

Кинетика и катализ, 2020, T. 61, № 5, стр. 664-674
Влияние кислорода на окисление метана до метанола пероксидом водорода в присутствии стабилизированных глутатионом нанокластеров золота
С. А. Голованова a, А. П. Садков a, А. Ф. Шестаков a, b, *
a ФГБУН Институт проблем химической физики РАН
142432 Черноголовка, просп. Акад. Семенова, 1, Россия
b ФГБОУ ВО Московский Государственный Университет им. М.В. Ломоносова,
Факультет фундаментальной физико-химической инженерии
119991 Москва, Ленинские горы, 1, стр. 5, Россия
* E-mail: a.s@icp.ac.ru
Поступила в редакцию 30.05.2019
После доработки 24.12.2019
Принята к публикации 05.03.2020
Аннотация
Изучена каталитическая активность тиолатных нанокластеров золота (1–2 нм), стабилизированных глутатионом, в реакции окисления метана H2O2 и кислородом воздуха в водной среде при 30 атм СН4 и 70°C. Соотношение и выход продуктов окисления СН3ОН и СН3ООН зависят от содержания в реакторе H2O2 и кислорода. Повышение парциального давления кислорода приводит к увеличению общего выхода продуктов при 5 атм воздуха по сравнению с суммой выходов независимых реакций окисления Н2О2 или О2, но к его снижению при 10 атм. Предложен молекулярный механизм окисления метана, который хорошо описывает кинетические кривые накопления и расходования СН3ОН и СН3ООН и влияние кислорода на их выход. На основании литературных данных о строении стабилизированных глутатионом кластеров Au25 и результатов квантово-химического моделирования предложена молекулярная модель активного центра. Эксперименты с повторным введением газовой фазы и возобновлением содержания H2O2 продемонстрировали 100%-е сохранение каталитической акивности. Выход СН3ОН достигает 60 молей в расчете на моль кластеров Au25.
ВВЕДЕНИЕ
Селективное окисление CH4 до CH3OH является предметом интенсивных исследований на протяжении последних десятилетий. Несмотря на практическую важность задачи получения ценных продуктов из природного газа, пока отсутствует ее решение, приемлемое для внедрения в производство [1, 2]. Это делает актуальным поиск новых эффективных катализаторов для низкотемпературного окисления метана [3–9]. Открытие маталлокомплексной активации алифатических С–Н-связей в мягких условиях (реакция Шилова) [4] указывает на возможность непосредственной низкотемпературной конверсии углеводородов в функционализированные продукты с использованием новых координационных катализаторов на основе переходных металлов [10, 11].
Пионерские работы Харуты (Haruta) по низкотермпературному окислению СО выявили химическую активность золота в высокодисперсном состоянии [12]. Как он и предполагал [13], в дальнейшем кластерная химия золота привела к открытию новых впечатляющих областей исследования с акцентом на драматические изменения реакционной способности частиц Au c магическим числом атомов. Cегодня уже известно, что соединения золота – комплексы, кластеры и наночастицы (НЧ) – катализируют широкий спектр химических реакций [14].
Изучение химической активности тиолатных кластеров Aun(SR)m, в том числе и в окислительных процессах [15, 16], представляет новую активно развивающуюся область исследований [17–19]. Важной особенностью строения этих кластеров является наличие вокруг металлического остова Aun золото-тиолатных “скрепочных” фрагментов ‒SR–Au–SR– или –SR–Au–SR–Au–SR–.
Согласно работам [20, 21] по окислению стирола активность и селективность систем, содержащих Aun(SR)m, где n = 25, 38, 144, определяются Au-остовом, а не лигандной оболочкой. Большое количество золото-тиолатных “скрепочных” фрагментов создает стерические препятствия для взаимодействия реагирующих молекул с активными центрами. С помощью процедур нагревания и восстановления удается осуществить частичное или полное удаление тиолатного покрытия, и это приводит к изменениям в каталитическом поведении Au-нанокластеров [15, 16, 22]. Сравнительное изучение нанокластеров Au25 (диаметр 1.0 нм), Au38 (1.3 нм) и Au144 (1.6 нм), иммобилизованных на подложке из гидроксиапатита, показало более высокую каталитическую активность Au25 в селективном окислении стирола, а данные рентгеновской абсорбционной спектроскопии подтвердили, что состав и размер кластеров не менялся во время окислительных процессов [23].
В кластерах Au25(SR)18 геометрические размеры заместителя R влияют на стерическую доступность каталитически активных центров, как было установлено при изучении гидрирования α,β-ненасыщенных кетонов [24]. Лиганды защитной оболочки нанокластера могут непосредственно модулировать расположение активных участков и контролировать координацию субстратов, воздействуя таким образом на селективность каталитических реакций [25]. Это позволяет с высокой избирательностью осуществлять окислительные процессы. В частности, реализуется 100% конверсия стирола при 92% селективности по эпоксидному продукту [26].
Доступность поверхности нанокластеров Au для субстратов чрезвычайно важна для катализа, и она может быть обеспечена также в результате воздействия окислителя. Мягкий метод снятия стабилизирующих лигандов с поверхности алкилтиолатных кластеров на подложке из гидроксиапатита с помощью Н2О2 или трет-бутилгидропероксида при умеренных температурах (50–60°С) был использован для получения катализаторов эпоксидирования стирола и аэробного окисления бензилового спирта [27].
В недавней работе [28] было показано, что стабилизированные поливинилпирролидоном коллоидные частицы Au–Pd со средним диаметром порядка 4 нм катализируют окисление метана в водной среде с помощью H2O2 при 50°С. СН3ООН образуется вместе с СН3ОН. Авторы склоняются к радикальному механизму реакции на основании обнаружения метильных радикалов в системе. В его пользу говорит некоторое увеличение выхода СН3ООН при уменьшении выхода СН3ОН в присутствии кислорода. Однако другие данные [28] находятся в противоречии с этим механизмом. Так, при использовании меченого 18О2 только 50% изотопа 18О обнаружено в СН3ОН, несмотря на значительный первоначальный избыток кислорода (5 атм) по отношению к H2O2 (0.05 мМ) в реакционной системе. Из полученных кинетических данных следует, что метилгидропероксид является промежуточным продуктом. Однако механизм образования метилового спирта в этой работе даже не обсуждается. Интересно, что в отсутствие Pd наночастицы золота размером ~4 нм практически неактивны. В этой связи представляют интерес растворимые в воде нанокластеры золота (НК Au), стабилизированные глутатионом (GSH). Поскольку они имеют небольшой размер, можно ожидать усиления их каталитических свойств. Также изменение строения поверхностного слоя металлического остова может повлиять на вклад радикального маршрута при окислении метана в присутствии H2O2. Отметим только что появившуюся работу [29], в которой было обнаружено окисление метана до метанола H2O2 в неводных средах, катализируемое нанокластером Au24 со смешанной оболочкой из PPh3- и SCH2CH2Ph-лигандов. Механизм образования СН3ОН по реакции СН4 с окисленной формой кластера исследован с помощью квантово-химических расчетов. Выход СН3ОН достигает 65 молей на моль кластера.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Получение катализатора
Нанокластеры Au были синтезированы с использованием ранее сообщенного протокола с небольшими модификациями [30, 31]. К 50 мл метанольного раствора HAuCl4 ⋅ 3H2O (0.5 мМ) добавляли GSH (1.0 мМ) и выдерживали в течение 2–3 ч на ледяной бане до образования прозрачного бледно-желтого раствора. Полученную смесь обрабатывали избытком водного, охлажденного до 0°C раствора NaBH4 (0.2 М, 12.5 мл) при интенсивном перемешивании. Об образовании НК Au свидетельствовало мгновенное отчетливое образование темно-коричневого раствора. Смесь выдерживали в течение 12 ч для завершения формирования защитной оболочки кластеров Aun(SG)m с небольшим разбросом числа атомов металла (n = 18–25), затем центрифугировали при 2000 об./мин в течение 10 мин с помощью центрифуги (“Heinz Janetzki”, Германия). Образовавшийся осадок многократно промывали метанолом, затем сушили при комнатной температуре в течение суток и собирали в виде темно-коричневого порошка.
Исследование физико-химических свойств катализатора
Синтезированные НК Au были охарактеризованы методами оптической спектроскопии и динамического рассеяния света (ДРС). Спектры поглощения образцов снимали на спектрометре Specord M40 (“Carl Zeiss Industrielle Messtechnik GmbH”, Германия). Распределение частиц по размерам получены при обработке результатов измерения ДРС с помощью анализатора Photocor Complex (ООО “Фотокор”, Россия) с программным обеспечением DynaLS. Содержание золота в пробах определяли методом атомно-адсорбционной спектроскопии с применением спектрометра A.A.S. 3 (“Carl Ceiss Jena”, Германия).
Исследование каталитической реакции
Активность катализатора в реакциях окисления метана H2O2 исследовали в автоклаве из нержавеющей стали с внутренним объемом 50 мл. Стеклянный реактор, содержащий 5 мл жидкой фазы с определенным количеством окислителя (0.01–0.4 М Н2О2) и катализатора (3 × 10–4 М Au), помещали в автоклав, герметизировали его и продували три раза метаном. Затем систему заполняли метаном под давлением 30 атм и выдерживали при температуре 70°С в течение заданного времени.
Для изучения окислительных реакций с кислородом использовали сжатый воздух. В этом случае реактор с жидкой реакционной средой помещали в автоклав, заполняли воздухом под соответствующим давлением, а затем метаном до достижения его парциального давления 30 атм. Для проведения экспериментов с повторным напуском газовой фазы через 6 ч автоклав разгерметизировали, а реакционный газ удаляли. В одном варианте после герметизации автоклав вновь заполняли воздухом и метаном (1 : 30), в другом – в жидкую фазу предварительно добавляли необходимое количество Н2О2.
После окончания реакции автоклав охлаждали до 10°C, чтобы минимизировать потери летучих продуктов. Продукты первичного окисления СН4 регистрировали методом газовой хроматографии. Для этого проводили анализ жидкой фазы на газовом хроматографе HР 58880A (“Hewlett-Paсkard”, США), используя капиллярную колонку “DW-WAX” длиной 60 м. СН3ОН в реакционной среде идентифицировали, сравнивая со стандартным раствором спирта. Для определения содержания алкилгидропероксида в образцы добавляли избыток NaBH4, при этом хроматографический анализ проводили дважды – до и после введения восстановителя. Изменение площади пика, соответствующего спирту, указывало на вклад от СН3ООН. Количество H2O2 определяли титрованием раствором КMnO4 (0.01 M) в подкисленной среде.
Квантово-химическое моделирование
Для квантово-химических вычислений применяли функционал плотности РВЕ [32]. Глутатионовые остатки в кластере Au25(SG)18 моделировали простейшим лигандом SМе. Ввиду важности релятивистских эффектов для атома золота при описании системы расчеты проводили в скалярном релятивистском приближении, которое основано на полном четырехкомпонентном одноэлектронном уравнении Дирака с исключенными спин-орбитальными эффектами [33]. Был использован оптимизированный по энергии расширенный гауссовый базис для больших компонент: Au (30s,29p,20d,14f,6g)/[9s,8p,6d,3f,1g ] , S (15s,11p,3d)/ [4s,3p,1d ] , C, O (10s,7p,3d)/[3s,2p,1d ] и H (6s,2p)/[2s,1p ] , и соответствующий кинетически сбалансированный базис для малых компонент. Все расчеты осуществляли с помощью программного комплекса “ПРИРОДА” [34, 35] с привлечением вычислительных возможностей МСЦ РАН.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
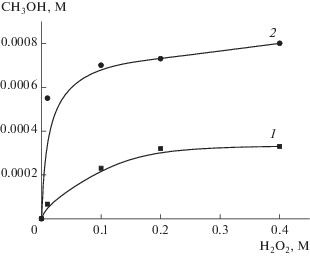
НК Au, стабилизированные глутатионом, катализируют окисление СН4 до СН3ОН в присутствии пероксида водорода. Для оптимизации количества окислителя в реакционной среде была исследована зависимость выхода продукта реакции за 6 ч от концентрации H2O2 при наличии в газовой фазе в одном случае только метана, в другом – дополнительно 1 атм воздуха (рис. 1). В интервале концентраций 0.1–0.4 М H2O2 выход метанола существенно не меняется, поэтому для изучения реакции в дальнейшем использовали 0.2 М окислителя. Дополнительные эксперименты позволили выявить эффект значительного увеличения выхода метанола в исследуемой системе уже при введении небольшого количества кислорода (0.2 атм) в газовую фазу. Это дает основание предполагать преимущественный вклад молекулярного механизма при каталитическом окислении метана, и экспериментальные данные будут трактоваться с этих позиций.
Рис. 1.
Зависимость образования СН3ОН от концентрации H2O2 в процессе активации метана НК Au: 1 – без добавления воздуха; 2 – при 1 атм воздуха. Условия реакции: 3 × 10–4 М катализатора, 5 мл водной фазы, 30 атм СН4, Т = 70°C, 6 ч.
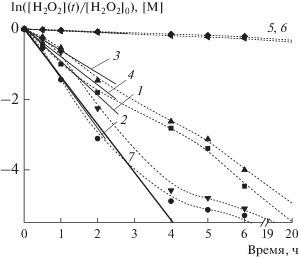
Одновременно с метанолом образуется метилгидропероксид, количество которого на начальном этапе превращения алкана заметно превышает содержание спирта (рис. 2). Концентрация СН3ООН достигает максимального значения за 1 ч, затем снижается примерно в два раза к 6 ч и далее медленно уменьшается примерно на 25% на протяжении последующих 18 ч реакции. Изучение скорости разложения Н2О2, проведенное в различных условиях, показывает, что Н2О2 практически отсутствует в реакционной смеси после 6 ч реакции (рис. 3). Поэтому сокращение суммарного содержания СН3ОН и СН3ООН в этот период указывает на их окисление кислородом, образовавшимся при разложении Н2О2.
Рис. 2.
Кинетические кривые накопления продуктов реакции в процессе активации метана НК Au: 1 – метанол, 2 – метилгидропероксид, 3 – сумма первичных продуктов окисления метана. Условия реакции: 3 × × 10–4 М катализатора, 0.2 М H2O2, 5 мл водной фазы, 30 атм СН4, Т = 70°C.
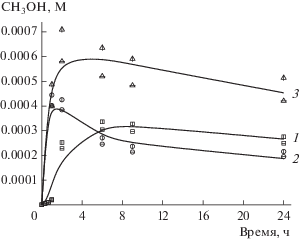
Рис. 3.
Расход H2O2 при добавлении НК Au: 1 атм аргона (1), 30 атм СН4 (2), 1 атм воздуха (3), 1 атм воздуха + 30 атм СН4 (4); без добавления НК Au: на воздухе (5), в присутствии 1.6 × 10–4 М GSH (6). Условия реакции: 3 × 10–4 М катализатора, 0.2 М H2O2, 5 мл водной фазы, Т = 70°C.
В начальный период времени скорость накопления метанола близка к нулю, и при образовании СН3ООН из СН4 расходуются 2 эквивалента Н2О2:
(I)
$\begin{gathered} {\text{С}}{{{\text{Н}}}_{4}} + 2{{{\text{Н}}}_{2}}{{{\text{О}}}_{2}} \to {\text{С}}{{{\text{Н}}}_{3}}{\text{ООН}} + \\ + \,\,2{{{\text{Н}}}_{2}}{\text{О}} + 43.7\,\,{{{\text{ккал}}} \mathord{\left/ {\vphantom {{{\text{ккал}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}. \\ \end{gathered} $Здесь и далее тепловые эффекты реакций приведены для стандартных условий в газовой фазе, поскольку для СН3ООН известна стандартная энтальпия образования только в газовой фазе [36], ‒31.0 ккал/моль. Область максимальной скорости накопления спирта соответствует максимальной концентрации метилгидропероксида (рис. 2). Это означает, что СН3ООН является промежуточным продуктом при образовании метанола. Однако после исчерпания Н2О2 процесс практически останавливается. Это дает основание полагать, что реакция прямого превращения СН3ООН в СН3ОН отсутствует, и НК Au катализируют как реакцию разложения Н2О2:
(II)
$2{{{\text{Н}}}_{2}}{{{\text{О}}}_{2}} \to 2{{{\text{Н}}}_{2}}{\text{О}} + {{{\text{О}}}_{2}} + 31.6\,\,{{{\text{ккал}}} \mathord{\left/ {\vphantom {{{\text{ккал}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}},~$(III)
$\begin{gathered} {{{\text{Н}}}_{2}}{{{\text{О}}}_{2}} + {\text{С}}{{{\text{Н}}}_{3}}{\text{ООН}} \to {\text{С}}{{{\text{Н}}}_{3}}{\text{ОН}} + \\ + \,\,{{{\text{Н}}}_{2}}{\text{О}} + {{{\text{О}}}_{2}} + 33.2\,\,{{{\text{ккал}}} \mathord{\left/ {\vphantom {{{\text{ккал}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}. \\ \end{gathered} $Для этого достаточно, чтобы Au-катализатор обладал активным центром М, способным к связыванию атома О и молекулы О2:
Каталитическое разложение Н2О2 опишем простейшей схемой:
(IV)
$\begin{gathered} {{{\text{Н}}}_{2}}{{{\text{О}}}_{2}} + {\mathbf{М}} \to {\mathbf{М}}{\text{О}} + {{{\text{Н}}}_{2}}{\text{О}} + \\ + \,\,{{Е}_{1}}--35.5\,\,{{{\text{ккал}}} \mathord{\left/ {\vphantom {{{\text{ккал}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}, \\ {{{\text{Н}}}_{2}}{{{\text{О}}}_{2}} + {\mathbf{М}}{\text{О}} \to {\mathbf{М}}{{{\text{О}}}_{2}} + {{{\text{Н}}}_{2}}{\text{О}} + {{Е}_{2}}-- \\ - \,\,{{Е}_{1}} + 82.7\,\,{{{\text{ккал}}} \mathord{\left/ {\vphantom {{{\text{ккал}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}, \\ {\mathbf{М}}{{{\text{О}}}_{2}} \to {\mathbf{М}} + {{{\text{О}}}_{2}}--{{Е}_{2}}, \\ \end{gathered} $Из анализа экспериментальных данных в [37] для энергии связи атома О с поверхностью Au (111) рекомендовано значение 56 ккал/моль. Для сравнения, энергия связи Au–O в двухатомной молекуле AuO равна 54 ккал/моль [38]. Таким образом, можно полагать, что величина Е1 составляет порядка 55 ккал/моль и, следовательно, первые две стадии схемы (IV) являются экзотермическими. Для протекания последней стадии десорбции молекулы кислорода с активного центра величина Е2 не должна превышать 15 ккал/моль, учитывая большую поступательную энтропию О2 в газовой фазе. Из дальнейшего будет ясно, что эта стадия является обратимой при 350 К, поэтому Е2 равна порядка 15 ккал/моль. При теоретическом изучении взаимодействия О2 с безлигандным НК Au55 [39] было найдено, что энергия адсорбции составляет 10.8 ккал/моль, а диссоциативной хемосорбции – –16.3 ккал/моль.
В приближении квазистационарных концентраций
Промежуточные продукты МО и МО2 постоянно образуются и расходуются при разложении ${{{\text{H}}}_{2}}{\text{O}}_{2}^{{}}$, и поэтому они ответственны за каталитическое окисление метана. Соединение МО2 может содержать или две оксогруппы AuO, или одну пероксогруппу. В последнем случае оно является предшественником образования кислорода. Для альтернативной структуры с двумя оксогруппами мы предполагаем реакцию активации С–Н-связи метана:
(V)
$\begin{gathered} {\text{С}}{{{\text{Н}}}_{4}} + {\mathbf{М}}{{{\text{О}}}_{2}} \to {\mathbf{М}}({\text{ОH}})({\text{OC}}{{{\text{H}}}_{{\text{3}}}}) \to \\ \to {\mathbf{М}} + {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ООН}}--{{E}_{2}} + 26.8\,\,{{{\text{ккал}}} \mathord{\left/ {\vphantom {{{\text{ккал}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}, \\ \end{gathered} $Для образования метилового спирта по суммарной реакции (III) необходимо включить элементарную стадию
(VI)
$\begin{gathered} {\text{С}}{{{\text{Н}}}_{3}}{\text{ООН}} + {\mathbf{М}}{\text{О}} \to {\mathbf{М}}{{{\text{О}}}_{2}} + {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ОН}}-- \\ - \,\,{{E}_{1}} + {{E}_{2}} + 83.1\,\,{{{\text{ккал}}} \mathord{\left/ {\vphantom {{{\text{ккал}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}} \\ \end{gathered} $С учетом значительного избыточного начального содержания Н2О2 по отношению к максимальному выходу СН3ООН и СН3ОН можно пренебречь влиянием реакций окисления метана как на текущую концентрацию [Н2О2] ≡ ох(t) = ox0ехр(–λt) (где λ = 2k1 [M]; ox0 = [H2O2] при t = 0 ), так и на концентрацию активных промежуточных продуктов МО и МО2. В результате имеем систему кинетических уравнений для текущих концентраций [СН3ООН] = = х и [СН3ОН] = у:
(1)
$\begin{gathered} \frac{{{\text{d}}x}}{{{\text{d}}t}} = {{k}_{4}}[{\text{C}}{{{\text{H}}}_{4}}]\frac{{{{k}_{1}}[{\mathbf{M}}]{\text{ox}}(t)}}{{{{k}_{3}}}} - {{k}_{5}}x\frac{{{{k}_{1}}[{\mathbf{M}}]{\text{ox}}(t)}}{{{{k}_{2}}}} - {{k}_{6}}x, \\ \frac{{{\text{d}}y}}{{{\text{d}}t}} = {{k}_{5}}x\frac{{{{k}_{1}}[{\mathbf{M}}]{\text{ox}}(t)}}{{{{k}_{2}}}} - {{k}_{6}}y. \\ \end{gathered} $Здесь k4 и k5 – эффективные константы скорости для стадий (V) и (VI). Система уравнений (1) также учитывают медленные реакции окисления СН3ООН и СН3ОН, которые приводят к снижению их концентраций на больших временах, когда уже отсутствует Н2О2. Для описания этих процессов используется кинетическое уравнение первого порядка с одинаковой эффективной константой скорости k6.
Уравнение для суммарного содержания продуктов z = х + у,
(2)
$\frac{{{\text{d}}z}}{{{\text{d}}t}} = {{k}_{4}}[{\text{C}}{{{\text{H}}}_{4}}]\frac{{{{k}_{1}}[{\mathbf{M}}]{\text{ox}}(t)}}{{{{k}_{3}}}} - {{k}_{6}}z,$(3)
$z(t) = {{k}_{4}}[{\text{C}}{{{\text{H}}}_{4}}]\frac{{{{k}_{1}}[{\mathbf{M}}]}}{{{{k}_{3}}}}\frac{{[\exp ( - {{k}_{6}}t) - \exp ( - \lambda t)]}}{{\lambda - {{k}_{6}}}}.$Наилучшее описание экспериментальной кривой (рис. 2) дают константы λ = 1.356 ч–1 и k6 = 1.5 × × 10–2 ч–1. Несмотря на ряд сделанных допущений, величина λ практически точно описывает наклон начального участка кинетической кривой разложения Н2О2 в полулогарифмических координатах (рис. 3), подчеркнем, из независимого эксперимента. С помощью уравнения (3) можно получить выражение для у(t) в виде интеграла, который не сводится к элементарным функциям. Поэтому мы используем приближенную формулу для описания у(t), основанную на асимптотических решениях:
Это дает возможность аппроксимировать у(t) выражением
где $a = {{k}_{4}}{{k}_{5}}[{\text{C}}{{{\text{H}}}_{4}}]\frac{{{{{({{k}_{1}}[{\mathbf{M}}]{\text{o}}{{{\text{x}}}_{0}})}}^{2}}}}{{2{{k}_{2}}{{k}_{3}}}}$и b – подгоночный параметр.
При ранее найденном значении k6 наилучшее описание кинетической кривой образования и расходования СH3ОH получено при a = 1.07 × 10–4 моль л–1 ч–2 и b = 0.28 ч–2. Эвристическое выражение (4) вполне удовлетворительно описывает экспериментальные данные (рис. 2).
Для понимания молекулярной природы каталитических реакций проведено квантово-химическое моделирование строения активного центра М и одного из промежуточных продуктов образования О2. Согласно исследованиям [41] НК Au25(SG)18 с глутатионовыми лигандами имеют такое же строение, как и аналогичные кластеры Au25(SR)18, структура которых установлена методом рентгеноструктурного анализа (РСА): к икосаэдрическому металлоостову Au13 через мостиковые лиганды SaR присоединены 12 внешних атомов Au, попарно связанных через мостиковые лиганды SbR в “скрепочные” фрагменты –Sа–Au–Sb–Au–Sa. Шесть глутатионовых лигандов, находящихся в середине “скрепочного” фрагмента в позиции Sb, наиболее лабильны и уходят при обработке Н2О2 или нагревании. В результате образуются метастабильные кластеры Au25(SG)12 [41]. Мы их обозначим LAu12, явно выделяя внешние атомы Au.
При выполнении квантово-химического расчета глутатионовый лиганд SG заменен на SМе-группу; полученная структура модельного кластера Au25(SМе)12 приведена на рис. 4а, из которого видно, что после ухода слабо связанных тиолатных лигандов атомы золота в каждом “скрепочном” фрагменте формируют новую связь Au–Au длиной 2.66 Å за счет аурофильных взаимодействий. Эта трансформация, по-видимому, и объясняет относительную стабильность такой структуры. Каждый фрагмент Au2 является наружным ребром тетраэдра Au4, внутреннее ребро которого принадлежит к металлическому остову и формирует связи Au–Au с внешним ребром длиной 2.79–2.81 Å. Таким образом, каждый тетраэдр имеет одну лабильную Au–Au-связь и две доступные грани, сопряженные с ней. Поэтому есть все основания полагать, что именно эти атомы золота принимают участие в каталитических превращениях субстратов в системе.
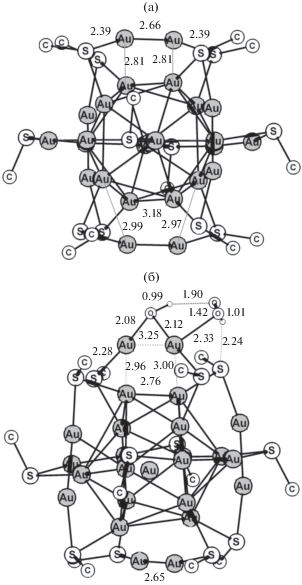
Известно, что гомогенное разложение Н2О2 происходит только при высоких температурах из-за значительной энергии активации, равной 32 ккал/моль [42]. Однако каталитическое разложение в присутствии других агентов происходит уже при более мягких условиях. Разрыв непрочной связи О–О компенсируется образованием двух новых связей между ОН-группами и атомами металла лабильной связи Au–Au. В результате этого взаимодействия Н2О2 с LAu12 образуется промежуточное соединение LAu10(AuОН)2. Такой тип реакции Н2О2 с кластерами Au24 и Au25 с лигандной оболочкой был постулирован и теоретически исследован в [29]. Энергетически менее выгодную реакцию с выходом гидроксильных радикалов в объем:
Действительно, очень активный ОН-радикал при его образовании, прежде всего, реагировал бы с С–Н-связями глутатионовой оболочки, что неизбежно привело бы к деградации системы. Имеются прямые данные об укрупнении алкилтиолатных нанокластеров золота [Au23(SR)16]– под действием H2O2 [43]. Однако эксперименты с повторным напуском газовой фазы показывают (рис. 5), что возобновление содержания Н2О2 в системе через 6 ч реакции приводит к удвоению выхода метанола за последующие 6 ч, что соответствует 100% сохранению активности катализатора. Более того, дополнительные эксперименты продемонстрировали, что предварительное выдерживание катализатора в течение этого же периода времени в условиях реакции при отсутствии метана не влияет на его последующую активность.
Рис. 5.
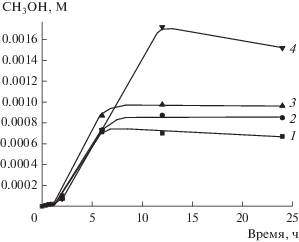
Кинетические кривые накопления CH3ОН в процессе активации метана НК Au: 0.2 М H2O2 (1); при повторном напуске газовой фазы через 6 ч без добавления H2O2: 0.2 М H2O2 (2), 0.4 М H2O2 (3); при добавлении к 0.2 М H2O2 свежей порции 0.2 М H2O2 (4). Условия реакции: 3 × 10–4 М катализатора, 5 мл водной фазы, 1 атм воздух + 30 атм СН4 на начало реакции, Т = 70°C, 24 ч.
Образование аквакомплекса в результате переноса атома Н от одной координированной ОН-группы к другой в активном центре и последующая десорбция молекулы воды,
Согласно расчетам [39] оксигенированный безлигандный кластер Au55(μ3-О) реагирует с метаном с умеренной энергией активации, причем при отрыве атома Н образующийся метильный радикал сначала связывается с кластером:
(VII)
$\begin{gathered} {\text{A}}{{{\text{u}}}_{{55}}}({{\mu }_{3}}{\text{ - О}}) + {\text{С}}{{{\text{Н}}}_{4}} = {\text{A}}{{{\text{u}}}_{{55}}}({\text{О}}{{{\text{Н}}}^{\centerdot }})({\text{СН}}_{3}^{\centerdot }) = \\ = {\text{A}}{{{\text{u}}}_{{55}}} + {\text{С}}{{{\text{Н}}}_{3}}{\text{ОН}}. \\ \end{gathered} $Можно полагать, что в условиях проводимого нами эксперимента атом О в оксигенированном кластере МО встраивается в лабильную Au–Au-связь вместо ранее располагавшегося тиолатного лиганда SbR. Так как в ближайшем окружении группировки Au–О–Au в кластере LAu12(μ-О) координационно ненасыщенные атомы Au отсутствуют, то оксокомплексы LAu12(О) не могут быть активными в реакции гидроксилирования метана, аналогичной (VI). Это вполне соответствует практически нулевой начальной скорости образования спирта. Согласно расчету, атом О в модельном кластере LAu12(μ-О) обладает значительным сродством к протону. В результате его взаимодействия с молекулой Н2О2 произойдет перенос протона на мостиковый атом О с последующим присоединением гидропероксид-аниона к катиону LAu12(μ-ОH)+:
Оптимизированная структура кластера LAu12(μ-ОН)(ОOН) приведена на рис. 4б. Наличие короткой водородной связи ОН…О(Н)О с длиной 1.9 Å благоприятно для процесса отщепления воды от гидропероксо-группы. В этом случае образуется бисоксопроизводное LAu10(Au2О)(О), промежуточный продукт МО2 на схеме (IV) . По сравнению с исходным кластером (рис. 4б) в его структуре наиболее сильно меняется расстояние до терминального атома О, которое сокращается до 1.92 Å. Длины мостиковых Au–O-связей уменьшаются с 2.08 и 2.12 Å до 2.00 и 1.98 Å соответственно. По данным расчета превращение МО2 в М + О2 требует небольших энергетических затрат – 13.7 ккал/моль. Более подробное теоретическое исследование механизма разложения Н2О2 в этой системе отражено в [45].
Предлагаемая молекулярная модель LAu10(Au2О)(О) для МО2 позволяет детализировать механизм реакции (V), в которой промежуточным продуктом является LAu10(Au2ОН)(ОСН3) или LAu10(Au2ОСН3)(ОН). Последующее восстановительное элимирование с рекомбинаций ОН- и ОСН3-групп дает СН3ООН.
Можно полагать, что активная частица LAu10(Au2О)(О) образуется и при непосредственном взаимодействии LAu12 с О2. Действительно, в отсутствие Н2О2 происходит окисление метана кислородом воздуха и выход СН3ООН за 6 ч сопоставим с его величиной (рис. 6) при начальных анаэробных условиях. С учетом незначительной величины константы Генри для растворения кислорода в воде можно найти, что при полном разложении 0.2 М Н2О2 в газовом объеме системы появляется кислород – эквивалент 1.2 атм воздуха.
Рис. 6.
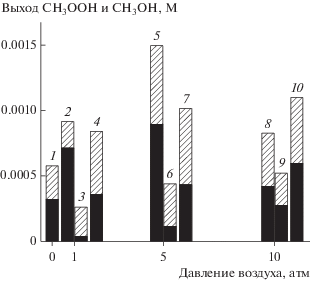
Выход СН3ООН ( ) и СН3ОН(◼) при окислении метана с использованием НК Au: в присутствии 0.2 М H2O2 в атмосфере метана (1) и при давлении воздуха (2), 5 (5) и 10 атм (8); в отсутствие H2O2 при давлении воздуха 1 (3), 5 (6) и 10 атм (9); сумма выхода первичных продуктов окисления в присутствии и без добавления 0.2 М
H2O2 в атмосфере метана при давлении воздуха 1 (4), 5 (7) и 10 атм (10). Условия реакции: 3 × 10–4 М катализатора, 5 мл водной фазы, 30 атм СН4, Т = 70°C, 6 ч.
) и СН3ОН(◼) при окислении метана с использованием НК Au: в присутствии 0.2 М H2O2 в атмосфере метана (1) и при давлении воздуха (2), 5 (5) и 10 атм (8); в отсутствие H2O2 при давлении воздуха 1 (3), 5 (6) и 10 атм (9); сумма выхода первичных продуктов окисления в присутствии и без добавления 0.2 М
H2O2 в атмосфере метана при давлении воздуха 1 (4), 5 (7) и 10 атм (10). Условия реакции: 3 × 10–4 М катализатора, 5 мл водной фазы, 30 атм СН4, Т = 70°C, 6 ч.
Таким образом, при окислении метана в присутствии воздуха в системе следует учесть обратную реакцию,
с константой скорости k–3. В этом случае концентрация МО2 будет напрямую определяться содержанием кислорода. Зависимость начальной концентрации МО2 от давления воздуха Ропределяется константой равновесия K3= k–3/k3. Эта концентрация будет слабо зависеть от времени, особенно при больших Р. Поэтому мы примем ее постоянной. В результате кинетическое уравнение для суммы продуктов упрощается:(6)
$\frac{{{\text{d}}z}}{{{\text{d}}t}} = {{k}_{4}}[{\text{C}}{{{\text{H}}}_{4}}]\frac{{{{K}_{3}}[{\mathbf{M}}]P}}{{1 + {{K}_{3}}P}} - {{k}_{6}}\left( {1 + \frac{P}{{1.2}}} \right)z.$Здесь для описания процессов окисления продуктов на больших временах учтено наличие дополнительного кислорода воздуха. Уравнение (6) решается тривиально, и с учетом ранее найденного значения k6 = 1.5 × 10–2 ч–1 можно найти отношение суммарного выхода продуктов за 6 ч при давлениях Р и 1:
(7)
$\frac{{z(P)}}{{z(1)}} = \frac{{P(1 + {{K}_{3}})}}{{1 + {{K}_{3}}P}}\frac{{\left( {1 + \frac{1}{{1.2}}} \right)}}{{\left( {1 + \frac{P}{{1.2}}} \right)}}\frac{{1 - \exp \left[ { - 0.09\left( {1 + \frac{P}{{1.2}}} \right)} \right]}}{{1 - \exp \left[ { - 0.09\left( {1 + \frac{1}{{1.2}}} \right)} \right]}}.$Это выражение качественно соответствует экстремальному выходу суммарного количества продуктов от давления воздуха (рис. 6). Для количественного сопоставления необходимо знать величину K3. Ее значение оценим из независимого эксперимента по разложению пероксида водорода в присутствии 1 атм воздуха, который показывает уменьшение начальной скорости разложения Н2О2 в 1.62 раза по сравнению с 1 атм аргона. Блокирование части активных центров кислородом (5) приводит к уменьшению стационарной концентрации МО:
(8)
$[{\mathbf{M}}{\text{O}}] = \frac{{{{k}_{1}}}}{{{{k}_{2}}}}\frac{{[{\mathbf{M}}]}}{{1 + {{K}_{3}}P}}$В условиях, когда концентрация комплекса MO2 определяется давлением воздуха, кинетическое уравнение для образования СH3OH сильно упрощается:
Интегрируя его с учетом (5) и (8), можно получить зависимость относительной доли метилгидропероксида от давления воздуха:
(9)
$\begin{gathered} \frac{{x(P)}}{{z(P)}} = \frac{{1 + \frac{P}{{1.2}}}}{{\frac{\alpha }{{1 + {{K}_{3}}P}} + 1 + \frac{P}{{1.2}}}} \times \\ \times \,\,\frac{{1 - \exp \left[ { - 0.09\left( {1 + \frac{P}{{1.2}} + \frac{\alpha }{{1 + {{K}_{3}}P}}} \right)} \right]}}{{1 - \exp \left[ { - 0.09\left( {1 + \frac{P}{{1.2}}} \right)} \right]}}, \\ \end{gathered} $Выбор значения α = 10 обеспечивает равенство теоретического значения (9) экспериментальной величине 0.40 при 5 атм. При этом x(10)/z(10) = 0.59, что выше экспериментального значения 0.49, а рассчитанная величина x(1)/z(1) = 0.17, что ниже экспериментального значения 0.25. Таким образом, кинетическая схема обратимого связывания кислорода с активным центром качественно правильно описывает заметное увеличение выхода метанола при добавлении воздуха и полуколичественно – снижение его доли с ростом Р.
На рис. 6 также приведена сумма продуктов окисления метана пероксидом водорода в анаэробных условиях и кислородом воздуха. Во всех случаях соотношение выходов СН3ОН и СН3ООН при его совместном окислении Н2О2 и кислородом воздуха в условиях исследуемого давления заметно отличается от данных, полученных методом суммирования. Небольшой выход метилового спирта при окислении кислородом, по-видимому, связан с катализируемым процессом гидролиза:
Метан ускоряет разложение Н2О2, в том числе, и в присутствии воздуха (см. рис. 3), при этом более 97% дополнительного расхода Н2О2 идет на образование кислорода. Именно наличие кислорода является причиной дальнейшего окисления как метилового спирта, так и метилгидропероксида в присутствии Au-НК на больших временах реакции (рис. 2). Медленный характер этой реакции обуславливает возможность накопления первичных продуктов окисления. Ранее проведенное исследование каталитических свойств HAuCl4 при окислении метана пероксидом водорода в воде при 90оС показало выход метанола на порядок ниже [46]. Основным продуктом был диоксид углерода, и наблюдалось неконтролируемое образование коллоидного золота. Ускорение разложения Н2О2 в присутствии метана, по-видимому, также обусловлено эффектом синергизма при параллельно протекающих процессах окисления метана и разложения Н2О2 на одном НК золота, стабилизированном глутатионом. В пользу этого предположения свидетельствуют наблюдение с помощью спектральных методов образования координированных метоксигрупп на нанокластерах Au при окислении метана, а также увеличение реакционной способности кластера с повышением средней степени окисления атомов Au в нем [29].
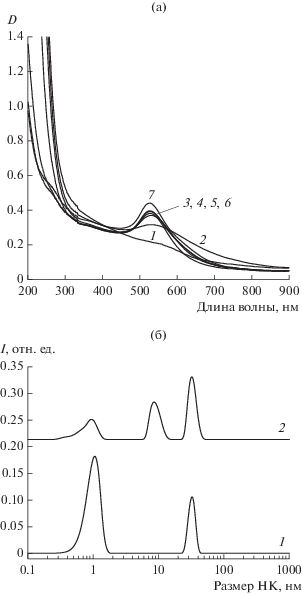
Для получения дополнительной информации о составе исследуемой системы были изучены оптические спектры поглощения жидкой фазы реакционной среды (рис. 7а). Поглощение исходного раствора НК Au монотонно увеличивается, начиная с 700 нм, и резко возрастает при 300 нм. Это свидетельствует о присутствии в нем нанокластеров, состоящих из 18–25 атомов Au [30]. В реакционной смеси через 30 мин появляется новая полоса поглощения, которая потом сужается до пика плазмонного резонанса с λмах = 525 нм, характерного для сферических НЧ золота размером ~20 нм. Остаточное поглощение в области 600–700 нм указывает на присутствие незначительного количества более крупных НЧ. За 2 ч формирование состава НЧ практически заканчивается, о чем свидетельствует малое изменение спектров на рис. 7а. Дополнительные исследования методом динамического рассеяния света каталитически активной системы показали, что после проведения реакции в системе уширяется пик распределения НК по размерам, и появляются НЧ золота размером 20 нм. Самые большие наночастицы являются агрегатами молекул глутатиона и имеют размер 60 нм (рис. 7б). С учетом данных о сохранении активности системы при повторном запуске реакции можно сделать вывод, что наблюдаемое укрупнение наночастиц, скорее всего, не затрагивает каталитически активные НК Au. Если принять, что все атомы золота в системе находятся в составе кластеров Au25, то выход метанола составляет порядка 25 на моль кластера, а в присутствии 1 атм воздуха он возрастает до 60 молей. Так как из данных динамического рассеяния света следует, что в каталитически активных частицах содержится менее 100% Au, то вышеприведенная оценка число оборотов катализатора является оценкой снизу.
Рис. 7.
а – Спектры поглощения реакционной среды с НК Au + Н2О2 во времени: исходный раствор (1) и через 0.5 (2), 1 (3), 2 (4), 4 (5), 6 (6) и 24 ч (7) реакции; б – гистограмма распределения частиц по величине их радиуса в растворе реакционной среды с НК Au с газовой фазой 30 атм СН4: исходный раствор (1) и через 24 ч реакции (2). Условия: 3 × 10–4 М катализатора, 0.2 М H2O2, Т = 70°C.
ЗАКЛЮЧЕНИЕ
Таким образом, показано, что нанокластеры Au (1–2 нм), стабилизированные глутатионом, катализируют окисление метана до СН3ОН и СН3ООН в водной среде при мягких условиях. На выход продуктов окисления существенно влияет не только содержание H2O2, но и присутствие кислорода воздуха. При окислении метана в аэробных условиях в отсутствие H2O2 повышается выход СН3ООН. Предложенный молекулярный механизм позволяет удовлетворительно описать кинетические кривые накопления и расходования СН3ОН и СН3ООН, а также увеличение выхода метанола в присутствии кислорода воздуха. На основании результатов квантово-химического моделирования предложена химическая структура окисленной формы кластера Au25, ответственная за активацию С–Н-связи молекулы метана. Эксперименты с повторным введением газовой фазы и возобновлением содержания H2O2 продемонстрировали 100% сохранение каталитической активности.
Список литературы
Ravi M., Ranocchiari M., van Bokhoven J.A. // Angew. Chem. Int. Ed. 2017. V. 56. № 52. P. 16464.
Zakaria Z., Kamarudin S.K. // Renewable and Sustainable Energy Rev. 2016. V. 65. P. 250.
Gesser H.D., Hunter N.R., Prakash C.B. // Chem. Rev. 1985. V. 85. № 4. P. 235.
Shilov A.E., Shul’pin G.B. // Chem. Rev. 1997. V. 97. № 8. P. 2879.
Otsuka K., Wang Y. // Appl. Catal. A: General. 2001. V. 222. № 1–2. P. 145.
Raynes S., Shah M.A., Taylor R.A. // Dalton. Trans. 2019. V. 48. P. 10364.
Gunsalus N.J., Koppaka A., Park S.H., Bischof S.M., Hashiguchi B.G., Periana R.A. // Chem. Rev. 2017. V. 117. № 13. P. 8521.
Cui W.G., Zhang G.Y., Hu T.L., Bu X.H. // Coord. Chem. Rev. 2019. V. 387. P. 79.
Bao J., Yang G.H., Yoneyama Y., Tsubaki N. // ACS Catal. 2019. V. 9. № 4. P. 3026.
Conley B.L., Tenn W.J., Young K.J.H., Ganesh S.K., Meier S.K., Ziatdinov V.R., Mironov O., Oxgaard J., Gonzales J., Goddard W.A., Periana R.A. // J. Mol. Cat. A: Chem. 2006. V. 251. P. 8.
Чепайкин Е.Г., Менчикова Г.Н., Помогайло С.И. // Изв. Акад. Наук. Сер. хим. 2019. Т. 68. № 8. С. 1465.
Haruta M. // Chem. Rec. 2003. V. 3. № 2. P. 75.
Haruta M. // Gold bull. 2004. V. 37. № 1–2. P. 27.
Hashmi A.S.K., Blanco M.C., Fischer D., Bats J.W. // Eur. J. Org. Chem. 2006. V. 2006. № 6. P. 1387.
Yamazoe S., Koyasu K., Tsukuda T. // Acc. Chem. Res. 2014. V. 47. № 3. P. 816.
Zhang B., Kaziz S., Li H., Hevia M.G., Wodka D., Mazet C., Bürgi T., Barrabés N. // J. Phys. Chem. C. 2015. V. 119. № 20. P. 11193.
Li G., Jin R.C. // RSC Catalysis Series. 2014. № 18. P. 27.
Nasaruddin R.R., Chen T.K., Yan N., Xie J.P. // Coord. Chem. Rev. 2018. V. 368. P. 60.
Chen Y.X., Zeng C.J., Jin R.C. // SPR-Catal. 2016. V. 28. P. 51.
Zhu Y., Qian H.F., Zhu M.Z., Jin R.C. // Adv. Mater. 2010. V. 22. № 17. P. 1915.
Taketoshi A., Haruta M. // Chem. Lett. 2014. V. 43. P. 380.
Yoskamtorn T., Yamazoe S., Takahata R., Nishigaki J., Thivasasith A., Limtrakul J., Tsukuda T. // ACS Catal. 2014. V. 4. № 10. P. 3696.
Zhu Y., Qian H.F., Jin R.C. // J. Mat. Chem. 2011. V. 21. P. 6793.
Zhu Y., Qian H., Drake B.A., Jin R. // Angew. Chem. Int. Ed. 2010. V. 49. № 7. P. 1295.
Nasaruddin R.R., Chen T.K., Yan N., Xie J.P. // Coord. Chem. Rev. 2018. V. 368. P. 60.
Yan Z., Huifeng Q., Rongchao J. // J. Mater. Chem. 2011. V. 21. P. 6793.
Zhang B., Fang J., Li J., Lau J.J., Mattia D., Zhong Z., Xie J., Yan N. // Chem. Asian. J. 2016. V. 11. № 4. P. 532.
Agarwal N., Freakley S.J., McVicker R.U., Althahban S.M., Dimitratos N., He Q., Morgan D.J., Jenkins R.L., Willock D.J., Taylor S.H., Kiely Ch.J., Hutchings G.J. // Science. 2017. V. 358. № 6360. P. 223.
Cai X., Saranya G., Shen K., Chen M., Si R., Ding W., Zhu Y. // Angew. Chem. Int. Ed. 2019. V. 58. P. 9964.
Negishi Y., Nobusada K., Tsukuda T. // J. Am. Chem. Soc. 2005. V. 127. № 14. P. 5261.
Zhu M., Lanni E., Garg N., Bier M.E., Jin R. // J. Am. Chem. Soc. 2008. V. 130. № 4. P. 1138.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Dyall K.G. // J. Chem. Phys. 1994. V. 100. P. 2118.
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. P. 151.
Laikov D.N. // Chem. Phys. Lett. 2005. V. 416. P. 116.
Wang H., Bozzelli J.W. // J. Chem. Eng. Date. 2016. V. 61. № 5. P. 1836.
Saliba N., Parker D.H., Koel B.E. // Surf. Sci. 1998. V. 410. № 2–3. P. 270.
Molecular spectra and molecular structure. IV Constants of diatomic molecules / Eds. Huber K.P., Herzberg G. New York: Van Nostrand Reinhold company. 1979. 732 p.
Staykov A., Miwa T., Yoshizawa K. // J. Catal. 2018. V. 364. № 13. P. 141.
Coperet C. // Chem. Rev. 2010. V. 110. № 2. P. 656.
Wu Z., Jin R. // ACS Nano. 2009. V. 3. № 7. P. 2036.
Giguere P.A., Liu L.D. // Can. J. Chem. 1957. V. 35. № 4. P. 283.
Higaki T., Liu C., Chen Y., Zhao S., Zeng C., Jin R., Wang S., Rosi N.L., Jin R. // J. Phys. Chem. Lett. 2017. V. 8. №4. P. 866.
Rosca D.A., Wright J.A., Hughes D.L., Bochmann M. // Nature Commun. 2013. V. 4. P. 2167.
Никитенко Н.Г., Шестаков А.Ф. / Теоретическое изучение механизма разложения H2O2, катализируемого кластером золота Au25(SCH3)12. Тез. докл. XXI Менделеевского съезда по общей и прикладной химии, Санкт-Петербург, 9–13 сентября, 2019. Т. 1. С. 248.
Yuan Q., Deng W.P., Zhang Q.H., Wang Y. // Adv. Synt. Catal. 2007. V. 349. № 7. P. 1199.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ