

Кинетика и катализ, 2020, T. 61, № 4, стр. 541-548
Исследование процесса парофазной дегидратации глицерина в акролеин в присутствии полиоксометаллатов
А. С. Белоусов a, *, А. Л. Есипович a, К. В. Отопкова a, Е. А. Канаков a, В. Д. Уварова a, А. В. Шишулина a, А. В. Воротынцев a
a ФГБОУ ВО Нижегородский государственный технический университет им. Р.Е. Алексеева
603950 Нижний Новгород, ул. Минина, 24, Россия
* E-mail: artem_belousov17@mail.ru
Поступила в редакцию 20.12.2019
После доработки 13.01.2020
Принята к публикации 14.01.2020
Аннотация
Исследована активность каталитических систем на основе полиоксометаллатов в процессе парофазной дегидратации глицерина в акролеин. Синтезированные катализаторы охарактеризованы с помощью методов ИК-спектроскопии, рентгенофазового анализа, электронной микроскопии, низкотемпературной адсорбции–десорбции азота (метод БЭТ). Показано, что природа кислотных центров на поверхности катализаторов оказывает существенное влияние на направление превращения глицерина. Увеличение доли центров Бренстеда от общего количества кислотных центров катализатора приводит к росту выхода акролеина, повышение доли центров Льюиса – к возрастанию выхода ацетола, образующегося по конкурирующему маршруту. Среди рассмотренных каталитических систем наилучшие результаты получены при использовании аниона кремнемолибденовой кислоты, нанесенного на оксид алюминия (SiMo/A). В присутствии системы SiMo/A изучены основные закономерности процесса парофазной дегидратации глицерина, определены условия, обеспечивающие наибольший выход акролеина.
ВВЕДЕНИЕ
Основным промышленным способом получения акролеина и акриловой кислоты является парциальное окисление пропилена в присутствии гетерогенных мультикомпонентных катализаторов [1, 2]. Промышленные технологии характеризуются высоким выходом диоксида углерода, оказывающего существенное негативное воздействие на окружающую среду, и образованием продуктов глубокого окисления (уксусная кислота, ацетальдегид, малеиновый ангидрид и др.), обуславливающих усложнение стадии выделения и очистки товарных продуктов. Указанные недостатки, а также повышенный спрос на полипропилен и материалы на его основе требуют поиска новых альтернативных технологий получения акролеина и акриловой кислоты, в том числе на основе экологичных возобновляемых источников сырья.
В то же время из-за увеличения мирового производства биодизельного топлива на рынке появился избыток глицерина-сырца, образующегося в качестве побочного продукта [3, 4]. В связи с этим в последние годы было предложено большое количество способов направленного превращения глицерина в ценные химические продукты, среди которых особое внимание уделяется получению акролеина и акриловой кислоты [5–7]. Процесс дегидратации глицерина предлагается проводить в жидкой или паровой фазе с использованием катализаторов кислотного типа: цеолитов [8, 9], оксидов металлов [10, 11] и гетерополикислот [12, 13].
В настоящее время ведутся активные исследования процесса окислительной дегидратации глицерина в акриловую кислоту в присутствии бифункциональных каталитических систем на основе полиоксометаллатов [14–18]. Однако большинство из предлагаемых катализаторов характеризуются либо низкой активностью и селективностью, либо имеют малый срок службы за счет дезактивации. Следует также отметить, что в литературе практически не рассматриваются вопросы активности, селективности и стабильности таких каталитических систем на первой стадии процесса – дегидратации глицерина в акролеин. Более полное понимание закономерностей протекания этой стадии позволит оптимизировать состав бифункциональных каталитических систем и условия проведения окислительной дегидратации глицерина.
Настоящая работа посвящена исследованию закономерностей процесса парофазной дегидратации глицерина в акролеин в присутствии каталитических систем на основе полиоксометаллатов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и материалы
В работе использовали: глицерин квалификации “ЧДА” (99.74 мас. %, ГОСТ 6259-75); акролеин (95 мас. %), ацетол (95 мас. %) и ацетальдегид (не менее 99 мас. %) фирмы “SIGMA-ALDRICH ChemieGmbH”, Германия; аллиловый спирт (не менее 99.9 мас. %, ТУ 6-09-3879-75); метанол квалификации “ХЧ” (не менее 99.5 мас. %, ГОСТ 6995-77); активный оксид алюминия (А-64, АО “Ангарский завод катализаторов и органического синтеза”); гетерополикислоты (ГПК): фосфорновольфрамовую (HPW), фосфорномолибденовую (HPMo) и кремнемолибденовую (HSiMo) квалификации “ХЧ” (АО “ВЕКТОН”). Все реагенты и материалы применяли без специальной подготовки.
Методики приготовления полиоксометаллатов, нанесенных на оксид алюминия
Для приготовления образцов PW/A, PMo/A, SiMo/A, где А – γ-Al2O3, раствор соответствующей ГПК в метаноле (10 г/л) добавляли к 3 г предварительно прокаленного при 400°С порошкообразного γ-Al2O3 и перемешивали в течение 10 ч при комнатной температуре. Полученный порошок отделяли, промывали метанолом и сушили в вакуумном шкафу при температуре 80°С до прекращения изменения массы.
Методика проведения процесса парофазной дегидратации глицерина
Дегидратацию глицерина в акролеин осуществляли в паровой фазе на лабораторной установке непрерывного действия. Схема установки представлена в работе [10]. Эксперименты проводили при температуре 280–350°С и атмосферном давлении в течение 5 ч. В реактор с постоянным расходом (0.1–0.4 г/мин) подавали водный раствор глицерина (5–20 мас. %). Масса катализатора в экспериментах составляла 0.5–2.0 г. Паровой поток, выходящий из реактора, конденсировали в холодильнике и собирали в пробоотборнике.
Методики анализов
Качественный состав продуктов анализировали методом хромато-масс-спектрометрии на газовом хроматографе GC–2010 Plus (“Shimadzu”, Япония), оснащенного МАСС- и ПИД-детекторами на капиллярной колонке VertiBond 1701 (длина – 30 м, внутренний диаметр – 0.25 мм, толщина пленки – 0.25 мкм). Температура термостата колонок – 100°С, испарителя – 250°С, детектора – 250°С. Газ-носитель – гелий.
Количественный состав продуктов определяли с использованием газовой хроматографии на Хромос ГХ–1000 (“Хромос Инжиниринг”, Россия) с пламенно-ионизационным детектором на капиллярной колонке VertiBond 1701 (длина – 30 м, внутренний диаметр – 0.25 мм, толщина пленки – 0.25 мкм). Температура термостата колонок – 100°С, испарителя – 250°С, детектора – 250°С. Газ-носитель – гелий.
Измерение удельной поверхности полученных образцов проводили с помощью прибора Sorbi MS (“META”, Россия). Пробоподготовка заключалась в обезвоживании образца в течение 2 ч при 110°С и предварительной термотренировке при 100°С. Для получения полной изотермы адсорбции–десорбции использовали абсолютную градуировку. Удельную поверхность (SBET) определяли методом БЭТ [19].
Кислотные свойства каталитических систем изучали методом ИК-спектроскопии адсорбированного пиридина на Фурье-спектрометре IRAffinity-1 (“Shimadzu”, Япония) при 150°С. Перед адсорбцией пиридина образцы вакуумировали при 250°С, после чего снижали температуру до 150°С и адсорбировали пиридин в течение 15 мин. Избыточный физически связанный пиридин удаляли вакуумированием при 150°С в течение 30 мин. Количество центров каждого типа (N, мкмоль/г) рассчитывали по формуле (1):
где A – интегральное поглощение ИК-полос пиридина на соответствующем кислотном центре, см–1; ρ – масса образца, отнесенная к площади таблетки, г/см2; A0 – молярный интегральный коэффициент поглощения, равный 3.0 и 3.5 для кислотных центров Бренстеда и Льюиса соответственно, см/мкмоль [20, 21].Состав образцов катализатора исследовали рентгенофазовым методом на дифрактометре XRD-6100 (“Shimadzu”, Япония) с CuKα-излучением в диапазоне углов 2θ от 5° до 80° с шагом сканирования 0.02°.
Поверхность катализаторов изучали с использованием растрового электронного микроскопа JSM-IT300LV (“JEOL”, Япония), снабженного энергодисперсионной приставкой.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исследование активности полиоксометаллатов в процессе парофазной дегидратации глицерина в акролеин
Хромато-масс-спектрометрический анализ реакционных масс показал, что основными продуктами дегидратации глицерина в присутствии исследуемых каталитических систем являются акролеин, ацетол (гидроксиацетон) и ацетальдегид (схема 1). В ходе процесса также образуются продукты осмоления, и происходит зауглероживание поверхности катализатора, о чем свидетельствует расхождение материального баланса по глицерину.
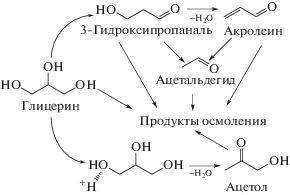
Схема 1 . Схема превращения глицерина при его парофазной дегидратации в присутствии катализаторов кислотного типа.
Каталитические испытания ГПК и полиоксометаллата (ПОМ), нанесенных на оксид алюминия, проводили при температуре 330°С и атмосферном давлении. Расход водного раствора глицерина (10 мас. %) составлял 0.2 г/мин. Масса катализатора – 1 г. Результаты испытаний представлены в табл. 1. Степень дезактивации катализатора D (%) рассчитывали по формуле (2):
где Xгл, 1 и Xгл, 5 – степень превращения глицерина после 1 и 5 ч реакции соответственно.Таблица 1.
Основные показатели процесса дегидратации глицерина в присутствии полиоксометаллатов после 5 ч реакции
| Катализатор | Удельная площадь поверхности, м2/г | Общая концентрация кислотных центров, мкмоль/г | B/(B + L) | Xгл, % | D, % | Yа, % | Yац, % | Yаа, % | Yас, % | Yост, % |
|---|---|---|---|---|---|---|---|---|---|---|
| HPW | 4 | 1042 | 1.00 | 26.2 | 53.2 | 12.9 | 1.4 | 1.3 | 0.1 | 10.5 |
| HPMo | 5 | 904 | 1.00 | 4.4 | 26.2 | 1.0 | 0.4 | 0.4 | 1.2 | 1.4 |
| HSiMo | 9 | 823 | 1.00 | 7.5 | 42.4 | 1.7 | 0.5 | 0.6 | 2.0 | 2.7 |
| PW/A | 223 | 620 | 0.40 | 74.8 | 14.2 | 30.5 | 10.7 | 4.1 | 0.3 | 29.2 |
| PMo/A | 194 | 462 | 0.46 | 81.6 | 15.4 | 43.1 | 8.9 | 5.6 | 0.7 | 23.3 |
| SiMo/A | 211 | 425 | 0.49 | 89.9 | 5.9 | 46.1 | 7.8 | 4.0 | 0.6 | 31.4 |
| γ-Al2O3 | 190 | 105 | 0.03 | 87.8 | 7.3 | 41.3 | 8.4 | 4.0 | 0.6 | 33.5 |
Примечание. Хгл – конверсия глицерина, %; Yа, Yац, Yаа, Yас – выходы акролеина, ацетола, ацетальдегида и аллилового спирта соответственно, %; Yост – выход остальных продуктов, образующихся в ходе процесса (продукты осмоления), %; D – степень дезактивации катализатора, %; B – количество кислотных центров Бренстеда, мкмоль/г; L – количество кислотных центров Льюиса, мкмоль/г; B/(B + L) – доля центров Бренстеда от общего количества кислотных центров.
Элементный анализ поверхности образцов PW/A, PMo/A, SiMo/A методом энергодисперсионной спектроскопии (ЭДС) показал, что содержание нанесенных анионов ${\text{P}}{{{\text{W}}}_{{{\text{12}}}}}{\text{O}}_{{{\text{4}}0}}^{{{\text{3}}--}},$ ${\text{PM}}{{{\text{o}}}_{{{\text{12}}}}}{\text{O}}_{{{\text{4}}0}}^{{{\text{3}}--}}$ и ${\text{SiM}}{{{\text{o}}}_{{{\text{12}}}}}{\text{O}}_{{{\text{4}}0}}^{{{\text{4}}--}}$ в синтезированных каталитических системах составляет 7.5–8.0 мас. %.
Как видно из табл. 1, повышение общей кислотности катализатора приводит к снижению конверсии глицерина и выхода акролеина на 5-м часу проведения процесса. Такая зависимость обусловлена отложением продуктов осмоления на сильных кислотных центрах, в результате чего происходит блокирование поверхности и дезактивация катализаторов.
Кроме общей кислотности на селективность и стабильность катализатора также существенно влияет соотношение кислотных центров Бренстеда и Льюиса.
Наличие высокой кислотности Бренстеда способствует ускорению процессов конденсации альдегидов и полимеризации по двойной связи. В присутствии ГПК, являющихся сильными кислотами Бренстеда, уже в первые часы наблюдается резкое снижение конверсии глицерина при его дегидратации.
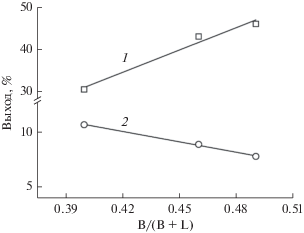
При близких значениях общей кислотности испытываемых каталитических систем наблюдается зависимость, согласно которой увеличение доли центров Бренстеда приводит к росту выхода акролеина, а уменьшение – к повышению выхода ацетола, образующегося по конкурирующему маршруту (рис. 1).
Рис. 1.
Влияние доли кислотных центров Бренстеда на выходы акролеина (1) и ацетола (2) в присутствии каталитических систем ПОМ/А.
Предполагаемые механизмы образования акролеина и ацетола из глицерина на бренстедовских и льюисовских кислотных центрах представлены на схемах 2 и 3 .
Дегидратация глицерина на кислотных центрах Бренстеда (схема 2 ) начинается с протонирования вторичной гидроксильной группы, атом кислорода которой имеет более высокий отрицательный заряд по сравнению атомами кислорода концевых групп. Затем полученный протонированный промежуточный продукт превращается в 1,3-дигидроксипропен с последующей его таутомеризацией до 3-гидроксипропаналя. Акролеин образуется на конечной стадии в результате реакции дегидратации 3-гидроксипропаналя.
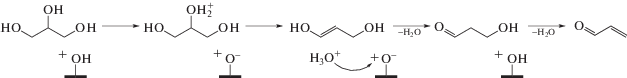
Схема 2 . Механизм дегидратации глицерина на кислотных центрах Бренстеда.
В то же время на кислотных центрах Льюиса (схема 3 ) происходит активация концевых гидроксильных групп. Взаимодействие концевой гидроксильной группы с льюисовским центром приводит к переносу протона вторичной гидроксильной группы, в результате чего образуется 2,3-дигидроксипропен. На последней стадии происходит образование ацетола и регенерация активного центра.
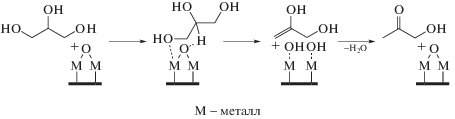
Схема 3 . Механизм дегидратации глицерина на кислотных центрах Льюиса.
Активный оксид алюминия – один из самых распространенных носителей. Он используется для получения катализаторов многих процессов основного органического и нефтехимического синтеза. При этом, как было показано в наших предыдущих работах [10, 11], гамма оксид алюминия сам по себе является достаточно активным катализатором дегидратации глицерина. Относительно высокая активность и стабильность γ-Al2O3, характеризующегося только льюисовской кислотностью после приготовления, обусловлена образованием центров Бренстеда средней силы в результате гидратирования поверхности катализатора в ходе процесса [22]. Гидратирование происходит, скорее всего, за счет того, что в исходном потоке при использовании 10% водного раствора глицерина присутствует большое количество воды.
Как видно из табл. 1, использование ПОМ, нанесенных на активный оксид алюминия, приводит к увеличению степени превращения глицерина и выхода акролеина по сравнению с ГПК. Более высокая активность и стабильность ПОМ, нанесенных на оксид алюминия, связаны, по-видимому, с их меньшей общей кислотностью, а также с оптимальным для рассматриваемого процесса соотношением центров Льюиса и Бренстеда. За счет наличия полиоксометаллатного аниона такой катализатор может эффективно применяться для дальнейшего окисления акролеина в акриловую кислоту.
Исследование закономерностей процесса парофазной дегидиратации глицерина в акролеин
Для исследования закономерностей процесса парофазной дегидратации глицерина был выбран образец SiMo/A, обеспечивающий наибольший выход акролеина после 5 ч реакции.
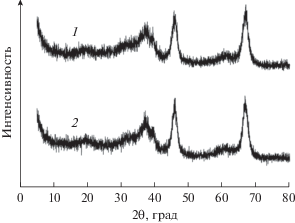
На рис. 2 представлены спектры рентгеновской дифрактометрии свежеприготовленного и отработанного катализатора SiMo/A. Спектры характеризуются двумя интенсивными линиями при углах 2θ ≈ 46° и 67°, соответствующими фазе γ-Al2O3. На дифрактограммах отсутствуют пики, характерные для ГПК. Это свидетельствует, что активная фаза каталитической системы находится в высоко диспергированном состоянии на поверхности носителя [14].
Результаты исследования кислотных свойств катализатора SiMo/A методом ИК-спектроскопии адсорбированного пиридина приведены на рис. 3.
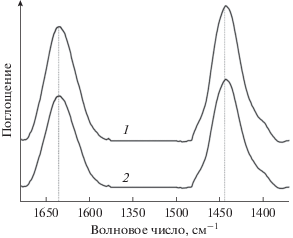
Кислотным центрам Бренстеда соответствует полоса поглощения ~1635 см–1, а центрам Льюиса – ~1450 см–1 [20]. Как видно из рис. 3, содержание на поверхности каталитической системы SiMo/A льюисовских и бренстедовских кислотных центров практически одинаково. Интенсивность пиков для отработанного катализатора (кривая 2) несколько ниже, чем для свежеприготовленного (кривая 1), что объясняется частичным блокированием активных центров продуктами осмоления.
На рис. 4 представлены микрофотографии образца SiMo/A до и после проведения в его присутствии реакции дегидратации.

Как видно из рис. 4, порошок синтезированного катализатора сильно агломерирован и состоит из зерен нерегулярной формы размером от 5 до 30 мкм. Элементный анализ поверхности свежеприготовленного образца SiMo/A показал содержание атомов (мас. %): O – 52.73; Al – 45.94; Si – 0.55; Mo – 0.78, что соответствует содержанию аниона кремнемолибденовой кислоты ~7.5 мас. %. После проведения процесса дегидратации наблюдается незначительное зауглероживание поверхности катализатора. В составе поверхности методом ЭДС были определены (мас. %): C – 6.06; O – 49.52; Al – 43.16; Si – 0.52; Mo – 0.74.
Необходимо отметить, что структура поверхности и фазовый состав отработанного катализатора практически не изменились по сравнению со свежеприготовленным образцом, о чем свидетельствуют данные рентгеновской дифрактометрии и электронной микроскопии.
В табл. 2 представлены результаты исследования закономерностей процесса парофазной дегидратации в присутствии катализатора SiMo/A.
Таблица 2.
Основные показатели процесса дегидратации глицерина в присутствии катализатора SiMo/A после 5 ч реакции
| № | T, °С | ω, мас. % | G, г/мин | mкат, г | g, ггл${\text{г}}_{{{\text{кат}}}}^{{ - 1}}$ мин–1 | Xгл, % | D, % | Yа, % | Yац, % | Yаа, % | Yас, % | Yост, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 280 | 10 | 0.2 | 1.0 | 0.02 | 32.6 | 35.1 | 11.3 | 2.2 | 1.4 | 1.8 | 15.9 |
| 2 | 300 | 10 | 0.2 | 1.0 | 0.02 | 60.6 | 26.5 | 22.8 | 3.2 | 2.3 | 3.0 | 29.3 |
| 3 | 330 | 10 | 0.2 | 1.0 | 0.02 | 89.9 | 5.9 | 46.1 | 7.8 | 4.0 | 0.6 | 31.4 |
| 4 | 350 | 10 | 0.2 | 1.0 | 0.02 | 49.4 | 37.1 | 20.2 | 2.5 | 5.5 | 2.1 | 19.1 |
| 5 | 330 | 5 | 0.2 | 1.0 | 0.01 | 94.2 | 1.5 | 40.3 | 3.4 | 6.5 | 2.3 | 41.7 |
| 6 | 330 | 10 | 0.2 | 1.0 | 0.02 | 89.9 | 5.9 | 46.1 | 7.8 | 4.0 | 0.6 | 31.4 |
| 7 | 330 | 15 | 0.2 | 1.0 | 0.03 | 65.2 | 24.2 | 26.6 | 6.5 | 4.3 | 2.4 | 25.4 |
| 8 | 330 | 20 | 0.2 | 1.0 | 0.04 | 53.5 | 33.0 | 18.3 | 5.5 | 3.7 | 2.1 | 23.9 |
| 9 | 330 | 10 | 0.1 | 1.0 | 0.01 | 92.3 | 4.3 | 36.6 | 4.2 | 8.0 | 3.2 | 40.3 |
| 10 | 330 | 10 | 0.2 | 1.0 | 0.02 | 89.9 | 5.9 | 46.1 | 7.8 | 4.0 | 0.6 | 31.4 |
| 11 | 330 | 10 | 0.3 | 1.0 | 0.03 | 70.7 | 27.1 | 29.3 | 5.2 | 3.9 | 3.2 | 29.1 |
| 12 | 330 | 10 | 0.4 | 1.0 | 0.04 | 51.5 | 39.3 | 18.2 | 3.1 | 2.9 | 2.0 | 25.3 |
| 13 | 330 | 10 | 0.2 | 0.5 | 0.04 | 54.6 | 33.8 | 17.6 | 5.1 | 3.5 | 2.4 | 26.0 |
| 14 | 330 | 10 | 0.2 | 1.0 | 0.02 | 89.9 | 5.9 | 46.1 | 7.8 | 4.0 | 0.6 | 31.4 |
| 15 | 330 | 10 | 0.2 | 2.0 | 0.01 | 94.1 | 4.6 | 37.6 | 4.0 | 6.7 | 2.3 | 43.5 |
Как видно из представленных данных, повышение температуры в интервале 280–330°С (опыты 1–3) привело к росту степени превращения глицерина и выхода целевого продукта. При дальнейшем нагреве (опыт 4) основные показатели процесса падают, что объясняется низкой термической стабильностью ПОМ в таких условиях и, как следствие, разрушением активных центров [23].
Основные характеристики процесса дегидратации глицерина существенно зависят от нагрузки на катализатор по чистому глицерину. С целью изучения влияния этого параметра на протекание реакции варьировали концентрацию глицерина в исходном потоке (опыты 5–8), скорость подачи водного раствора глицерина (опыты 9–12) и массу загружаемого в реактор катализатора (опыты 13–15). Нагрузку на катализатор (g, ггл${\text{г}}_{{{\text{кат}}}}^{{ - {\text{1}}}}$ мин–1) рассчитывали по формуле (3):
где ω – концентрация глицерина, мас. %; G – расход водного раствора глицерина, г/мин; mкат – масса катализатора, г.Во всех случаях увеличение нагрузки при температуре 330°С приводило к снижению конверсии глицерина после 5 ч проведения процесса (рис. 5), что обусловлено уменьшением времени пребывания реакционной массы в каталитической зоне.
Рис. 5.
Зависимости конверсии глицерина (1), выходов акролеина (2) и тяжелых продуктов (3) от нагрузки на катализатор по чистому глицерину при 330°С после 5 ч реакции.
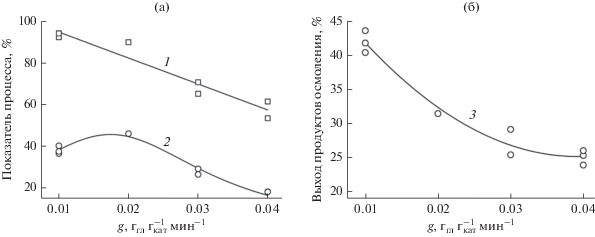
Кривая 2, описывающая зависимость выхода целевого продукта от нагрузки на катализатор, имеет экстремальный характер с максимумом при g ~ 0.02 ггл${\text{г}}_{{{\text{кат}}}}^{{ - {\text{1}}}}$ мин–1. При низком значении g (~0.01 ггл${\text{г}}_{{{\text{кат}}}}^{{ - {\text{1}}}}$ мин–1) и, соответственно, длительном времени пребывания реакционной массы в каталитической зоне на сильных кислотных центрах протекают побочные превращения акролеина в продукты осмоления (кривая 3). При высокой нагрузке на катализатор (g ≈ 0.03–0.04 ггл${\text{г}}_{{{\text{кат}}}}^{{ - {\text{1}}}}$ мин–1), по-видимому, не достигается эффективная адсорбция глицерина на активных центрах, что ведет к снижению выхода акролеина.
Стоит отметить, что увеличение концентрации глицерина до значений более 10 мас. % в исходном потоке способствует более быстрой дезактивации катализатора. Вероятно, это связано с блокированием активных центров катализатора продуктами конденсации акролеина при его высокой концентрации. Использование водного раствора глицерина с концентрацией менее 10 мас. % также нецелесообразно из-за низкой производительности по целевому продукту и роста затрат на выделение продуктов реакции.
Таким образом, на основании проведенных исследований для проведения процесса конверсии глицерина в присутствии каталитической системы SiMo/A определены следующие оптимальные условия: температура – 330°С; концентрация водного раствора глицерина – 10 мас. %; нагрузка на катализатор по чистому глицерину – 0.02 ггл${\text{г}}_{{{\text{кат}}}}^{{ - {\text{1}}}}$ мин–1. Такие условия позволяют достичь конверсии глицерина 90% и выхода акролеина 46%. Степень дезактивации катализатора за 5 ч работы не превышает 6%.
ЗАКЛЮЧЕНИЕ
В представленной работе проведена сравнительная оценка эффективности каталитических систем на основе ПОМ в процессе парофазной дегидратации глицерина в акролеин. Показано, что повышение общей кислотности катализатора приводит к снижению конверсии глицерина и выхода акролеина за счет отложения продуктов осмоления на сильных кислотных центрах, в результате чего происходит блокирование поверхности и дезактивация катализатора. Увеличение количества кислотных центров Бренстеда способствует росту выхода акролеина, в то время как повышение количества льюисовских центров ведет к изменению направления процесса и возрастанию выхода побочного продукта – ацетола. Наилучшие результаты получены при использовании каталитической системы SiMo/A, в присутствии которой изучены основные закономерности процесса парофазной дегидратации глицерина и определены условия, обеспечивающие наибольший выход акролеина.
Список литературы
Алхазов Т.Г., Аджамов К.Ю., Ханмамедова А.К. // Успехи химии. 1982. Т. 51. С. 950.
Кейко Н.А., Воронков М.Г. // Успехи химии. 1993. Т. 62. С. 796.
Katryniok B., Paul S., Capron M., Dumeignil F. // ChemSusChem. 2009. V. 2. P. 719.
Wang Y., Xiao Y., Xiao G. // Chin. J. Chem. Eng. 2019. V. 27. P. 1536.
Talebian-Kiakalaieh A., Aishah Saidina Amin N., Hezaveh H. // Renewable Sustainable Energy Rev. 2014. V. 40. P. 28.
Galadima A., Muraza O.J. // Taiwan Inst. Chem. Eng. 2016. V. 67. P. 29.
Sun D., Yamada Y., Sato S., Ueda W. // Green Chem. 2017. V. 19. P. 3186.
Gu Y., Cui N., Yu Q., Li C., Cui Q. // Appl. Catal. A. Gen. 2012. V. 429–430. P. 9.
Shan J., Li Z., Zhu S., Liu H., Li J., Wang J., Fan W. // Catalysts. 2019. V. 9. 121.
Danov S.M., Esipovich A.L., Belousov A.S., Rogozhin A.E. // Russ. J. Appl. Chem. 2014. V. 87. P. 461.
Danov S.M., Esipovich A.L., Belousov A.S., Rogozhin A.E. // Chin. J. Chem. Eng. 2015. V. 23. P. 1138.
Alhanash A., Kozhevnikova E.F., Kozhevnikov I.V. // Appl. Catal. A. Gen. 2010. V. 378. P. 11.
Liu R., Wang T., Liu C., Jin Y. // Chin. J. Catal. 2013. V. 34. P. 2174.
Thanasilp S., Schwank J.W., Meeyoo V., Pengpanich S., Hunsom M. // J. Mol. Catal. A: Chem. 2013. V. 380. P. 49.
Liu R., Wang T., Cai D., Jin Y. // Ind. Eng. Chem. Res. 2014. V. 53. P. 8667.
Thanasilp S., Schwank J.W., Meeyoo V., Pengpanich S., Hunsom M. // Chem. Eng. J. 2015. V. 275. P. 113.
Li X., Zhang Y. // ACS Catal. 2016. V. 6. P. 2785.
Suganuma S., Hisazumi T., Taruya K., Tsuji E., Katada N. // Mol. Catal. 2018. V. 449. P. 85.
Lowell S., Shields J.E., Thomas M.A., Thommes M. Characterization of Porous Materials and Powders: Surface Area, Pore Size and Density. Netherlands: Springer, 2004. 350 p.
Barzetti T., Selli E., Moscotti D., Forni L. // J. Chem. Soc. Faraday Trans. 1996. V. 92. P. 1401.
Волкова Г.Г., Буднева А.А., Шалыгин А.С., Саланов А.Н., Петров Р.В., Решетников С.И., Паукштис Е.А. // Химия в интересах устойчивого развития. 2012. № 2. С. 189.
Escribano V.S., Garbarino G., Finocchio E., Busca G. // Top. Catal. 2017. V. 60. P. 1554.
Kozhevnikov I.V. // Chem. Rev. 1998. V. 98. P. 171.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ