHont R., Münck E., Que L., Jr. // Nature Chem. 2010. V. 2. P. 400.
Xue G., Wang D., DeHont R., Fiedler A.T., Shan X., Münck E., Que L., Jr. // Proc. Natl. Acad. Sci. USA. 2007. V. 104. P. 20713.
Tinberg C.E., Lippard S.J. // Acc. Chem. Res. 2011. V. 44. P. 280.
Do L.H., Hayashi T., Moënne-Loccoz P., Lippard S.J. // J. Am. Chem. Soc. 2010. V. 132. P. 1273.
Kodera M., Kawahara Y., Hitomi Y., Nomura T., Ogura T., Kobayashi Y. // J. Am. Chem. Soc. 2012. V. 134. P. 13236.
Kodera M., Itoh M., Kano K., Funabiki T., Reglier M. // Angew Chem, Int Ed. 2005. V. 44. P. 7104.
Friedle S., Reisner E., Lippard S.J. // Chem. Soc. Rev. 2010. V. 39. P. 2768.
Do L.H., Lippard S.J. // J. Inorg. Biochem. 2011. V. 105. P. 1774.
Sievert I., Limberg C. // Chem. Eur. J. 2009. V. 15. P. 10 316.
Sankaralingam M., Palaniandavar M. // Polyhedron. 2014. V. 67. P. 171.
Kodera M., Shimakoshi H., Kano K. // Chem. Commun. 1996. V. 15. P. 1737.
Kodera M., Tsuji T., Yasunaga T., Kawahara Y., Hirano T., Hitomi Y., Nomura T., Ogura T., Kobayashi Y., Sajith P.K., Shiota Y., Yoshizawa K. // Chem. Sci. 2014. V. 5. P. 2282.
Kulikova V.S., Gritsenko O.N., Shteinman A.A. // Mendeleev Comm. 1996. P. 119.
Costas M., Chen K., Que L., Jr. // Coord. Chem. Revs. 2000. V. 200–202. P. 517.
Gutkina E.A., Trukhan V.M., Pierpont C.G., Mkoyan S., Strelets V.V., Nordlander E., Shteinman A.A. // Dalton Trans. 2006. P. 492.
Трухан В.М., Полухов В.В., Сулименков И.В., Ованесян Н.С., Ковальчук Н.А., Додонов А.Ф., Штейнман А.А. // Кинетика и катализ. 1998. T. 39. C. 858.
Гуткина Е.А., Трухан В.М., Штейнман А.А. // Кинетика и катализ. 2003. T. 44. C. 821.
Турицына Е.А., Трухан В.М., Штейнман А.А. // Изв. АН. Сер. хим. 2011. Т. 60. C. 2051.
Das B., Al-Hunaiti A., Haukka M., Demeshko S., Meyer S., Shteinman A.A., Meyer F., Repo T., Nordlander E. // Eur. J. Inorg. Chem. 2015. V. 21. P. 3590.
Nam W. // Acc. Chem. Res. 2007. V. 40. P. 522.
Que L., Jr. // Acc. Chem. Res. 2007. V. 40. P. 493.
Friedle S., Reisner E., Lippard S.J. // Chem. Soc. Rev. 2010. V. 39. P. 2768.
Jasniewski A.J., Que L.Jr. // Chem. Rev. 2018. V. 118. P. 2554.
Xue G., Fiedler A.T., Martinho M., Münck E., Que L., Jr. // Proc. Natl. Acad. Sci. USA. 2008. V. 105. P. 20 615.
Xue G., Pokutsa A., Que L. // J. Am. Chem. Soc. 2011. V. 133. P. 16 657.
Kal S., Que L. // Angew. Chem. Int. Ed. 2019. https://doi.org/10.1002/anie.201903465
Bernasconi L., Belanzoni P., Baerends E.J. // Phys. Chem. Chem. Phys. 2011. V. 13. P. 15 272.
Banerjee R., Proshlyakov Y., Lipscomb J., Proshlyakov D.A. // Nature. 2015. V. 518. P. 431.
Grzyska P.K., Appelman E.H., Hausinger R.P., Proshllyakov D.A. // Proc. Natl. Acad. Sci. USA. 2010. V. 107. P. 3982.
Wilkinson E.C., Dong Y.H., Zang Y., Fujii H., Fraczkiewicz R., Fraczkiewicz G., Czernuszewicz R.S., Que L., Jr. // J. Am. Chem. Soc. 1998. V. 120. P. 955.
Shteinman A.A. // J. Biol. Inorg. Chem. 1998. V. 3. P. 325.
Bauer M. // Phys. Chem. Chem. Phys. 2014. V. 16. P. 13 827.
Proux O., Lahera E., Del Net W., Kieffer I., Rovezzi M., Testemale D., Irar M., Thomas S., Aguilar-Tapia A., Bazarkina E.F., Prat A., Tella M., Auffan M., Rose J., Hazemann J.L. // J. Env. Qual. 2017. V. 46. P. 1146.
Xue G., Geng C., Ye S., Fiedler A.T., Neese F., Que L. // Inorg Chem. 2013. V. 52. V. 3976.
Castillo R.G., Banerjee R., Allpress C.J., Rohde G.T., Bill E., Que L.Jr., Lipscomb J.D., DeBeer S. // J. Am. Chem. Soc. 2017. V. 139. P. 18 024.
Cutsail III G.E., Banerjee R., Zhou A., Que L.Jr., Lipscomb D., DeBeer S. // J. Am. Chem. Soc. 2018. V. 140. P. 16 807.
Balasubramanian R., Smith S.M., Rawat S., Yatsunyk L.A., Stemmler T.L., Rosenzweig A. // Nature. 2010. V. 465. P. 115.
Ross M.O., MacMillan F., Wang J., Nisthal A., Lawton T.J., Olafson B. D., Mayo S.L., Rosenzweig A.C., Hoffman B.M. // Science. 2019. V. 364. P. 566.
Chan S.I., Yu S.S.-F. // Acc. Chem. Res. 2008. V. 41. P. 969.
Martinho M., Choi D.W., DiSpirito A.A., Anttholine W.E., Semrau J.D., Münck E. // J. Am. Chem. Soc. 2007. V. 129. P. 15783.
Lieberman R.L., Rosenzweig A.C. // Nature. 2005. V. 434. P. 177.
Culpepper M.A., Cutsail G.E., Gunderson W.A., Hoffman B.M., Rosenzweig A.C. // J. Am. Chem. Soc. 2014. V. 136. P. 11767.
Chan S.I., Yu S.S.-F. // Nature Catal. 2019. V. 2. P. 286.
Shimokawa C., Teraoka J., Tachi Y., Itoh S. // J. Inorg. Biochem. 2006. V. 100. P. 1118.
Haack P., Limberg C. // Angew. Chem. Int. Ed. 2014. V. 53. P. 4282.
Liu C.C., Lin T.S., Chan S.I., Mou C.Y. // J. Catal. 2015. V. 322. P. 139.
Salvadeo E., Dubois L., Latour J.-M. // Coord. Chem. Rev. 2018. V. 374. P. 345.
Chen P.P.Y., Chan S.I. // J. Inorg. Biochem. 2006. V. 100. P. 801.
Chan S.I., Lu Y.J., Nagababu P., Maji S., Hung M.C., Lee M.M., Hsu I.J., Minh P.D., Lai J.C.H., Ng K.Y. // Angew. Chem. Int. Ed. 2013. V. 52. P. 3731.
Liu C.C., Mou C.Y., Yu S.S.F., Chan S.I. // Energ. Environ. Sci. 2016. V. 9. P. 1361.
Ikbal S.A., Colomban C., Zhang D., Delecluse M., Brotin T., Dufaud V., Dutasta J.-P., Sorokin A.B., Martinez A. // Inorg. Chem. 2019. V. 58. P. 7220.
Zhang D., Bousquet B., Mulatier J.C., Pitrat D., Jean M., Vanthuyne N., Guy L., Dutasta J.P., Martinez A. // J. Org. Chem. 2017. V. 82. P. 6082.
Raynes S., Shan M.A., Taylor R.A. // Dalton Trans. 2019. V. 48. P. 10 364.
Kirchon A., Feng L., Drake H.F., Joseph E.A., Zhou H.-C. // Chem. Soc. Rev. 2018. V. 47. P. 8611.
Starokon E.V., Parfenov M.V., Pirutko L.V., Abornev S.I., Panov G.I. // J. Phys. Chem. C. 2011. V. 115. P. 2155.
Panov G.I., Sobolev V.I., Dubkov K.A., Parmon V.N., Ovanesyan N.S., Shilov A.E., Shteinman A.A. // React. Kinet. Catal. Lett. 1997. V. 61. P. 251.
Dubkov K.A., Sobolev V.I., Talsi E.P., Rodkin M.A., Watkins N.H., Shteinman A.A., Panov G.I. // J. Mol. Catal. A. 1997. V. 123. P. 155.
Ованесян Н.С., Штейнман А.А., Дубков К.А., Соболев В.И., Панов Г.И. // Кинетика и катализ. 1998. V. 39. P. 863.
Ovanesyan N.S., Shteinman A.A Dubkov K.A., Pyalling A.A. // J. Ragioanalyt. Nucl. Chem. 2000. V. 246. P. 149.
Dubkov K.A., Ovanesyan N.S., Shteinman A.A., Starokon E.V., Panov G.I. // J. Catal. 2002. V. 207. P. 341.
Xia H., Sun K. (1), Sun K. (2), Feng Z., Li W.X., Li C. // J. Phys. Chem. C. 2008. V. 112. P. 9001.
Snyder B.E.R., Vanelderen P., Bols M.L., Hallaert S.D., Böttger L.H., Ungur L., Pierloot K., Schoonheydt R.A., Sels B.F., Solomon E.I. // Nature. 2016. V. 536. P. 317.
Hammond C., Forde M.M., Ab Rahim M.H., Thetford A., He Q., Jenkins R.L., Dimitratos N., Lopes-Sanchez J.A., Dummer N.F., Murphy D.M., Carley A.F., Taylor S.H., Willock D.J., Stangland E.E., Kang J., et al. // Angew. Chem. Int. Ed. 2012. V. 51. P. 5129.
Hammond C., Hermans I., Dimitratos N. // ChemCatChem. 2015. V. 7. P. 434.
Tabor E., Lemishka M., Sobalik Z., Mlekodaj K., Andrikopoulos P.C., Dedecek J., Sklenak S. // Commun. Chem. 2019. V. 2. P. 1.
Xiao P., Wang Y., Nishitoba T., Kondo J.N., Yokoi T. // Chem. Commun. 2019. V. 55. № 20. P. 2896.
Snyder B.E.R., Bols M.L., Schoonheydt R.A., Sels B.F., Solomon E.I. // Chem. Rev. 2018. V. 118. P. 2718.
Arutyunov V. // Catal. Today. 2013. V. 215. P. 243.
Parfenov M.V., Starokon E.V., Pirutko L.V., Panov G.I. // J. Catal. 2014. V. 318. P. 14.
Grundner S., Luo W., Sanchez-Sanchez M., Lercher J.A. // Chem. Commun. 2016. V. 52. P. 2553.
Tomkins P., Mansouri A., Bozbag S.E., Krumeich F., Park M.B., Alayon E.M., Ranocchiari M., Bokhoven J.A. // Angew. Chem. Int. Ed. 2016. V. 55. P. 5467.
Pappas D.K., Borfecchia E., Dyballa M., Pankin I.A., Lomachenko K.A., Martini A., Signorile M., Teketel S., Arstad B., Berlier G., Lamberti C., Bordiga S., Olsbye U., Lillerud K.P., Svelle S., Beato P. // J. Am. Chem. Soc. 2017. V. 139. P. 14 961.
Pappas D., Martini A., Dyballa M., Kvande K., Teketel S., Lomachenko K., Baran R., Glatzel P., Arstad B., Berlier G., Lamberti C., Bordiga S., Olsbye U., Svelle S., Beato P., Borfecchia E. // J. Am. Chem. Soc. 2018. V. 140. P. 15 270.
Knorpp A.J., Pinar A.B., Newton M., Sushkevich V., Bokhoven J.A. // ChemCatChem. 2018. V. 10. P. 5593.
Sushkevich V.L., Bokhoven J.A. // ACS Catal. 2019. V. 9. P. 6293.
Osadchii D.Y., Olivos-Suarez A.I., Szécsény Á., Li G., Nasalevich M.A., Dugulan I.A., Crespo P.S., Hensen E.J.M., Veber S.L., Fedin M.V., Sankar G., Pidko E.A., Gascon J. // ACS Catal. 2018. V. 8. P. 5542.
Baek J., Rungtaweevoranit B., Pei X.K., Park M., Fakra S.C., Liu Y.S., Matheu R., Alshmimri S.A., Alshehri S., Trickett C.A., Somorjai G.A., Yaghi O.M. // J. Am. Chem. Soc. 2018. V. 140. P. 18 208.
Zheng J., Ye J., Ortuño M.A., Fulton J.L., Gutiérrez O.Y., Camaioni D.M., Motkuri R.K., Li Z., Webber T.E., Mehdi B.L., Browning N.D., Penn R.L., Farha O.K., Hupp J.T., Truhlar D.G., Cramer C.J., Lercher J.A. // J. Am. Chem. Soc. 2019. V. 141. P. 9292.
Smeets P.J., Groothaert M.H., Schoonheydt R.A. // Catal. Today. 2005. V. 110. P. 303.
Woertink J.S., Smeets P.J., Groothaert M.H., Wance M.A., Sels B.F., Schoonheydt R.A., Solomon E.I. // Proc. Natl. Acad. Sci. 2009. V. 106. P. 18 908.
Smeets P.J., Woertink B.F., Sels B.F., Solomon E.I., Schoonheydt R.A. // Inorg. Chem. 2010. V. 49. P. 3573.
Smeets P.J., Hadt R.G., Woertink J.S., Vanelderen P., Schoonheydt R.A., Sels B.F., Solomon E.I. // J. Am. Chem. Soc. 2010. V. 132. P. 14 736.
Ravi M., Ranocchiari M., Bokhoven J.A. // Angew. Chem. Int. Ed. 2017. V. 56. P. 16 464.
Tomkins P., Ranocchiari M., Bokhoven J.A. // Acc. Chem. Res. 2017. V. 50. P. 418.
Narsimhan, K., Iyoki, K., Dinh, K., Román-Leshkov Y. // ACS Cent. Sci. 2016. V. 2. P. 424.
Dinh K.T., Sullivan M.M., Serna P., Meyer R.J., Dinca M., Roman-Leshkov Y. // ACS Catal. 2018. V. 8. P. 8306.
Zhou H.-C., Long J.R., Yaghi O.M. // Chem. Rev. 2012. V. 112. P. 673.
Nath I., Chakraborty J., Verpoort F. // Chem. Soc. Rev. 2016. V. 45. P. 4127.
Verma P., Vogiatzis K.D., Planas N., Borycz J., Xiao D.J., Long J.R., Gagliardi L., Truhlar D.G. // J. Am. Chem. Soc. 2015. V. 137. P. 5770.
Szecsenyi A., Li G., Gascon J., Pidco E.A. // Chem. Sci. 2018. V. 9. P. 6765.
Vitillo J.G., Bhan A, Cramer C.J., Lu C.C., Gagliardi L. // ACS Catal. 2019. V. 9. P. 2870.
Ikuno T., Zheng J., Vjunov A., Sanchez-Sanchez M., Ortuño M.A., Pahls D.R., Fulton J.L., Camaioni D.M., Li Z., Ray D., Lercher J.A. // J. Am. Chem. Soc. 2017. V. 139. P. 10 294.
Yang D., Gates B.C. // ACS Catal. 2019. V. 9. P. 1779.
Lange J.-P., Sushkevich V.L., Knorpp A.J., Bokhoven J.A. // Ind. Eng. Chem. Res. 2019. V. 58. P. 8674.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ