

Кинетика и катализ, 2020, T. 61, № 4, стр. 526-539
Палладий-фосфорные наночастицы как эффективные катализаторы хемоселективного гидрирования алкинолов
Н. И. Скрипов a, Л. Б. Белых a, *, Т. П. Стеренчук a, К. Л. Гвоздовская a, В. В. Жердев a, Т. М. Дашабылова a, Ф. К. Шмидт a
a ФГБОУ ВО Иркутский государственный университет
664003 Иркутск, К. Маркса, 1, Россия
* E-mail: belykh@chem.isu.ru
Поступила в редакцию 18.09.2019
После доработки 08.11.2019
Принята к публикации 24.12.2019
Аннотация
Рассмотрено влияние состава каталитической системы и условий проведения реакции на свойства палладиевых катализаторов, модифицированных фосфором, в гидрировании алкинолов. Показано, что модифицирование фосфором приводит к росту активности и производительности палладиевых катализаторов в гидрировании модельного соединения 2-метил-3-бутин-2-ола (MBY) без снижения селективности по аллиловому спирту при 95–98% конверсии MBY. Промотирующий эффект фосфора на свойства палладиевого катализатора обусловлен не только повышением дисперсности, но и, вероятно, изменением энергии взаимодействия реагентов с активными центрами. Кинетическими методами с применением в качестве основного измеряемого параметра дифференциальной селективности каталитических систем в условиях конкуренции двух алкинолов дискриминированы гипотезы о природе носителей каталитической активности в Pd–P-частицах. Показано, что из двух потенциально активных форм в Pd–P-наночастицах в гидрировании ацетиленовых спиртов участвует только одна – кластеры Pd(0); в гидрировании образующихся аллиловых спиртов возможно участие и кластеров Pd(0), и фосфидов палладия.
ВВЕДЕНИЕ
Селективное гидрирование кратных связей, включающее хемоселективное гидрирование алкинов до алкенов [1–4], а также алкинолов [5, 6] или ненасыщенных карбонильных соединений [7] до аллиловых спиртов без их дальнейшего гидрирования до алканов или алканолов соответственно, относится к важным реакциям химической промышленности, которые используются, главным образом, в производстве полимеров, фармацевтических препаратов, жирорастворимых витаминов (например, Е и К) и агрохимикатов. Так, уровень содержания фенилацетилена в мономере – стироле полимеризационной степени чистоты – должен быть ниже 10 ppm [8]. Однако в синтезе многих фармацевтических препаратов, витаминов, ароматизаторов и агрохимикатов применяются катализаторы, разработанные более 60 лет назад [1]. Типичные катализаторы для реакций селективного гидрирования чаще всего получают путем “частичного отравления активного металла” (Pd, Pt, Rh или Ni), при этом активный в катализе металл защищается и селективно дезактивируется различными соединениями: Pb, V, Cu, Ag, CO, хинолином и т.д. [1]. Повышение селективности, как правило, достигается за счет снижения каталитической активности. Присутствие в составе хемоселективного катализатора токсичных элементов (прежде всего, Pb) и невысокая каталитическая активность накладывают серьезные ограничения на использование таких материалов в будущем. В связи с современными стандартами экологической безопасности [9] ставится задача создания высокоселективных и более экологически чистых катализаторов, которые отвечают стратегии устойчивого развития и характеризуются разумным соотношением селективности и активности [10]. Поэтому исследования, проводимые в данной области, в последнее десятилетие получили новый импульс [1].
Современные подходы, направленные на создание селективных катализаторов хемоселективного гидрирования алкинов и алкинолов, базируются на контроле размера и формы наночастиц переходного металла [10–12], изолировании активных центров на поверхности гибридных материалов и разработке катализаторов на основе отдельных атомов палладия [1, 13], применении новых биметаллических сплавов [5, 14–16], а также на модифицировании поверхности активного компонента катализатора [6, 17] или носителя [18].
В качестве модификаторов палладиевых катализаторов гидрирования наиболее широкое распространение получили основания Льюиса [1] и катионы щелочных металлов в виде солей и оснований [6]. Если введение азотсодержащих оснований Льюиса приводит к повышению селективности за счет снижения каталитической активности в результате блокирования активных центров [1], то катионы щелочных металлов при определенных концентрациях оказывают промотирующее действие и на селективность, и на активность палладиевых катализаторов в гидрировании алкинолов [6]. Гидроксиды натрия и калия облегчают восстановление прекурсоров Pd(II) до Pd(0). Катионы щелочных металлов могут взаимодействовать с поверхностью Pd-наночастиц, приводя к “эффекту разделения участков” и изменяя энергию адсорбции реагентов, а также влиять на подвижность наночастиц Pd на твердых носителях, предотвращая спекание во время термической обработки. При этом активность гетерогенных палладиевых катализаторов, модифицированных катионами щелочных металлов, в гидрировании алкинолов не превышает 426 мин–1 при достаточно высокой (90°С) температуре [6].
Третичные, вторичные и первичные фосфины в отличие от азотсодержащих оснований Льюиса подвергаются деструкции в координационной сфере Pd(0), приводящей к появлению полиядерных комплексов палладия с фосфидными, фосфиниденовыми лигандами и даже фосфидов палладия [19, 20]. Учитывая образование фосфидов палладия в процессе формирования и функционирования активных частиц на основе фосфиновых комплексов палладия, для разработки эффективных катализаторов гидрирования в качестве модификатора вместо фосфинов нами была предложена наиболее реакционная форма элементного фосфора – белый фосфор [21].
Восстановление соединений палладия водородом в присутствии P4 в мягких условиях позволяет получать высокодисперсные палладиевые катализаторы, частицы которых содержат кластеры Pd(0) и фосфиды палладия. Размер, состав и состояние поверхностного слоя частиц можно изменять, варьируя соотношение P : Pd [21], природу ацидолиганда при палладии [22, 23] или растворитель [24]. Из двух химических форм палладия (Pd0, Pdδ+) при малых исходных отношениях P : Pd (≤ 0.3) на поверхности наночастиц преобладает Pd(0), энергия связи электронов которого на 0.7 эВ ниже, чем у массивного палладия [25]. Т. к. гидрирование алкинолов относится к размерно-чувствительным реакциям [11, 12], то перечисленные выше отличительные физические характеристики Pd–P-наночастиц могут оказать положительный эффект на их свойства в гидрировании алкинолов.
Для разработки эффективных катализаторов хемоселективного гидрирования ацетиленовых соединений и установления природы носителей каталитической активности в настоящей работе исследованы закономерности гидрирования 2-метил-3-бутин-2-ола и конкурентное гидрирование алкинолов под действием Pd–P-частиц, формируемых низкотемпературным способом.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Растворители (бензол, N,N-диметилформамид) очищали по стандартным методикам [26]. N,N-диметилформамид (ДМФА) выдерживали над безводным сульфатом меди до получения раствора зеленого цвета с целью обезвоживания и удаления примесей аминов и дважды подвергали вакуумной разгонке (8 мм рт. ст.) при температуре не выше 42°С. Для более глубокой осушки бензол дополнительно перегоняли над LiAlH4 с использованием ректификационной колонки. Растворители хранили в атмосфере аргона в запаянных ампулах над молекулярными ситами 4А.
Бис-(ацетилацетонат) палладия Pd(acac)2 получали согласно методике [27] с последующей перекристаллизацией из ацетона. Диацетат палладия (Pd(OAc)2) и дихлорид палладия (PdCl2) квалификации “х. ч.” применяли без дополнительной очистки.
Белый фосфор непосредственно перед использованием механически очищали от поверхностных продуктов окисления и промывали в безводном бензоле. Раствор белого фосфора в бензоле готовили и хранили в инертной атмосфере в сосуде типа “палец”, предварительно вакуумированном и заполненным аргоном.
2-Метил-3-бутин-2-ол (MBY) (“Sigma-Aldrich”, >98%), 3-метил-1-пентин-3-ол (MPY) (“Sigma-Aldrich”, >98%) применяли без дополнительной очистки. Фенилацетилен выдерживали над предварительно прокаленным дихлоридом кальция в течение 2 недель и перегоняли над гидридом кальция (40°С/10 мм рт. ст.). Стирол очищали путем встряхивания его с 5%-ным раствором щелочи до тех пор, пока порция щелочи не становилась бесцветной. Затем промывали дистиллированной водой, сушили над безводным хлоридом кальция и перегоняли в вакууме (32°С/10 мм рт. ст.).
Триэтилалюминий (AlEt3) фирмы “Sigma-Aldrich” перегоняли в вакууме, собирая фракцию, кипящую при 48–49°C/1 мм рт. ст., и использовали в виде раствора в бензоле. ЯМР 1H: δ(СН3) = 1.22 м. д. (т, 3Н, 1J = 8.24 Гц); δ(СН2) = = 0.45 м. д. (кв., 2Н, 1J = 8.24 Гц).
Примеры проведения экспериментов
Гидрирование алкинолов проводили в стеклянном термостатируемом сосуде типа “утка” при 30°C и начальном давлении водорода 2 атм в присутствии Pd–P-катализатора, сформированного in situ.
К раствору Pd(аcac)2 (0.00304 г, 1 × 10–5 моль) в 9 мл ДМФА, помещенному в термостатируемый сосуд “утка”, в потоке водорода добавляли по каплям 1 мл раствора фосфора в бензоле (0.1 × 10–5–1.0 × 10–5 моль в расчете на атомную форму фосфора) и перемешивали в течение 7 мин при комнатной температуре. Затем температуру поднимали до 80°C и перемешивали реакционную смесь в водороде в течение 40–45 мин до количественного превращения Pd(аcac)2. Полученный коллоидный раствор черно-коричневого цвета охлаждали до 30°C, создавали давление водорода 2 атм и шприцом через тефлоновую пробку с резиновой прокладкой вводили субстрат – 2-метил-3-бутин-2-ол. Гидрирование MBY осуществляли при интенсивном перемешивании, исключающем протекание реакции в диффузионной области, периодически отбирая аликвоты реакционной смеси для ГЖХ-анализа.
Таким же образом формировали каталитические системы при концентрациях Pd(аcac)2 более 1 ммоль/л. Свойства Pd–P-частиц при концентрациях катализатора 0.25 и 0.5 ммоль/л исследовали, разбавляя предварительно сформированную каталитическую систему (${{С}_{{{\text{Pd}}{{{\left( {{\text{асас}}} \right)}}_{{\text{2}}}}}}}$ = 1 ммоль/л). Каталитические эксперименты с применением других палладиевых прекурсоров (PdCl2, Pd(OAc)2) проводили аналогичным образом. Отличия состояли в температуре и времени формирования катализаторов. В случае PdCl2 и Pd(OAc)2 каталитические системы формировали в водороде при 30°C в течение 5 мин.
Гидрирование алкинолов протекает в две стадии: на первой стадии преимущественно гидрируется тройная связь, на второй стадии образующийся аллиловый спирт восстанавливается до насыщенного спирта. Количество продукта полного гидрирования (алканола) на первой стадии не превышало 5%. Учитывая этот факт, скорость реакции r $\left( {{{{\text{мол}}{{{\text{ь}}}_{{{{{\text{H}}}_{2}}}}}} \mathord{\left/ {\vphantom {{{\text{мол}}{{{\text{ь}}}_{{{{{\text{H}}}_{2}}}}}} {{\text{мин}}}}} \right. \kern-0em} {{\text{мин}}}}} \right)$ на каждой стадии определяли из кинетической кривой следующим образом. Скорость гидрирования MBY до аллилового спирта рассчитывали по углу наклона прямых участков кривых в диапазоне поглощения водорода 0.1–0.5 моль H2 на моль MBY. Скорость гидрирования аллилового спирта до насыщенного спирта находили по углу наклона прямых участков кривых в диапазоне поглощения водорода 1.1–1.3 моль H2 на моль MBY. Селективность Pd–P-катализаторов по 2-метил-3-бутен-2-олу вычисляли при конверсии исходного ацетиленового спирта 95–98%. Чтобы определить наибольшее число оборотов (TON) и стабильность катализатора Pd–P, процедуру гидрирования повторяли, вводя аликвоту свежего субстрата в реакционный сосуд после завершения гидрирования предыдущей порции MBY до 2-метилбутан-2-ола.
Конкурентное гидрирование алкинолов в присутствии Pd–P-частиц, полученных из Pd(acac)2 и P4 в водороде, проводили аналогично неконкурентному гидрированию алкинолов. Для этого в сформированный in situ (как описано выше) коллоидный раствор Pd–P-частиц шприцом вводили смесь алкинолов – 2-метил-3-бутин-2-ол (MBY) (3.3535 ммоль, 0.31 мл) и 3-метил-1-пентин-3-ол (MPY) (3.3644 моль, 0.38 мл) и гидрировали при интенсивном перемешивании. Контроль осуществляли ГЖХ-анализом периодически отбираемых проб.
Расчет дисперсности (DПЭМ) и частоты оборотов (TOF) Pd-черни и Pd–P-частиц проводили по формулам (1)–(3):
(1)
${{D}_{{{\text{ПЭМ}}}}} = \frac{{6{{M}_{{Pd}}}}}{{{{\rho }_{{Pd}}}{{d}_{{ПЭМ}}}{{A}_{{Pd}}}{{N}_{A}}}},$(2)
${{d}_{{{\text{ПЭМ}}}}} = \frac{{\sum\limits_i {{{n}_{i}}d_{i}^{3}} }}{{\sum\limits_i {{{n}_{i}}d_{i}^{2}} }},$Вышеуказанный подход был применен нами только для Pd–P-частиц (исходное отношение P : : Pd ≤ 0.3), объем и поверхность которых по данным рентгенофазового анализа (РФА) и рентгеновской фотоэлекронной спектроскопии (РФЭС) обогащены палладием Pd(0) [25].
Методы исследования
УФ-спектры растворов катализатора снимали на спектрофотометре СФ-2000 (“ОКБ Спектр” Россия) в кварцевых кюветах с толщиной поглощающего слоя 0.1 см. Контроль за превращением Pd(аcac)2 проводили по полосе поглощения 330 нм (ε330 = 10630 л см–1 моль–1).
Продукты гидрирования анализировали методом ГЖХ на хроматографе Кристалл 5000 (“Хроматэк”, Россия), снабженном капиллярной колонкой длиной 30 м (фаза – 5% дифенил, 95% диметилполисилфениленсилоксан) и пламенно-ионизационным детектором (ДИП), по методу внутреннего стандарта, используя температурное программирование: 100°C (1.5 мин), 270°C (10 мин), скорость нагрева 30оC/мин. Давление газа-носителя (азот) – 20 кПа.
Дополнительно продукты гидрирования анализировали на хромато-масс-спектрометре GCMS-QP2010 Ultra (“Shimadzu”, Япония) с капиллярной колонкой GsBP⋅5MS длиной 30 м, фаза – поли(5% дифенил, 95% диметилполисилфениленсилоксан). Ионизацию проводили под действием электронного удара, энергия ионизации составляла 70 эВ. Полученные масс-спектры сравнивали с литературными данными (библиотеки сравнения Wiley, NIST, NIST05).
Снимки ПЭМ получали на электронном микроскопе Tecnai G2 (“FEI”, США) с ускоряющим напряжением 200 кВ. Каплю полученного реакционного раствора наносили на науглероженную медную сеточку, высушивали в боксе в инертной атмосфере при комнатной температуре. Изображения записывали с помощью CCD-камеры (“Soft Imaging System”, Германия). Для нахождения среднего размера частиц обрабатывали участок, содержащий не менее 100–150 частиц.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
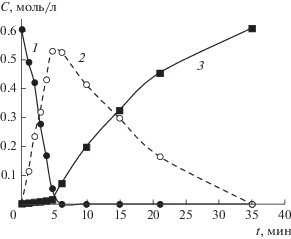
Гидрирование 2-метил-3-бутин-2-ола (MBY) в присутствии Pd-черни и Pd–P-частиц в среде ДМФА представляет собой пример типичной последовательной реакции. На первой стадии реакции (до поглощения 1 моль Н2 на моль MBY) происходит преимущественное гидрирование 2-метил-3-бутин-2-ола до 2-метил-3-бутен-2-ола (MBE), на второй стадии образуется продукт гидрирования 2-метил-3-бутен-2-ола – 2-метилбутан-2-ол (MBA) (рис. 1). Продукты димеризации алкинола С10 и гидрирования образовавшегося димера (2,7-диметил-3,5-октадиин-2,7-диола до 2,7-диметил-5-октен-3-ин-2,7-диола) методами хромато-масс-спектроскопии и ГЖХ обнаружены в следовых количествах. Продукты изомеризация 2-метил-3-бутин-2-ола до ненасыщенного альдегида отсутствовали.
Рис. 1.
Кинетические кривые гидрирования 2-метил-3-бутин-2-ола (1) и образования 2-метил-3-бутен-2-ола (2), 2-метилбутан-2-ола (3) в присутствии Pd–P-частиц, Р : Pd = 0.3. Условия реакции: СPd = = 0.25 ммоль/л, СMBY = 0.598 ммоль/л, Т = 30°С; ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм.
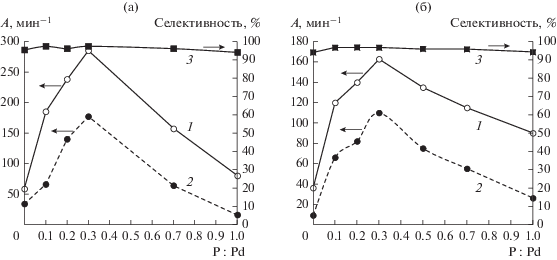
Активность Pd–P-частиц в гидрировании 2‑метил-3-бутин-2-ола зависит от состава каталитической системы и условий проведения реакции. В результате модифицирования палладиевого катализатора фосфором, вводимым до стадии восстановления Pd(acac)2 водородом, возрастает как скорость гидрирования тройной связи MBY, так и скорость гидрирования двойной связи образующегося 2-метил-3-бутен-2-ола (MBE) без снижения селективности по аллиловому спирту во всем исследованном диапазоне отношений P : Pd = 0.1–1 при конверсии субстрата 96–98% (рис. 2). Даже при введении малых количеств элементного фосфора (P : Pd = 0.1) активность Pd–P-частиц (А), рассчитанная на весь палладий, возрастает практически в 3 раза в сравнении с Pd-чернью: с 58.5 до 185 мин–1 (рис. 2а). При концентрации Pd(acac)2 равной 1 ммоль/л наибольший промотирующий эффект фосфора наблюдается при отношении P : Pd = 0.3: активность Pd–P-частиц в гидрировании MBY составляет 285 мин–1 (рис. 2а).
Рис. 2.
Влияние фосфорного модификатора на активность (1, 2) и селективность по МВЕ (3) Pd–Р-наночастиц в гидрировании 2-метил-3-бутин-2-ола (1), 2-метил-3-бутен-2-ола (2) при концентрации Pd(acac)2, равной 1 (а) или 2 ммоль/л (б). Условия реакции: νMBY = 6.64 × 10–3 моль, растворитель – ДМФА, 10 мл, Т = 30°С; ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм. Селективность измерена при конверсии MBY 97–98%.
При изотермических условиях проведения процесса активность Pd–P-частиц определяется не только отношением P : Pd, но и давлением водорода, концентрациями MBY и палладиевого прекурсора (рис. 3). Скорость гидрирования тройной связи MBY (r1) линейно зависит от давления водорода (рис. 3а); а зависимость скорости реакции от концентрации субстрата MBY проходит через максимум (рис. 3б). Скорость гидрирования MBY нелинейно возрастает с повышением концентрации Pd(acac)2 от 0.25 до 5 ммоль/л, выходя на плато при ${{С}_{{{\text{Pd}}{{{\left( {{\text{acac}}} \right)}}_{{\text{2}}}}}}}$ = 2 ммоль/л (рис. 3в). Зависимость активности Pd–P-частиц от ${{С}_{{{\text{Pd}}{{{\left( {{\text{acac}}} \right)}}_{{\text{2}}}}}}}$ в указанном диапазоне концентраций носит антибатный характер (рис. 3в). При оптимальной концентрации Pd(acac)2 (0.25–0.5 ммоль/л) активность Pd–P-частиц в гидрировании MBY достигает 450 мин–1 при 30°С и ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм. По активности в гидрировании MBY частицы Pd–Р превосходят кластеры палладия в форме куба или кубооктаэдра размерами 6 и 5.5 нм (АPd общий = 153 и 135 мин–1 соответственно; Т = 60°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 3 атм) [10], биметаллические сплавы: Pd7Bi/SiO2 (АPd общий = 12.6 мин–1; Т = 50°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 1 атм) [14], PdZn/TiO2 (АPd общий = = 180 мин–1; Т = 40°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 1 атм) [14] и сопоставимы с системами циглеровского типа (табл. 1). Для систем циглеровского типа, так же как и для Pd–P-частиц, наблюдается рост каталитической активности при разбавлении системы Pd(acac)2–n AlEt3 (n = 4, 6) (с 2 до 0.5 ммоль/л) (табл. 1). Причины влияния разбавления на активность Pd–P-частиц и нанокластеров палладия в циглеровских системах, на наш взгляд, могут быть следующие.
Рис. 3.
Зависимость скорости гидрирования 2-метил-3-бутин-2-ола (r) или активности (А) от давления водорода (а), концентрации субстрата (б) и Pd(acac)2 (в) в присутствии Pd–P-частиц (Р : Pd = 0.3). Условия реакции: СPd = 0.5 ммоль/л, СMBY = 0.598 моль/л, Т = = 20°С (а); СPd = 0.5 ммоль/л, Т = 30°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм (б); νMBY = 6.64 × 10–3 моль, Т = 30°С; ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм, растворитель – ДМФА, 10 мл (в).
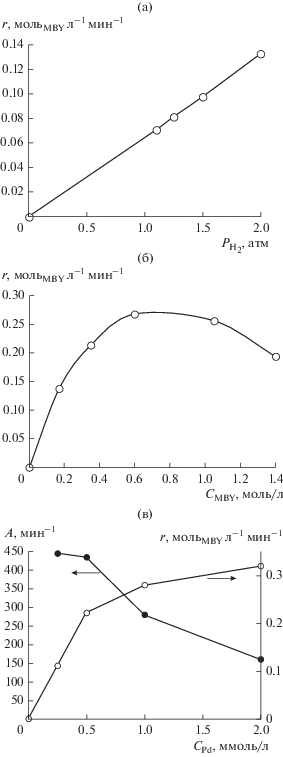
Таблица 1.
Каталитические свойства систем циглеровского типа Pd(acac)2–nAlEt3 в гидрировании 2-метил-3-бутин-2-ола*
| AlEt3 : Pd | СPd, ммоль/л | Активность (А), мин–1 | А1 : А2 | Конверсия, % | Селективность по MBE, % | |
|---|---|---|---|---|---|---|
| А1 −С≡С− |
А2 −С=С− |
|||||
| 4 | 2 | 194 | 441 | 0.4 | 94.2 | 93.9 |
| 6 | 2 | 184 | 373 | 0.5 | 95.8 | 92.7 |
| 4 | 1 | 395 | 528 | 0.7 | 91.8 | 95.6 |
| 4 | 0.5 | 370 | 772 | 0.5 | 96.6 | 94.1 |
| 6 | 0.5 | 367 | 744 | 0.5 | 97.8 | 93.7 |
| 10 | 0.5 | 332 | 557 | 0.6 | 97.1 | 92.9 |
Согласно данным ПЭМ наночастицы Pd–P представляют собой агрегаты частиц (рис. 4б–4д), в то время как нанокластеры палладия в циглеровских системах представлены отдельными высококонтрастными частицами (рис. 4е). Причиной агрегативной устойчивости нанокластеров палладия является их стабилизация AlEt2(acac) и AlEt3 [28–30]. Ингибирующее действие алюминийорганических соединений в результате адсорбции на активных центрах палладия ранее было доказано экспериментально [29, 30]. Исходя из этого, рост активности циглеровских систем в гидрировании MBY при разбавлении реакционного раствора обусловлен, преимущественно, устранением ингибирующего действия стабилизаторов (AlEt2(acac), AlEt3) в результате гидролиза поверхностных соединений примесями воды в растворителе. Причиной влияния разбавления на активность Pd–Р-частиц может быть также предотвращение агрегирования частиц, которое, как известно, происходит по реакции второго порядка [28]. Протекание реакции в кинетической области было доказано дополнительными экспериментами.
Рис. 4.
ПЭМ-снимки катализаторов: Pd-чернь (а); система Pd(acac)2–4AlEt3, СPd = 5 ммоль/л (е); Pd–P-частицы, сформированные в водороде: P : Pd = 0.3 (б); P : Pd = 1.0, СPd = 1 ммоль/л (в); P : Pd = 0.1, СPd = 1 ммоль/л (г); СPd = = 2 ммоль/л (д).

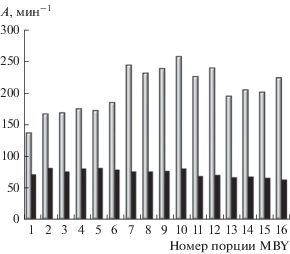
Введение фосфора до стадии восстановления Pd(acac)2 водородом значительно повышает не только активность, но и производительность палладиевых катализаторов. Число оборотов (TON) в гидрировании MBY под действием Pd–P-катализатора (P : Pd = 0.5) практически в два раза (10 650 моль MBY на моль Pd) превышает TON Pd-черни (5470 моль MBY на моль Pd) (рис. 5).
Рис. 5.
Гидрирование первых 16 порций 2-метил-3-бутин-2-ола (серый столбец) и образующегося 2-метил-3-бутен-2-ола (черный столбец) под действием Pd–P-катализатора, P : Pd = 0.5. Условия реакции: СPd = 2 ммоль/л; 1 порция MBY – 6.64 × 10–3 моль; растворитель – ДМФА, 10 мл, Т = 30°С; ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм.
Замена ацетилацетонатного лиганда при палладии на ацетат практически не оказывает влияние на свойства Pd–Р-наночастиц в гидрировании 2-метил-3-бутин-2-ола (табл. 2). Меньшее значение активности Pd–Р-частиц, полученных из PdCl2, является результатом отравления поверхностных атомов палладия анионами хлора, которые образуются в результате гидрогенолиза. Таким образом, основное воздействие фосфор оказывает на активность и производительность палладиевого катализатора в гидрировании 2-метил-3-бутин-2-ола, сохраняя при этом высокую селективность при 96–98% конверсии субстрата.
Таблица 2.
Влияние природы прекурсора на активность и селективность Pd−Р-наночастиц в гидрировании 2-метил-3-бутин-2-ола*
| Прекурсор | Активность (А), мин–1 | А1 : А2 | Конверсия, % | Селективность по MBE, % | |
|---|---|---|---|---|---|
| −С≡С− | −С=С− | ||||
| Pd(OAc)2 | 166 | 127 | 1.3 | 97.2 | 95.6 |
| PdСl2 | 91 | 91 | 1.0 | 98.0 | 95.5 |
| Pd(acac)2 | 163 | 113 | 1.4 | 96.2 | 96.1 |
Промотирующее действие фосфора на активность Pd–Р-наночастиц в гидрировании 2-метил-3-бутин-2-ола может быть как следствием увеличения доли активных центров, так и (или) образования новых более активных центров катализаторов. Для различения этих двух факторов необходимо, прежде всего, учесть долю поверхностных атомов у Pd-черни, кластеров палладия в циглеровских системах и Pd–P-частиц. Результаты определения методом ПЭМ размеров Pd-черни, сформированной из Pd(acac)2 в водороде, кластеров палладия в циглеровских системах и Pd–P-частиц в зависимости от начальной концентрации Pd(acac)2 и количества введенного модификатора представлены в табл. 3. Размер частиц Pd–P-катализатора в сравнении с Pd-чернью уменьшается практически в 4 раза (рис. 4). При этом среднечисленный размер Pd–P-частиц с ростом отношения P : Pd изменяется незначительно: от 5.9 ± 1.04 (P : Pd = 0.1) до 5.5 ± 0.92 (P : Pd = = 0.3) и 5.3 ± 1.01 нм (P : Pd = 1.0). С увеличением концентрации прекурсора (Pd(acac)2) размер Pd–P-частиц и Pd-черни повышается. Для Pd–P-катализатора (P : Pd = 0.1) он возрастает от 5.9 ± ± 1.04 нм (СPd = 1 ммоль/л) до 10.7 ± 2.3 нм (СPd = = 2 ммоль/л) (рис. 4).
Таблица 3.
Частоты оборотов различных палладиевых катализаторов, сформированных из Pd(acac)2, в гидрировании MBY*
| Катализатор | P : Pd | ${{С}_{{{\text{Pd}}{{{\left( {{\text{acac}}} \right)}}_{{\text{2}}}}}}},$ ммоль/л | Размер частиц, нм | DПЭМ | TOF, мин–1 | |||
|---|---|---|---|---|---|---|---|---|
| d | dПЭМ | MBY | ФА** | стирол | ||||
| Pd-чернь | 0 | 1 | 22.2 ± 4.5 | 23.4 | 0.045 | 1300 | 444 | 666 |
| Pd−P-частицы | 0.1 | 1 | 5.9 ± 1.0 | 6.0 | 0.169 | 1189 | 911 | 1118 |
| 0.1 | 2 | 10.7 ± 2.3 | 11.7 | 0.090 | 1444 | 942 | − | |
| 0.3 | 1 | 5.5 ± 0.9 | 5.65 | 0.187 | 1497 | 850 | 1497 | |
| 0.3 | 0.5 | 5.5 ± 0.9 | 5.65 | 0.187 | 2326 | − | − | |
| 0.3 | 0.25 | 5.5 ± 0.9 | 5.65 | 0.187 | 2380 | − | − | |
| Pd(acac)2−4AlEt3 | 0 | 5 | 2.45 ± 0.64 | 2.53 | 0.418 | 430 | 279 | 430 |
| 0 | 2 | 2.45 ± 0.64 | 2.53 | 0.418 | 464 | − | − | |
| 0 | 0.5 | 2.45 ± 0.64 | 2.53 | 0.418 | 794 | − | − | |
| Pd(acac)2−6AlEt3 | 0 | 5 | 1.55 ± 0.57 | 1.65 | 0.604 | 382 | 201 | 264 |
| 0 | 0.5 | 1.55 ± 0.57 | 1.65 | 0.604 | 607 | − | − | |
Прочерки означают, что гидрирование ФА и стирола при этих концентрациях не проводили. * Условия см. в табл. 1 и 2 и на рис. 2. ** ФА − фенилацетилен.
Значения частоты оборотов палладиевых катализаторов (TOF), рассчитанные на поверхностные атомы палладия, представлены в табл. 3. Наибольшие величины TOF, характерные для Pd‑черни и Pd–P-частиц, в гидрировании MBY различаются в 1.8 раз (TOFPd–P : TOFPd-чернь = 1.8). По частоте оборотов Pd–P-частицы превосходят не только близкие по размерам нанокластеры палладия в форме куба (TOF = 918 мин–1), октаэдра (TOF = = 1050 мин–1) и кубооктадра (TOF = 876 мин–1) [10], но и системы циглеровского типа (табл. 3).
Дополнительно проведенные эксперименты по гидрированию фенилацетилена и стирола в присутствии Pd–P-частиц показали аналогичные результаты (табл. 3). По частоте оборотов в гидрировании и фенилацетилена, и стирола Pd–P-частицы превосходят и Pd-чернь, и нанокластеры палладия в циглеровских системах. Значительное превышение частоты оборотов Pd–P-частиц (TOF = 2380 мин–1) над соответствующими величинами для Pd-черни (табл. 3) и нанокластеров палладия различной формы [10] указывает на бóльшую реакционную способность Pd–P-наночастиц (P : Pd = 0.3) в гидрировании алкинолов, алкинов и алкенов.
Исходя из полученных данных, мы полагаем, что модифицирующее действие фосфора на свойства Pd–P-частиц в гидрировании обусловлено не только геометрическим, но и электронным фактором, который заключается в изменении энергии взаимодействия активных центров Pd–P-частиц с участниками реакции. Это предположение основано на следующих фактах.
Сохранение высокой селективности Pd–P-наночастиц по алкенолу при 96–98% конверсии субстрата показывает, что она зависит не только от соотношения скоростей гидрирования конкурирующих субстратов, но и от соотношения констант адсорбционного равновесия конкурирующих субстратов (алкинол, алкенол) с катализатором. Как известно, селективность палладиевых катализаторов при гидрировании тройной связи обусловлена более прочной адсорбцией алкина, чем алкена или алкана [3]. Для протекания реакции гидрирования адсорбция реагирующих веществ не должна быть слишком сильной. По данным РФЭС, при малых отношениях P : Pd (P : Pd = 0.3) кластеры Pd(0), преобладающие на поверхности Pd–P-частиц, характеризуются меньшим значением энергии связи (Есв(Pd3d5/2) = 334.5 эВ) [25] в сравнении с массивным металлом (Есв(Pd3d5/2) = 335.2 эВ) [31]. Как правило, Pd-катализаторы с высокой электронной плотностью на палладии более активны в гидрировании ненасыщенных соединений [32, 33]. С одной стороны, избыток электронов на атомах палладия облегчает диссоциативную хемосорбцию молекулы водорода [32, 33]. С другой стороны, он снижает адсорбцию и алкинов, и алкенов, при этом адсорбция алкенов подавляется в большей степени, чем алкинов [3]. Монозамещенные алкины и алкинолы характеризуются слишком высоким адсорбционным сродством к палладию. Поэтому повышение электронной плотности на Pd(0) в Pd–P-частицах (P : Pd = 0.3) и, как следствие этого, уменьшение сорбционного сродства алкинов (алкинолов) должны благоприятствовать росту каталитической активности. Увеличение селективности и активности биметаллических сплавов Pd–Zn в гидрировании 2-метил-3-бутин-2-ола в результате снижения абсолютных значений констант адсорбционного равновесия KMBY и KMBE и роста отношения KMBY/KMBE в 1.4 раза в сравнении с катализатором Линдлара было отмечено в работе [34].
Данный вывод может объяснить высокие значения TOF частиц Pd–P только в том случае, если носителями каталитической активности являются кластеры Pd(0). Но Pd–P-частицы, получаемые низкотемпературным методом, содержат и кластеры Pd(0), и фосфиды палладия, в которых присутствует электронодефицитная форма палладия (Pdδ+). Фосфиды палладия, обогащенные палладием, также могут обладать активностью в гидропроцессах [35–37]. Для того чтобы установить, какая из этих двух форм (или они обе) ответственна за каталитические свойства Pd–P-наночастиц в гидрировании алкинолов в мягких условиях, проведены дополнительные эксперименты.
Результаты формально-кинетического исследования гидрирования MBY в присутствии наиболее активных Pd–P-частиц (P : Pd = 0.3) представлены выше на рис. 3. Экстремальный характер зависимости скорости гидрирования от концентрации MBY в области 0.6–1.4 ммоль/л свидетельствует о конкурентной адсорбции молекул MBY и водорода на активных центрах Pd–P-частиц. Близкий к нулевому концентрационный порядок реакции по MBY в области концентраций 0.6–1.05 моль/л, а также первый порядок по водороду (n(Н2) = 1.04) в мягких условиях проведения реакции (1–2 атм) могут указывать на насыщение поверхности катализатора субстратом и участие водорода в лимитирующей стадии реакции. Поэтому для описания гетерогенно-каталитического гидрирования 2-метил-3-бутин-2-ола в присутствии Pd–P-частиц нами применен механизм Ленгмюра–Хиншельвуда, согласно которому для протекания химической реакции необходима активация и водорода, и MBY. Гидрирование 2-метил-3-бутин-2-ола по механизму Ленгмюра–Хиншельвуда предполагает конкурентную [38, 39] или неконкурентную [34] адсорбцию между водородом и молекулами MBY на активных центрах катализатора. Механизм Ридила–Или, в рамках которого учитывается активация только одного реагента [40], в данном случае не рассматривался.
Различные варианты кинетических моделей механизма Ленгмюра–Хиншельвуда для реакции гидрирования представлены в работе [41]. Т.к. на поверхности Pd–P-наночастиц присутствуют две потенциально активные формы палладия, то наиболее вероятны следующие кинетические модели (табл. 4):
Таблица 4.
Кинетические модели гидрирования 2-метил-3-бутин-2-ола по механизму Ленгмюра−Хиншельвуда [41]
| № | Кинетическая модель | Кинетическое уравнение* | Примечание |
|---|---|---|---|
| 1 | ${\text{MBY}} + {{{\theta }}_{{\text{o}}}}\overset {{{K}_{{{\text{MBY}}}}}} \leftrightarrows {{{\theta }}_{{{\text{MBY}}}}};$ ${{{\text{H}}}_{2}} + 2{{{\theta }}_{{\text{o}}}}\overset {{{K}_{{{{{\text{H}}}_{2}}}}}} \leftrightarrows 2{{{\theta }}_{{\text{H}}}};$ ${{{\theta }}_{{{\text{MBY}}}}} + 2{{{\theta }}_{{\text{H}}}}\xrightarrow{{{{k}_{1}}}}{{{\theta }}_{{{\text{MBE\;}}}}} + 2{{{\theta }}_{{\text{o}}}};$ ${{r}_{1}} = {{k}_{1}}{{{\theta }}_{{{\text{MBY}}}}}{\theta }_{{\text{Н}}}^{2}$ |
${{r}_{1}} = {{k}_{1}}\frac{{{{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}{{K}_{{{{{\text{H}}}_{2}}}}}{{P}_{{{{{\text{H}}}_{2}}}}}}}{{{{{\left( {1 + K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}} + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}} \right)}}^{3}}}}$ | ${{{\theta }}_{{\text{Н}}}} = \frac{{K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}{{\left( {1 + K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}} + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}} \right)}}$ ${{{\theta }}_{{{\text{MBY}}}}} = \frac{{{{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}}}{{1 + K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}} + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}}}$ |
| 2 | ${\text{MBY}} + {{{\theta }}_{{\text{о}}}}\overset {{{K}_{{{\text{MBY}}}}}} \leftrightarrows {{{\theta }}_{{{\text{MBY}}}}};$ ${{{\text{H}}}_{2}} + 2{\theta }_{{\text{о}}}^{'}~\overset {K_{{{{{\text{H}}}_{2}}}}^{'}} \leftrightarrows 2{\theta }_{{\text{Н}}}^{'}$ ${{{\theta }}_{{{\text{MBY}}}}} + 2{\theta }_{{\text{Н}}}^{'}\xrightarrow{{{{k}_{1}}}}{{{\theta }}_{{{\text{MBE}}}}} + 2{\theta }_{{\text{о}}}^{'}$ ${{r}_{1}} = {{k}_{1}}{{{\theta }}_{{{\text{MBY}}}}}{{({\theta }_{{\text{H}}}^{'})}^{2}}$ |
${{r}_{1}} = {{k}_{1}}\frac{{{{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}K_{{{{{\text{H}}}_{2}}}}^{'}{{P}_{{{{{\text{H}}}_{2}}}}}}}{{\left( {1 + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}} + K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}} \right){{{\left( {1 + {{{(K_{{{{{\text{H}}}_{2}}}}^{'})}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}} \right)}}^{2}}}}$ | ${\theta }_{{\text{Н}}}^{'} = \frac{{{{{(K_{{{{{\text{H}}}_{2}}}}^{'})}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}{{\left( {1 + {{{(K_{{{{{\text{H}}}_{2}}}}^{'})}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}} \right)}}$ ${{{\theta }}_{{{\text{MBY}}}}} = \frac{{{{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}}}{{1 + K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}} + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}}}$ |
| 3 | ${\text{MBY}} + {{{\theta }}_{{\text{о}}}}\overset {{{K}_{{{\text{MBY}}}}}} \leftrightarrows {{{\theta }}_{{{\text{MBY}}}}};$ ${{{\text{H}}}_{2}} + 2{\theta }_{{\text{о}}}^{'}\overset {K_{{{{{\text{H}}}_{2}}}}^{'}} \leftrightarrows 2{\theta }_{{\text{Н}}}^{'}$ ${{{\theta }}_{{{\text{MBY}}}}} + 2{\theta }_{{\text{Н}}}^{'}\xrightarrow{{{{k}_{1}}}}{{{\theta }}_{{{\text{MBE}}}}} + 2{\theta }_{{\text{о}}}^{'}$ ${{r}_{1}} = {{k}_{1}}{{{\theta }}_{{{\text{MBY}}}}}{{({\theta }_{{\text{H}}}^{'})}^{2}}$ |
${{r}_{1}} = {{k}_{1}}\frac{{{{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}K_{{{{{\text{H}}}_{2}}}}^{'}{{P}_{{{{{\text{H}}}_{2}}}}}}}{{{{{\left( {1 + {{{(K_{{{{{\text{H}}}_{2}}}}^{'})}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}} + K_{{{\text{MBY}}}}^{'}{{С}_{{{\text{MBY}}}}}} \right)}}^{2}}\left( {1 + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}} \right)}}$ | ${\theta }_{{\text{Н}}}^{'} = \frac{{K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}{{\left( {1 + {{{(K_{{{{{\text{H}}}_{2}}}}^{'})}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}} + K_{{{\text{MBY}}}}^{'}{{С}_{{{\text{MBY}}}}}} \right)}}$ ${{{\theta }}_{{{\text{MBY}}}}} = \frac{{{{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}}}{{\left( {1 + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}} \right)}}$ |
| 4 | ${{{\text{H}}}_{2}} + 2{{{\theta }}_{{\text{o}}}}\xrightarrow{{{{k}_{{{{{\text{Н}}}_{2}}}}}}}2{{{\theta }}_{{\text{H}}}}$ ${\text{MBY}} + {{{\theta }}_{{\text{о}}}}\overset {{{K}_{{{\text{MBY}}}}}} \leftrightarrows {{{\theta }}_{{{\text{MBY}}}}}$ ${{r}_{1}} = {{k}_{{{{{\text{Н}}}_{2}}}}}{\theta }_{{\text{o}}}^{2}{{P}_{{{{{\text{H}}}_{2}}}}}$ |
${{r}_{1}} = \frac{{{{k}_{{{{{\text{Н}}}_{2}}}}}{{P}_{{{{{\text{H}}}_{2}}}}}}}{{{{{\left( {1 + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}} \right)}}^{2}}}}$ | ${{{\theta }}_{{\text{o}}}} = \frac{1}{{\left( {1 + {{K}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}} \right)}}$ |
| 5 | ${\text{MBY}} + {{{\theta }}_{{\text{o}}}}\xrightarrow{{{{k}_{{{\text{MBY}}}}}}}{{{\theta }}_{{{\text{MBY}}}}}$ ${{{\text{H}}}_{2}} + 2{{{\theta }}_{{\text{o}}}}\overset {{{K}_{{{{{\text{H}}}_{2}}}}}} \leftrightarrows 2{{{\theta }}_{{\text{H}}}}$ ${{r}_{1}} = {{k}_{{{\text{MBY}}}}}{{C}_{{{\text{MBY}}}}}{{{\theta }}_{{\text{o}}}}$ |
${{r}_{1}} = \frac{{{{k}_{{{\text{MBY}}}}}{{С}_{{{\text{MBY}}}}}}}{{\left( {1 + K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}} \right)}}$ | ${{{\theta }}_{{\text{o}}}} = \frac{1}{{\left( {1 + K_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}P_{{{{{\text{H}}}_{2}}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}} \right)}}$ |
1) реакция гидрирования протекает между предварительно адсорбированными молекулами MBY и водорода на активных центрах одной природы (конкурентная адсорбция); лимитирующей является стадия поверхностной реакции (модель 1);
2) реакция гидрирования происходит между предварительно адсорбированными молекулами MBY и водорода на активных центрах разной природы (неконкурентная адсорбция), но водород может адсорбироваться и на тех же центрах, что MBY; лимитирующей является стадия поверхностной реакции (модель 2);
3) реакция гидрирования протекает между предварительно адсорбированными молекулами MBY и водорода на активных центрах разной природы (неконкурентная адсорбция), но MBY может адсорбироваться и на тех же центрах, что водород; лимитирующей является стадия поверхностной реакции (модель 3);
4) лимитирующей является стадия хесоморбции молекулы водорода (модель 4);
5) лимитирующей является стадия адсорбции MBY (модель 5).
Снижение скорости реакции с ростом начальной концентрации MBY выше 0.6 моль/л позволяет исключить из рассмотрения кинетические модели 2 и 5, для которых начальная скорость реакции с увеличением начальной концентрации MBY должна непрерывно повышаться (линейно для модели 5 и нелинейно – для модели 2) (табл. 4). Для моделей 1, 3 и 4 начальная скорость реакции уменьшается в области определенных концентраций MBY [41]. Лимитирующей стадией гидрирования MBY в присутствии Pd–P-частиц, вероятно, не может быть стадия хемосорбции водорода (модель 4), т.к. скорость реакции в этом случае будет снижаться c ростом начальных концентрациях MBY. Таким образом, из пяти предложенных моделей остаются 1-я или 3-я.
Для ответа на вопрос, участвуют ли в реакции гидрирования 2-метил-3-бутин-2-ола обе потенциально активные формы палладия (нанокластеры палладия и фосфиды палладия) или только одна из них (модели 1 и 3), применена комбинация кинетических методов исследования, которые используют в качестве основного измеряемого параметра дифференциальную селективность каталитических систем в условиях конкуренции нескольких субстратов.
Дифференциальная селективность, в отличие от каталитической активности, при одних и тех же степенях конверсии не зависит от концентрации активного компонента катализатора и определяется его природой [42, 43]. Но это условие выполняется только тогда, когда образующиеся на одном катализаторе продукты имеют одинаковые порядки по его концентрации [43]. Выполнение постулата об идентичности активных центров, на которых происходит образование продуктов, реализуется в том случае, если проводить конкурентное гидрирование двух однотипных субстратов, которые отличаются заместителем, удаленным от реакционного центра [43]. В качестве простого метода оценки величины дифференциальной селективности авторами [42, 43] было предложено использовать фазовые траектории реакции, которые представляют зависимости выхода продуктов конкурирующих реакций друг от друга. Тангенс угла наклона к любой точке фазовой траектории равен отношению скоростей накопления продуктов конкурирующих реакций, которое однозначно характеризует дифференциальную селективность.
Анализ результатов конкурентного гидрирования двух однотипных субстратов, представленных в виде фазовых траекторий, с большой долей вероятности позволит различить предложенные выше модели 1 и 3. Совпадение фазовых траекторий при использовании различных палладиевых катализаторов указывает на совпадение величин дифференциальной селективности и с большой вероятностью будет свидетельствовать об одинаковой природе активных центров [43]. Однако необходимо учитывать, что такой результат все же не является однозначным доказательством одинаковой природы катализатора. Если селективность окажется малочувствительной к изменению природы катализатора, то тогда с учетом экспериментальной точности ее определения может наблюдаться кажущееся постоянство селективности. Если же при варьировании параметров процесса фазовые траектории вновь совпадают, то природа активных центров одинакова. Несовпадение фазовых траекторий конкурентного гидрирования двух однотипных субстратов указывает на то, что природа активных центров катализаторов различается [43]. Учитывая вышеизложенное, для конкурентного гидрирования были выбраны два близких по природе ацетиленовых спирта: 2-метил-3-бутин-2-ол и 3-метил-1-пентин-3-ол.
Предварительно проведенные параллельные эксперименты конкурентного гидрирования MBY и MPY в присутствии различных палладиевых катализаторов показали хорошую воспроизводимость. Практически полное совпадение фазовых траекторий параллельных экспериментов конкурентного гидрирования двух ацетиленовых спиртов в присутствии Pd-черни (рис. 6а), коллоидных растворов Pd–P-наночастиц (рис. 6б) или циглеровских систем (рис. 7) позволяет исключить влияние воспроизводимости гетерогенно-каталитических экспериментов на дискриминацию гипотез и перейти к анализу фазовых траекторий для различных палладиевых катализаторов. Следует подчеркнуть, что в параллельных экспериментах совпали фазовые траектории, построенные как по накоплению продуктов конкурирующих реакций друг от друга (образующихся аллиловых или насыщенных спиртов), так и по убыли концентраций конкурирующих ацетиленовых спиртов (рис. 6б). В то же время аналогичные фазовые траектории конкурентного гидрирования MBY и MPY в присутствии Pd–P-наночастиц и Pd-черни расходятся (рис. 8). На первый взгляд, можно было бы высказать предположение, что из двух потенциальных форм (кластеры Pd(0) и фосфиды палладия) в гидрировании алкинолов активность проявляют фосфиды палладия. Однако, учитывая, что гидрирование алкинолов относится к размерно-чувствительным реакциям [11, 12], а размеры Pd–P-наночастиц (d = 5.9 нм) и Pd-черни (d = 22 нм) существенно отличались, дополнительно были проверены эксперименты по конкурентному гидрированию MBY и MPY в присутствии нанокластеров палладия меньшего размера (рис. 4). В качестве таких катализаторов были выбраны циглеровские системы, в которых размер нанокластеров палладия относительно легко можно регулировать соотношением алюминийорганического сокатализатора к палладиевому прекурсору [30].
Рис. 6.
Фазовые траектории параллельных экспериментов конкурентного гидрирования двух алкинолов (MBY, MPY) (1, 2) и образования аллиловых спиртов (MBE, MPE) (3, 4) в присутствии Pd-черни (а) и Pd–Р-наночастиц, Р : Pd = = 0.3 (б). Условия реакции: CPd = 1.0 ммоль/л (а), CPd = 0.5 ммоль/л (б), Т = 30°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм, растворитель – ДМФА, 10 мл.
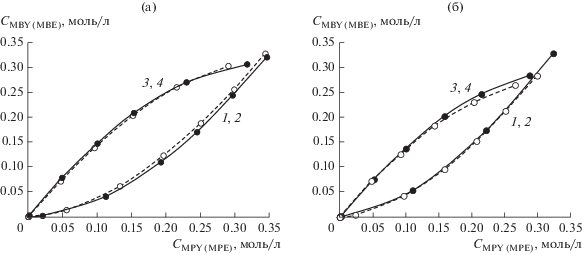
Рис. 7.
Фазовые траектории параллельных экспериментов конкурентного гидрирования двух ацетиленовых спиртов MBY и MPY (1, 2), образования аллиловых (MBE, MPE) (3, 4) и насыщенных спиртов (MBА, MPА) (5, 6) в присутствии системы Pd(acac)2–6AlEt3. Условия реакции: CPd = 0.5 ммоль/л, Т = 30°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = = 2 атм, растворитель – толуол, 10 мл.
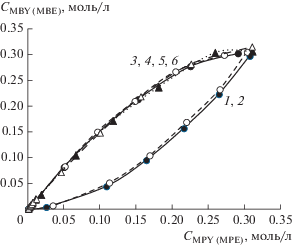
Рис. 8.
Фазовые траектории конкурентного гидрирования алкинолов (MBY, MPY) (1, 2) и образования аллиловых спиртов (MBE, MPE) (3, 4) в присутствии Pd-черни (1, 3), Pd–Р-наночастиц, Р : Pd = 0.3 (2, 4). Условия реакции: Т = 30°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм, растворитель – ДМФА, 10 мл.
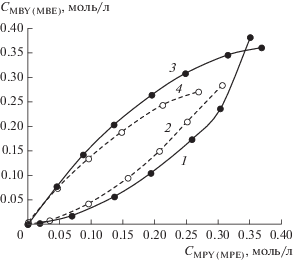
Сравнение фазовых траекторий конкурентного гидрирования ацетиленовых спиртов (MBY и MPY) в присутствии Pd–P-наночастиц и циглеровских систем дало неожиданный результат. Фазовые траектории конкурентного гидрирования, построенные по убыли концентраций MBY и MPY совпали, а по концентрациям продуктов реакции – разошлись, т.е. соотношения скоростей гидрирования алкинолов совпадают, а соотношения скоростей накопления (образования и расходования) этиленовых спиртов отличаются (рис. 9). Совпадение фазовых траекторий конкурентного гидрирования ацетиленовых спиртов с большой долей вероятности указывает на одинаковую природу активных центров в этих катализаторах. Следовательно, из двух потенциально активных центров, содержащихся в Pd–P-наночастицах, в гидрировании алкинолов активность проявляют кластеры Pd(0).
Рис. 9.
Фазовые траектории конкурентного гидрирования алкинолов (MBY, MPY) (1, 2), образования аллиловых (MBE, MPE) (3, 4) (а) и насыщенных спиртов (MBА, MPА) (5, 6) (б) в присутствии Pd–Р-наночастиц, Р : Pd = 0.3 (1, 3, 5) и системы Pd(acac)2–6AlEt3 (2, 4, 6). Условия реакции: Т = 30°С, ${{Р}_{{{{{\text{Н}}}_{{\text{2}}}}}}}$ = 2 атм.
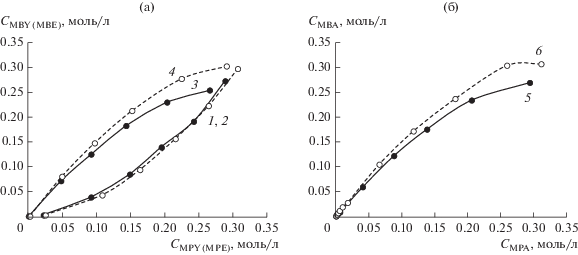
Несовпадение фазовых траекторий по образующимся этиленовым спиртам могло бы быть результатом превращения алкинолов в побочные продукты (димеризации и изомеризации). Однако совпадение фазовых траекторий, построенных по суммам продуктов превращения алкинолов (аллиловых и насыщенных спиртов), исключает эту версию. Следовательно, наблюдаемое различие фазовых траекторий конкурентного гидрирования ацетиленовых спиртов (MBY и MPY) по аллиловым спиртам связано с дальнейшим их насыщением. Если бы образующиеся ненасыщенные спирты оставались конечными продуктами, то фазовые траектории, построенные как по убыли концентрации ацетиленовых спиртов, так и по накоплению продуктов их гидрирования, совпадали бы.
Известно, что в реакции гидрирования может участвовать не только водород поверхностного слоя, но и водород из приповерхностного слоя, называемый “неселективным” [1], который насыщает алкины до алканов без стадии их десорбции. Учитывая меньшую растворимость водорода в малых кластерах палладия [44] и фосфидах палладия в сравнении с массивным палладием [45], а также невысокую долю 2-метилбутан-2-ола и 3-метилпентан-3-ола в ходе гидрирования алкинолов (рис. 1), вкладом “неселективного” водорода можно пренебречь.
Анализ фазовых траекторий конкурентного гидрирования MBY и MPY позволяет предполагать, что гидрирование алкинолов в присутствии Pd–P-наночастиц наиболее вероятно протекает на кластерах Pd(0), а дальнейшее насыщение аллиловых спиртов – на обоих типах активных центров: кластерах Pd(0) и фосфиде палладия. Из двух типов активных центров реакция преимущественно идет на тех, которые характеризуются меньшей энергией активацией. Фосфиды d-металлов в гидрогенизационном катализе обычно менее активны, чем металлические катализаторы, но более устойчивы к дезактивации [46, 47] Для протекания реакции гидрирования необходима активация и молекулярного водорода, и субстрата. Так как частичный положительный заряд на палладии в фосфиде палладия (Есв(Pd3d5/2) = = 336.1 эВ) [25] неблагоприятен для активации молекулярного водорода, то вероятнее всего хемосорбция водорода происходит преимущественно на кластерах Pd(0), а адсорбция аллиловых спиртов – как на кластерах Pd(0), так и на фосфидах палладия. Причиной проявления активности фосфидов палладия в присутствии более активной формы – кластеров Pd(0) – может быть частичный перенос водорода по спилловер механизму от кластеров Pd(0) к аллиловому спирту, адсорбированному на фосфиде палладия, и последующему его насыщению.
ЗАКЛЮЧЕНИЕ
Предложен способ получения эффективного катализатора гидрирования алкинолов, превосходящего по активности известные нанокластеры палладия и сохраняющего при этом высокую селективность (95–96%) при 97–98% конверсии субстрата. Промотирующий эффект фосфора на свойства палладиевого катализатора обусловлен не только повышением дисперсности, но и большей реакционной способностью Pd–P-частиц. Анализ дифференциальной селективности различных палладиевых катализаторов показывает, что из двух типов активных центров, содержащихся в Pd–P-частицах, гидрирование алкинолов с наибольшей вероятностью протекает на кластерах Pd(0), а дальнейшее насыщение аллиловых спиртов – и на кластерах Pd(0), и на фосфиде палладия.
Список литературы
Vile G., Albani D., Almora-Barrios N., Lopez N., Perez-Ramirez J. // ChemCatChem. 2016. V. 8. P. 21.
McCue A.J., Anderson J.A. // Front. Chem. Sci. Eng. 2015. V. 9. № 2. P. 142.
Николаев С.А., Занавескин Л.Н., Смирнов В.В., Аверьянов В.А., Занавескин К.Л. // Успехи химии. 2009. Т. 78. № 3. С. 248.
Stolarov I.P., Yakushev I.A., Churakov A.V., Cherkashina N.V., Smirnova N.S., Khramov E.V., Zubavichus Y.V., Khrustalev V.N., Markov A.A., Klyagina A.P., Kornev A.B., Martynenko V.M., Gekhman A.E., Vargaftik M.N., Moiseev I.I. // Inorg. Chem. 2018. V. 57. № 18. P. 11 482.
Johnston S.K., Cherkasov N., Pérez-Barrado E., Aho A., Murzin D.Y., Ibhadon A.O., Francesconi M. G. // Appl. Catal. A. Gen. 2017. V. 544. P. 40.
Nikoshvili L., Bykov A., Khudyakova T., LaGrange T., Héroguel F., Luterbacher J.S., Matveeva V.G., Sulman E.M., Dyson P.J., Kiwi-Minsker L. // Ind. Eng. Chem. Res. 2017. V. 56. P. 45.
Maki-Arvela P., Hajeк I., Salmi T., Murzin D.Yu. // Appl. Cata1. A. Cen. 2005. V. 292. P. 1.
Wu W., Zhang W., Long Y., Qin J., Wen H., Ma J. // J. Ccolloid Interf. Sci. 2018. V. 531. P. 642.
Directive 2002/95/EC of the European Parliament and of the Council of 27 January 2003 on the restriction of the use of certain hazardous substances in electrical and electronic equipment // Official Journal L. 2002. V. 37. P. 13.
Crespo-Quesada M., Yarulin A., Jin M., Xia Y., Kiwi-Minsker L. // J. Am. Chem. Soc. 2011. V. 133. P. 12 787.
Markov P.V., Mashkovsky I.S., Bragina G.O., Warn J., Gerasimov E.Yu., Bukhtiyarov V.I., Stakheev A.Yu., Murzin D.Yu. // Chem. Eng. J. 2019. V. 358. P. 520.
Ярулин А.Э., Креспо-Кесада М.P., Егорова Е.В., Киви-Минскер Л.Л. // Кинетика и катализ. 2012. Т. 53. № 2. С. 263.
Vorobyeva E., Chen Z., Mitchell S., Leary R.K., Midgley P., Thomas J.M., Hauert R., Fako E., Lopez N., Perez-Ramırez J. // J. Mater. Chem. A. 2017. V. 5. P. 16 393.
Okhlopkova L.B., Cherepanova S.V., Prosvirin I.P., Kerzhentsev M.A., Ismagilov Z.R. // Appl. Catal. A: Gen. 2018. V. 549. P. 245.
Shen L., Mao S., Li J., Li M., Chen P., Li H., Chen Z., Wang Y. // J. Catal. 2017. V. 350. P. 13.
Mashkovsky I.S., Baeva G.N., Stakheev A.Y., Vargaftik M.N., Kozitsyna N.Y., Moiseev I.I. // Mendeleev Commun. 2014. V. 24. P. 355.
Шляпин Д.А., Глыздова Д.В., Афонасенко Т.Н., Темерев В.Л., Цырульников П.Г. // Кинетика и катализ. 2019. Т. 60. № 4. С. 479.
Guo M., Li H., Ren Y., Ren X., Yang Q., Li C. // ACS Catal. 2018. V. 8. P. 6476.
Варгафтик M.H., Козицына Н.Ю., Черкашин Н.В., Рудый Р.И., Кочубей Д.И., Новгородов Б.Н., Моисеев И.И. // Кинетика и катализ. 1998. Т. 39. № 6. С. 806.
Шмидт Ф.К., Белых Л.Б., Черенкова Т.В. // Кинетика и катализ. 2001. Т. 42. № 2. С. 182.
Белых Л.Б., Скрипов Н.И., Белоногова Л.Н., Уманец В.А., Шмидт Ф.К. // Кинетика и катализ. 2010. Т. 51. № 1. С. 47.
Скрипов Н.И., Белых Л.Б., Белоногова Л.Н., Уманец В.А., Рыжкович Е.Н., Шмидт Ф.К. // Кинетика и катализ. 2010. Т. 51. № 5. С. 739.
Скрипов Н.И., Белых Л.Б., Стеренчук Т.П., Акимов В.В., Таусон В.Л., Шмидт Ф.К. // Кинетика и катализ. 2017. Т. 58. № 1. С. 36.
Белых Л.Б., Стеренчук Т.П., Скрипов Н.И., Акимов В.В., Таусон В.Л., Романченко А.С., Гвоздовская К.Л., Санжиева К.Л., Шмидт Ф.К. // Кинетика и катализ. 2019. Т. 60. №. 6. С. 788.
Белых Л.Б., Скрипов Н.И., Cтеренчук Т.П., Акимов В.В., Таусон В.Л., Шмидт Ф.К. // Журн. общей химии. 2016. Т. 86. № 9. С. 1454.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976. 572 с.
Matthews J.C., Nashua N.H., Wood L.L. USA, Patent 3474464, 1969.
Ott L.S., Finke R.G. // Coord. Chem. Rev. 2007. V. 251. № 9–10. P. 1075.
Шмидт Ф.К., Титова Ю.Ю., Белых Л.Б. // Кинетика и катализ. 2015. Т. 56. № 5. С. 582.
Белых Л.Б., Титова Ю.Ю., Уманец В.А., Шмидт Ф.К. // Журн. прикл. химии. 2006. Т. 79. № 8. С. 1285.
Цырульников П.Г., Афонасенко Т.Н., Кощеев С.В, Боронин А.И. // Кинетика и катализ. 2007. Т. 48. № 5. С. 778.
Мироненко Р.М., Бельская О.Б., Лавренов А.В., Лихолобов В.А. // Кинетика и катализ. 2018. Т. 59. № 3. С. 347.
Maccarrone M.J., Lederhos C.R., Torres G., Betti C., Coloma-Pascual F., Quiroga M.E., Yori J.C. // Appl. Catal. A. Gen. 2012. V. 441–442. P. 90.
Vernuccio S., Goy R., Rohr Ph.R., Medlock J., Bonrath W. // React. Chem. Eng. 2016. V. 1. P. 445.
Carenco S., Leyva-Perez A., Concepciyn P., Boissiere C., Mezailles N., Sanchez C., Corma A. // Nano Today. 2012. V. 7. P. 21.
Oyama S.T., Gott T., Zhao H., Lee Y.K. // Catal. Today. 2009. V. 143. № 1–2. P. 94.
d’Aquino A.I., Danforth S.J., Clinkingbeard T.R., Ilic B., Pullan L., Reynolds M.A., Murray B.D., Bussel M.E. // J. Catal. 2016. V. 335. P. 204.
Semagina N., Grasemann M., Xanthopoulos N., Renken A., Kiwi-Minsker L. // J. Catal. 2007. V. 251. P. 213.
Crespo-Quesada M., Grasemann M., Semagina N., Renken A., Kiwi-Minsker L. // Catal. Today. 2009. V. 147. № 3–4. P. 247.
Protasova L.N., Rebrov E.V., Choy K.L., Pung S.Y., Engels V., Cabaj M., Wheatley A.E.H., Schouten J.C. // Catal. Sci. Technol. 2011. V. 1. P. 768.
Serna P., Concepción P., Corma A. // J. Catal. 2009. V. 265. P. 19.
Schmidt A.F., Kurokhtina A.A., Larina E.V. // Cat. Sci. Tech. 2014. V. 4. P. 3439.
Шмидт А.Ф., Курохтина А.А., Ларина Е.В. // Кинетика и катализ. 2019. Т. 60. № 5. С. 555.
Kobayashi H., Yamauchi M., Kitagawa H., Kubota Yo., Kato K., Takata M. // J. Am. Chem. Soc. 2008. V. 130. P. 1818.
Stojewski M., Kowalska J., Jurczakowski R. // J. Phys. Chem. C. 2009. V. 113. № 9. P. 3707.
Prins R., Bussell M.E. // Catal Lett. 2012. V. 142. № 12. P. 1413.
Zhao M. // Chem. Asian J. 2016. V. 11. P. 461.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ