

Кинетика и катализ, 2020, T. 61, № 5, стр. 688-699
Гетерогенные катализаторы типа SILP с фосфорновольфрамовой кислотой для окислительной десульфуризации: влияние ионной жидкости
А. А. Брыжин a, А. К. Буряк b, М. Г. Гантман c, В. М. Зеликман a, М. И. Шилина a, И. Г. Тарханова a, *
a ФГБОУ ВО Московский государственный университет им. М.В. Ломоносова, химический факультет
119991 Москва, Ленинский горы, 1, стр. 3, Россия
b ФГБУН Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, корп. 4, Россия
c Helmholtz Institut Erlangen-Nürnberg for renewable Energy
Erlangen, Egerlandstraße 3 – Zimmer T00.10791058, Deutschland
* E-mail: itar_msu@mail.ru
Поступила в редакцию 07.11.2019
После доработки 28.01.2020
Принята к публикации 06.03.2020
Аннотация
Для пероксидного окисления тиофена получены катализаторы типа SILP (supporting ionic liquid phase) на силикагеле, содержащие имидазольные катионы с сульфокислотной группой и фосфорновольфрамат-анионы. Состав активной фазы и структура поверхности катализаторов охарактеризованы набором современных физико-химических методов, в том числе хроматомасс-спектрометрией в технике ПАЛДИ. Установлено, что строение органического катиона ионной жидкости влияет на стабильность гетерополианионов и деструктивные процессы в SILP, а также на каталитические свойства образцов. Катализаторы SILP эффективны в десульфуризации дизельной фракции и позволяют получать топливо с содержанием S менее 10 ppm.
ВВЕДЕНИЕ
Гетерополикислоты (ГПК) и полиоксометаллатные соединения на их основе в сочетании с ионными жидкостями (ИЖ) широко используются для создания каталитически активных гибридных материалов. Благодаря высокой кислотности, окислительному потенциалу, а также совместимости с “зелеными окислителями” – кислородом и пероксидом водорода, такие композиции эффективно применяются в ряде окислительных процессов [1], в том числе, в обессеривании углеводородного сырья [2–4].
Десульфуризация является важной областью нефтеперерабатывающей промышленности и направлена на получение топлива, отвечающего современным экологическим стандартам (содержание общей серы менее 10 ppm) [5]. Проблема переработки углеводородного сырья напрямую связана с удалением гетероатомных соединений, оказывающих негативное воздействие на окружающую среду, вызывающих коррозию оборудования и травление дорогостоящих катализаторов. Традиционный способ очистки топлива от серосодержащих соединений – гидроочистка – требует высоких температур, давления и большого расхода водорода [6]. Процесс эффективен в отношении тиолов, сульфидов и тиофена, но для удаления гетероциклических соединений, например, дибензотиофена и его алкильных производных, необходимо ужесточение условий его проведения, что зачастую приводит к повышению себестоимости конечного продукта [7]. Поэтому в дополнение к гидроочистке используют адсорбцию [8, 9], экстракцию [10, 11] и окислительную десульфуризацию (ОДС) [2, 4]. Среди перечисленных методов наиболее многообещающим является ОДС, благодаря мягким условиям и возможности реализации на традиционном нефтеперерабатывающем оборудовании [12].
При окислительной десульфуризации обычно образуются полярные сульфоны и сульфоксиды, которые далее удаляют методами адсорбции или экстракции [3, 13]. Катализатор процесса является ключевым компонентом любой окислительной системы. Как правило, применяют композиции, содержащие оксиды в индивидуальном и смешанном виде, а также соли переходных металлов [2, 12, 14, 15]. Особое внимание уделяют гетерогенным катализаторам на основе ГПК и полиоксометаллатных соединений, нанесенным на пористые материалы с большой удельной поверхностью и обладающим высокой активностью в окислении сероорганических соединений [2, 4, 16]. Однако актуальная проблема создания таких систем – низкая стабильность анионов, связанная с сильным взаимодействием между гетерополикислотой и поверхностью минеральных носителей [17–19].
Известно, что ИЖ применяют в сероочистке как гомогенные катализаторы [20, 21] и экстрагенты [10, 22]. Тем не менее, их высокая стоимость ограничивает возможность масштабного использования в данном направлении, поэтому для снижения расхода ионных жидкостей был предложен путь их иммобилизации на поверхности [4, 23, 24]. Катализатор SILP представляет собой слой ионной жидкости, нанесенный на поверхность пористых материалов путем физической адсорбции [25], при этом каталитической активностью может обладать как сама ИЖ (катион и/или анион), так и дополнительные металлсодержащие компоненты, находящиеся в ИЖ-слое. В результате полученные гибридные композиции обладают достоинствами гомогенных (высокая активность и селективность в мягких условиях) и гетерогенных систем (стабильность, простота отделения продуктов) [26]. Вследствие чего гетерогенные системы типа SILP широко используются в ряде каталитических процессов, в том числе окислительном обессеривании [4, 24, 27–29].
В нашей предыдущей работе [4] мы предложили эффективный способ синтеза гетерогенных катализаторов на основе фосфорновольфрамовой или фосфорномолибденовой кислот и 4-(3'-этилимидазолий)-бутансульфоната. Цель настоящей работы – установление влияния строения имидазольного катиона на стабильность фосфорновольфрамат-анионов на поверхности силикагеля и активность полученных гибридных материалов типа SILP в окислении тиофена пероксидом водорода. Это соединение было выбрано в качестве модельного субстрата из-за его наибольшей устойчивости к окислению среди других серосодержащих гетероциклов [30]. Образцы типа SILP получали с использованием имидазолийбутансульфоната c этильным или винильным заместителями и фосфорновольфрамовой кислоты (схема 1 ). Выбор обусловлен литературными данными, согласно которым винильные ИЖ по сравнению с этильными обладают свойством образовывать внутри- и межмолекулярные водородные связи [31]. Мы полагали, что такое дополнительное взаимодействие будет способствовать стабилизации как ИЖ на поверхности силикагеля, так и анионов. Кроме того, для сравнения тестировали образец, синтезированный в аналогичных условиях из фосфорновольфрамовой кислоты и силикагеля без использования органических добавок.
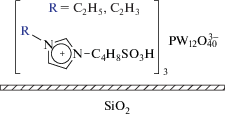
Схема 1 . Структура синтезированных композиций SILP.
Применение тиофена в качестве модельного субстрата вызывает интерес по двум причинам: во-первых, он имеет низкую электронную плотность на атоме серы и, как следствие, является наиболее трудноокисляемым среди сероорганических соединений тиофенового ряда [2, 32]. Во-вторых, окисление тиофена может приводить к образованию сульфат-аниона, стирола, карбоновых кислот, углекислого газа [2, 33–35]. Эти продукты, особенно серная кислота, способны оказывать негативное влияние на катализатор и понижать его стабильность. Таким образом, катализаторы проходили тестирование в заведомо жестких условиях. Кроме модельного процесса, все гетерогенные композиции были испытаны в ОДС дизельной фракции, предоставленной ОАО “Варьеганнефть”.
Одним из факторов, препятствующих глубокому окислению субстрата, является побочная реакция разложения пероксида водорода, протекающая под действием металлсодержащих катализаторов. Для повышения эффективности использования окислителя мы применили подход, заключающийся в дробной загрузке пероксида водорода. Этот прием основан на том, что согласно исследованиям [36, 37] порядок реакции каталитического разложения H2O2 меняется в зависимости от его начальной концентрации, а порядок основной реакции по окислителю остается неизменным. Поэтому для увеличения эффективности использования пероксид водорода необходимо добавлять небольшими порциями, т.е. понижать его начальную концентрацию и тем самым замедлять побочную реакцию разложения. Этот подход известен из литературы [38, 39] и был успешно реализован в наших предыдущих работах [4, 40–42].
Выполненные исследования позволили провести сравнительный анализ катализаторов типа SILP и аналогичного гетерогенного образца без ионной жидкости, а также оценить влияние строения имидазольного катиона на стабильность гетерополианионов, текстурные характеристики образцов, их кислотность и активность в окислении модельного субстрата и ОДС дизельной фракции.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы и вспомогательные материалы
В работе использованы реактивы производства фирмы “Sigma Aldrich”: тиофен (99%), пероксид водорода (50%), 1,4-бутансультон (99%), 1-этилимидазол (95%), 1-винилимидазол (95%), фосфорновольфрамовая кислота (99%). Растворители получены из коммерческих источников без дополнительной очистки. Носитель для приготовления катализаторов – силикагель марки BASF Perlkat 97-0.
Приготовление катализаторов
Синтез цвиттер-ионных соединений. Цвиттер-ионные соединения (ЦИ) синтезированы согласно методике (схема 2 ), описанной нами ранее [4]. Производное имидазола (винилимидазол, 9.4 г или этилимидазол 9.6 г) растворили в ацетонитриле (100 мл) и перемешали с 15 г 1,4-бутан сультона (мольное соотношение имидазол : сультон 1 : 1.1). Смесь продували аргоном, после чего кипятили с обратным холодильником при интенсивном перемешивании в течение 24 ч. Выпавший осадок отфильтровали, трижды промыли ацетоном и сушили под вакуумом при 100° в течение 16 ч. Структуру цвиттер-ионов подтверждали при помощи спектроскопии ЯМР на приборе JNM ECX-400 (“JEOL”, Япония), 400 МГц. Данные по двум образцам ЦИ приведены ниже:
4-(3'-этилимидазолий)-бутансульфонат: 1H ЯМР (400 МГц, D2O) 1.51 (3.0H, t, J = 7.2 Гц, NCH2CH3), 1.83–1.72 (2.0H, m, NCH2CH2CH2–CH2SO3), 2.11–2.03 (2.0H, m, NCH2CH2CH2CH2SO3), 2.93 (2.0H, t, J = 7.1 Гц, NCH2CH2CH2CH2SO3), 4.32–4.24 (4H, m, NCH2CH3, NCH2CH2CH2CH2SO3), 7.81 (2.0H, s, NCHNCHCH), 9.03 (1H, s, NCHNCHCH). 13C ЯМР (D2O) 14.3 (NCH2CH3), 23.5 (NCH2CH2CH2CH2SO3), 29.7 (NCH2CH2CH2CH2SO3), 45.7 (NCH2CH2CH2CH2SO3), 51.3 (NCH2CH2CH2CH2SO3, NCH2CH3), 51.6 (NCH2CH2CH2CH2SO3, NCH2CH3), 124.9 (NCHNCHCH), 125.3 (NCHNCHCH), 134.6 (NCHNCHCH);
4-(3'-винилимидазолий)-бутансульфонат: 1H ЯМР (400 МГц, D2O) 1.58–1.67 (2.0H, m, NCH2CH2CH2–CH2SO3), 1.87–1.96 (2.0H, m, NCH2CH2CH2CH2SO3), 2.91 (2.0H, t, J = 7.6 Гц, NCH2CH2CH2CH2SO3), 4.16 (2.0H, t, J = 8.4 Гц, NCH2CH2CH2CH2SO3), 5.26–5.30, (1H dd, N‒CH=CHH), 5.63–5.69, (1H dd, N–CH=CHH), 6.96–7.04 (1H, dd, N–CH=CH2) 7.47 (1.0H, m, NCHNCHCH), 7.65 (1.0H, m, NCHNCHCH), 8.94 (1H, s, NCHNCHCH). 13C ЯМР (D2O) 21.0 (NCH2CH2CH2CH2SO3), 28.0 (NCH2CH2CH2CH2SO3), 49.3 (NCH2CH2CH2CH2SO3), 50.1 (NCH2CH2CH2CH2SO3), 109.4 (N–CH=CH2) 119.6 (NCHNCHCH), 122.9 (NCHNCHCH), 128.3 (NCHNCHCH), 134.5 (N–CH=CH2).
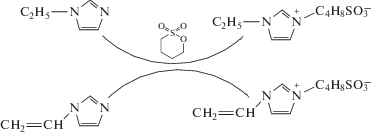
Схема 2 . Синтез цвиттер-ионных соединений.
Синтез ИЖ и получение образцов SILP. Для протонирования сульфоновой группы 0.7 г цвиттер-ионного соединения (4-(3'-этилимидазолий)-бутансульфонат или 4-(3'-винилимидазолий)-бутансульфонат) добавляли к концентрированному водному раствору 2.9 г фосфорновольфрамовой гетерополикислоты (ФВК) (мольное соотношение ЦИ : ФВК составляло 3 : 1). Смесь интенсивно перемешивали в течение 6 ч при комнатной температуре, полученные ИЖ, представляющие собой кристаллические вещества белого цвета, сушили на воздухе.
Силикагель выдерживали в водном растворе ИЖ (массовое соотношение носитель : ИЖ составляла 10 : 1) при комнатной температуре в течение 12 ч. Затем раствор декантировали, а каталитические образцы сушили в вакууме при 60°С до постоянной массы.
Таким образом получены образцы “vin-W” и “et-W” из 4-(3'-винилимидазолий)-бутансульфоната и 4-(3'-этилимидазолий)-бутансульфоната соответственно (схема 1 ).
Синтез катализатора без ионной жидкости. Катализатор “W” приготовлен путем нанесения фосфорновольфрамовой гетерополикислоты на силикагель из водно-спиртового раствора (массовое соотношение силикагель : кислота составляло 10 : 1) [17], процесс проводили в течение 12 ч, затем полученные гранулы сушили в вакууме, как описано выше.
Характеристика катализаторов
Идентификацию ИЖ проводили методом ИК-спектроскопии в таблетках с KBr на Фурье-ИК-спектрофотометре Infralum FT-801 (“СИМЕКС”, Россия) в диапазоне 4000–400 см–1.
Для установления состава гетерополианионов на поверхности носителя использовали метод масс-спектрометрии с поверхностно-активированной лазерной десорбцией/ионизацией (ПАЛДИ). Масс-спектры образцов регистрировали в режиме RN Pep Mix на приборе Ultraflex (“Bruker”, Германия), оборудованном азотным лазером (длина волны – 337 нм, энергия – 110 мкДж), масс-анализатор – времяпролетный. Запись спектров производили с применением рефлектрона в режиме регистрации отрицательных ионов. Идентификацию кластерных ионов по изотопному распределению осуществляли с помощью программы-симулятора IsoPro.
Элементный анализ проводили с использованием автоматического CHN-анализатора CE1106 (“Carlo Erba Instruments”, Италия).
Информация о кислотных свойствах каталитических композиций получена методом термопрограммированной десорбции аммиака (ТПД NH3) на сорбционном анализаторе УСГА-101 (“Унисит”, Россия). Предварительно образец прогревали в инертной атмосфере при 150°С, затем охлаждали до комнатной температуры. Адсорбцию NH3 осуществляли при 60°C в течение 30 мин, аммиак разбавляли азотом в соотношении 1 : 1. Физически сорбированный аммиак отдували в токе осушенного гелия при 100°С в течение 1 ч. Эксперименты по термопрограммированной десорбции аммиака проводили в интервале температур от 60 до 400°С в токе осушенного гелия (скорость 30 мл/мин). Скорость нагрева составляла 8°/мин.
Кислотные свойства водных растворов ЦИ определяли с использованием портативного pH-метра марки HI 8314 (“Hanna Instruments ”, Германия).
Топографию поверхности методом сканирующей электронной микроскопии (СЭМ) исследовали на электронном микроскопе JSM-6000 NeoScope (“JEOL”, Япония) со встроенным рентгеновским анализатором EX-230 для энергодисперсионного анализа распределения частиц. Микроскопию проводили в режиме высокого вакуума с ускоряющим напряжением 15 кВ. Режим детектирования сигнала – SEI (изображение во вторичных электронах).
Содержание вольфрама на поверхности катализатора определяли фотометрически с использованием пирокатехина, который образует устойчивое комплексное соединение с вольфраматом в присутствии сульфита натрия и едкого натрия [43]. Электронные спектры регистрировали на приборе UV-2101PC (“Shimadzu”, Япония), измеряя оптическую плотность при 350 нм.
Адсорбционные измерения осуществляли на автоматическом сорбтомере ASAP 20000N (“Micromeritics”, США). Перед измерением образцы вакуумировали при 150°С в течение 2 ч.
Каталитические эксперименты и анализ продуктов
Для приготовления модельной смеси рассчитанное количество тиофена растворяли в изооктане, чтобы получить концентрацию исследуемого субстрата 1 мас. %.
Эксперименты по окислению тиофена проводили в термостатируемом стеклянном реакторе с рубашкой. Модельную смесь (10 мл), 50% пероксид водорода (0.4 мл) и катализатор (0.1 г) помещали в реактор при комнатной температуре, затем смесь нагревали до 60°С и перемешивали в течение 4 ч. Оптимальная температура процесса (60°С) для катализаторов на основе гетерополикислот была определена в наших предыдущих работах [4, 40, 42]. При дробной загрузке H2O2 окислитель добавляли по 0.2 мл каждые два часа. Для количественного анализа органической фазы использовали метод газожидкостной хроматографии (ГЖХ), пробы (0.5 мкл) отбирали с интервалом в 1 ч, внутренним стандартом служил додекан. Анализ осуществляли на приборе Кристалл 2000 (“Хроматэк”, Россия), оснащенном капиллярной колонкой Zebron ZB-1 (“Phenomenex”, США, 30 м × 0.32 мм × 0.5 мкм). Идентификацию продуктов процесса проводили с помощью ЯМР 1H и 13C. Спектры регистрировали на приборе Avance-600 (“Bruker”, Германия, 600 МГц) при комнатной температуре. Для повторного использования катализаторов реакционную смесь декантировали, а образцы промывали изооктаном.
Для окислительного обессеривания дизельное топливо производства ОАО “Варьеганнефть” (20 мл, общее содержание серы – 1080 ppm), катализатор (0.04 г) и H2O2 (0.4 мл, 50%) термостатировали в стеклянном реакторе (60°С) при интенсивном перемешивании в течение 4 ч. Смесь охлаждали до комнатной температуры и промывали 5 мл диметилформамида (ДМФА) в делительной воронке. ДМФА применяли как наиболее эффективный экстрагент для удаления продуктов окисления сероорганических компонентов дизельного топлива [18]. В предварительных экспериментах при обработке топлива ДМФА без окисления было показано, что экстракция серы не превышала 20%. Затем топливо помещали в реактор, добавляли свежую порцию окислителя (0.4 мл) и использованный на предыдущей стадии катализатор, промытый изооктаном. Смесь перемешивали еще 4 ч при 60°С и повторяли процедуру экстракции продуктов окисления. Для сравнения процесс проводили в одну стадию с пероксидом водорода (0.8 мл). Остаточное содержание серы определяли на рентгеновском флуоресцентном спектрометре АСЭ-2 (“Буревестник”, Россия), предел погрешности измерения ±0.5%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристика катализаторов
ИК-спектры индивидуальных ИЖ, приготовленных с использованием двух ЦИ и фосфорнофольфрамовой кислоты в таблетках KBr, приведены на рис. 1. Отнесение полос и идентификацию соединений проводили с использованием данных литературы [44, 45].
Рис. 1.
ИК-спектры ИЖ на основе H3PW12O40 с 4-(3'-винилимидазолий)-бутансульфонатом (1) и 4-(3'-этилимидазолий)-бутансульфонатом (2).

Как видно из рис. 1, в обоих спектрах присутствуют полосы в областях 3200–2800 см–1, отвечающие валентным колебаниям С–H в ароматическом гетероцикле, алкильном и винильном фрагментах. Полосы в области 1600–1500 см–1 характеризуют валентные колебания имидазолиевого кольца. Сравнительно сильная полоса при 1650 см–1 в спектре vin-W может отвечать ν(С=С)-колебаниям в винильном фрагменте. Кроме того, полосы при 1173–1165 cм–1 в спектрах обоих образцов могут относиться и к деформационным колебаниям в гетероциклах δ(H–C–C) и δ(H–C–N) и к валентным колебаниям S–O и S=O сульфогруппы (1160–1170 и 1350–1380 см–1). Полосы валентных колебаний в гетерополианионе PW12 наблюдаются в области 1100–800 см–1. Для P–O-связи в характерны полосы поглощения при 1080 см–1 и 500 см–1, которые можно отнести к валентным и деформационным колебаниям соответственно. Полосы при 975, 895 и 806 см–1 характеризуют соответственно связи W c атомами O, входящими в группы WO6, а также мостиковые W–O–W, связывающие октаэдры WO6, обладающие общим углом или общей гранью. Таким образом, данные ИК-спектроскопии подтверждают присутствие в анализируемых образцах ионных жидкостей, соответствующих имидазолийсульфонатов и гетерополианионов кеггиновского типа.
Физико-химические параметры твердых образцов и носителя представлены в табл. 1. В результате синтеза катализаторов SILP происходит существенное уменьшение удельной поверхности и объема пор носителя; по данным СЭМ наблюдается формирование плотных слоeв ИЖ, равномерно распределенных на поверхности силикагеля (рис. 2). Напротив, модификация поверхности с ГПК без ИЖ (катализатор W) не приводит к заметному изменению удельной поверхности и объема пор. Мы полагаем, что это обусловлено разложением ФВК (табл. 2) с образованием низкомолекулярных вольфраматов и оксидов вольфрама WO3 и W2O6, обладающих значительно меньшим размером, чем молекулы гетерополикислоты, и не оказывающих существенного влияния на текстурные характеристики носителя. Тем не менее, поверхность этого образца, а также et-W, имеет схожую шероховатую рыхлую структуру в отличие от катализатора vin-W, на СЭМ-снимках которого отчетливо видно формирование крупных агломератов ИЖ (рис. 2 и 3). Такое различие в поведении ИЖ на поверхности может быть обусловлено склонностью винильных производных к образованию водородных связей между катионами и анионами как внутри, так и между молекулами [31].
Таблица 1.
Текстурные характеристики носителя, образцов и содержание в них вольфрама и азота по данным элементного анализа
| Образцы | Sуд, м2/г | Dпор, нм | Vпор, см3/г | W, мас. % | N, мас. % |
|---|---|---|---|---|---|
| W | 297 | 9 | 0.69 | 9 | 0 |
| vin-W | 150 | 11 | 0.39 | 6.5 | 0.78 |
| et-W | 104 | 10 | 0.32 | 2.8 | 0.45 |
| Силикагель | 300 | 10 | 0.75 | 0 | 0 |

Таблица 2.
Состав ионов на поверхности катализаторов, установленный методом ПАЛДИ в режиме регистрации отрицательных ионов*
| Катализатор | Ионы | m/z | Абсолютная интенсивность, усл. ед. |
|---|---|---|---|
| W | WO3 | 232 | 86 |
| W2O6 | 464 | 54 | |
| et-W | PWO6 | 311 | 15 |
| PW2O5 | 479 | 62 | |
| PW3O12 | 775 | 102 | |
| PW4O14 | 991 | 46 | |
| PW5O15 | 1191 | 23 | |
| PW6O17 | 1407 | 8 | |
| PW12O40 | 2879 | 12 | |
| vin-W | PWO6 | 311 | 510 |
| W2O6 | 464 | 5410 | |
| PW2O5 | 479 | 870 | |
| W3O9 | 696 | 5720 | |
| W4O12 | 928 | 5700 | |
| W5O15 | 1160 | 2830 | |
| W6O18 | 1392 | 2440 | |
| PW6O17 | 1407 | 115 | |
| PW7O19 | 1623 | 118 | |
| PW12O40 | 2879 | 130 |
* Данные хромато-масс спектрометрии ПАЛДИ по составу ионов индивидуальной ФВК приведены в нашей работе [42].
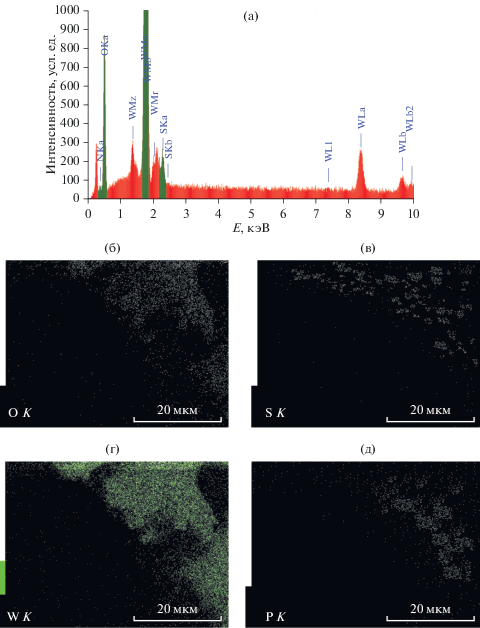
Согласно результатам исследования, полученным методом масс-спектрометрии в технике ПАЛДИ, катализаторы отличаются составом ионов, структура которых изменяется в зависимости от типа используемой ИЖ (табл. 2).
В результате синтеза катализатора W (без ионной жидкости) происходит полное разложение гетерополикислоты с образованием низкомолекулярных оксидов вольфрама (табл. 2). С другой стороны, в масс-спектрах образцов SILP (et-W и vin-W) присутствуют пики, соответствующие фрагментам ФВК: ${\text{PWO}}_{{\text{6}}}^{ - },$ ${\text{P}}{{{\text{W}}}_{{\text{2}}}}{\text{O}}_{{\text{5}}}^{ - },$ ${\text{P}}{{{\text{W}}}_{{\text{3}}}}{\text{O}}_{{{\text{12}}}}^{ - },$ ${\text{P}}{{{\text{W}}}_{{\text{4}}}}{\text{O}}_{{{\text{14}}}}^{ - },$ ${\text{P}}{{{\text{W}}}_{{\text{5}}}}{\text{O}}_{{{\text{15}}}}^{ - },$ ${\text{P}}{{{\text{W}}}_{{\text{6}}}}{\text{O}}_{{{\text{17}}}}^{ - },$ ${\text{P}}{{{\text{W}}}_{{\text{7}}}}{\text{O}}_{{{\text{19}}}}^{ - },$ ${\text{P}}{{{\text{W}}}_{{{\text{12}}}}}{\text{O}}_{{{\text{4}}0}}^{ - }.$ Несмотря на схожую методику синтеза катализаторов (et-W, vin-W и W), нарушение структуры гетерополикислоты может происходить за счет сильного взаимодействия между гетерополианионами и поверхностью носителя [4, 19, 46]. Кроме того, согласно [17, 47] стабильность ФВК на кремнеземе зависит от ее загрузки, и устойчивая фаза формируется только при загрузках выше 20 мас. %, а в нашей работе содержание активной фазы на поверхности катализаторов не превышало 10% (табл. 1). Образец et-W, полученный на основе 4-(3'-этилимидазолий)-бутансульфоната, обладает широким набором гетерополианионов с преимущественным сохранением структуры исходной фосфорновольфрамовой кислоты-предшественника. Однако на поверхности образца vin-W происходит частичное разложение ГПК с образованием высокомолекулярных поливольфраматов (табл. 2). Такое различие в стабильности, по нашему мнению, обусловлено кислотностью среды при синтезе ИЖ и твердых катализаторов. Согласно проведенным измерениям кислотность водного раствора 4-(3'-этилимидазолий)-бутансульфоната выше, чем у 4-(3'-винилимидазолий)-бутансульфоната, что может быть связано с низкой растворимостью последнего вследствие сильного межмолекулярного взаимодействия. Учитывая, что синтез ИЖ проводили в воде, а ГПК устойчива в водном растворе только в кислой среде в узком диапазоне pH [1, 17, 48], роль ЦИ заключалась в регулировании кислотности в процессе получения металлсодержащих ИЖ. Кроме того, разложение ГПК могло происходить и на стадии иммобилизации ИЖ на силикагеле, однако протонированные ИЖ с металлсодержащими анионами (схема 1 ) имеют, по-видимому, отличную от свободной ФВК природу взаимодействия с поверхностью кремнезема, что приводит к повышению стабильности гетерополианионов в ряду et-W > vin-W > W.
Кислотные характеристики образцов, измеренные методом ТПД NH3, приведены в табл. 3.
Таблица 3.
Кислотные свойства образцов и носителя согласно данным ТПД NH3
| Образцы | Количество десорбировавшегося аммиака, мкмоль NH3/гкат | |||
|---|---|---|---|---|
| <190°С | 190–250°С | >250°С | всего | |
| W | 47 | 10 | 17 | 74 |
| vin-W | 25 | 31 | 6 | 62 |
| et-W | 22 | 53 | 79 | 154 |
| Силикагель | 7 | 6 | 7 | 20 |
В результате формирования слоя активной фазы происходит увеличение общей кислотности, причем et-W отличается значительным количеством кислотных центров, что, по-видимому, связано с их доступностью благодаря равномерному распределению ионной жидкости на поверхности (по данным СЭМ). Из табл. 3 также следует, что для катализаторов можно выделить область температур до 190°С, в которой происходит десорбция аммиака со слабых бренстедовских центров, представляющих собой, вероятно, гидроксилированные оксиды вольфрама. Десорбцию NH3 в интервале 190–250°С можно связать с более сильными бренстедовскими центрами, относящимися к кислотным фрагментам ГПК или сульфогруппами (–SO3H) в ИЖ. Для катализаторов W и et-W также можно отметить присутствие льюисовских центров (Тдес выше 250°С), что, возможно, вызвано частичным разложением анионов ГПК в ходе анализа. Отсутствие этих центров в катализаторе vin-W может быть обусловлено островковой посадкой этой ИЖ, приводящей к большей стабильности металлсодержащих фрагментов и их меньшей доступностью для адсорбата. Как будет показано далее, катализатор et-W оказался наиболее активным, поскольку, как известно, кислотные центры играют важную роль в активации как сероорганических соединений, так и пероксида водорода. Мы полагаем, что основной вклад в каталитические свойства этого образца дают именно сильные бренстедовские кислотные центры.
Изотермы адсорбции азота на катализаторах представлены на рис. 4. Изотермы относятся к IV типу с петлей гистерезиса H1, что указывает на мезопористую структуру поверхности. Помимо этого резкий перегиб при достаточно высоком относительном давлении (P/P0 > 0.6) свидетельствует о капиллярной конденсации в однородных порах цилиндрической формы.
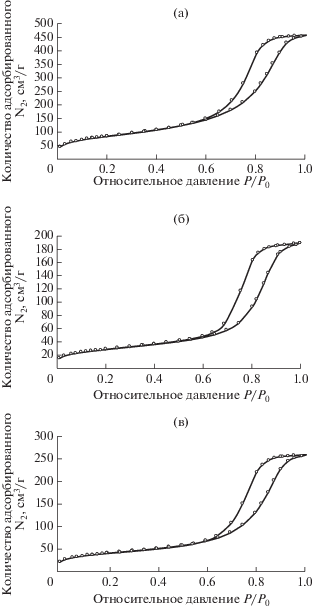
Каталитические свойства композиций
Каталитические свойства образцов определяли в реакциях пероксидного окисления тиофена и ОДС дизельной фракции, предоставленной ОАО “Варьеганнефть”.
На рис. 5 представлены результаты испытания катализаторов (W, et-W и vin-W) в окислении модельной смеси, содержащей 1 мас. % тиофена – наиболее трудноокисляемого сероорганического соединения [2, 32]. Очевидным преимуществом обладает et-W, для которого высокая конверсия субстрата коррелирует с количеством сильных бренстедовских кислотных центров и стабильностью гетерополианионов.
Рис. 5.
Зависимость конверсии тиофена от времени. Условия реакции: 4 ч, 60°С, 0.1 г катализатора, однократная загрузка 0.4 мл H2O2.
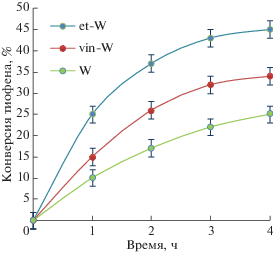
Из литературы известно о влиянии бренстедовской кислотности на активность гетерогенных катализаторов в окислении сероорганических соединений: нейтрализация кислых протонов влечет за собой снижение скорости реакции до полной остановки [49]. Кислотные группы могут быть также центрами адсорбции тиофена и его производных, что было показано в работе [50]. В общем случае катализатор или адсорбент с кислотными (электрофильными) характеристиками обладает большей реакционной способностью по отношению к соединениям серы, присутствующим в углеводородном сырье, в связи с их высокой нуклеофильностью [12], а добавление кислот в реакционную смесь способствует более глубокому протеканию процесса окисления сероорганических соединений в гомогенных и бифазных системах [51, 52]. Модель двойной активации тиофена была предложена на катализаторе [53], содержащем Бренстедовскую ионную жидкость и вольфрамат аммония: ароматичность субстрата нарушалась вследствие формирования водородной связи с ИЖ. В нашей работе, согласно результатам 1H, 13C ЯМР, а также данным хроматографического анализа, органических продуктов окисления тиофена обнаружено не было. Бариево-сульфатным методом установлено появление сульфат-анионов в водной фазе реакционного раствора. Таким образом, в результате процесса происходит разрушение ароматической структуры тиофена с образованием ${\text{SO}}_{{\text{4}}}^{{{\text{2}} - }}$ и, возможно, CO2. Из литературы хорошо известно, что в механизме окисления принимают участие пероксокомплексы, формирующиеся в результате взаимодействия пероксида водорода и гетерополианионов [2, 16, 54]. В ходе процесса ОДС происходит нуклеофильная атака сероорганического субстрата на пероксокомплекс (схема 3 ), в результате чего образуются неустойчивые сульфоксид и сульфон тиофена, которые далее превращаются в сульфат-анионы и различные оксигенаты: стирол, бензойную кислоту или CO2 [33–35, 53].
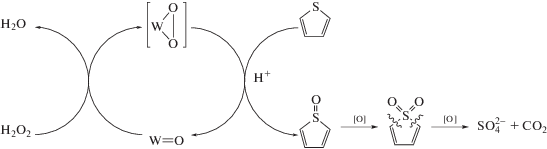
Схема 3 . Схема образования пероксокомплексов и их участие в механизме окисления тиофена.
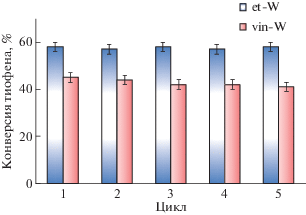
Несмотря на высокую активность катализатора et-W (конверсия тиофена 45%), одним из факторов, препятствующих глубокому окислению субстрата в его присутствии, является побочная реакция разложения пероксида водорода, протекающая под действием металлсодержащих катализаторов. Один из способов снижения расхода пероксида водорода – регулирование кислотности реакционной среды за счет кислотных центров Бренстеда [55]. Другой способ – дробная загрузка окислителя [38, 39], поэтому в настоящей работе добавляли H2O2 двумя порциями. Как видно из рис. 6, такой прием способствует увеличению конверсии тиофена, которая достигает 58% при испытании наиболее активного образца. Кроме того, следует отметить, что катализаторы SILP сохраняют свою активность на протяжении 5 циклов работы (рис. 6).
Рис. 6.
Конверсия тиофена на катализаторах SILP в 5 последовательных циклах окисления при дробной загрузке пероксида водорода 0.2 + 0.2 мл. Условия реакции: 4 ч, 60°С, 0.1 г катализатора.
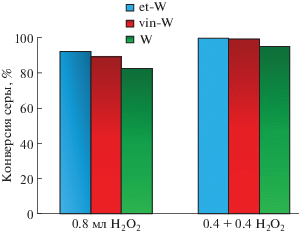
Результаты ОДС дизельной фракции (исходное содержание серы 1080 ppm) представлены на рис. 7. Все катализаторы проявили высокую эффективность при дробной загрузке пероксида водорода, однако только с помощью образцов SILP (et-W и vin-W) удалось получить дизельное топливо с содержанием серы 3 и 7 ppm соответственно. В сравнении со многими известными методиками проведения ОДС [3, 56–60], предложенный способ позволять получать дизельное топливо, соответствующее международным экологическим стандартам (общее содержание S менее 10 ppm).
ЗАКЛЮЧЕНИЕ
Таким образом, предложен способ синтеза гетерогенных катализаторов типа SILP на основе протонированных фосфорновольфрамовой гетерополикислотой цвиттер-ионных жидкостей – 4‑(3'-этилимидазолий)-бутансульфоната и 4-(3'-винилимидазолий)-бутансульфоната. Установлено, что применение протонированной ИЖ в составе активной фазы повышает стабильность гетерополианионов ФВК на поверхности силикагеля. Наиболее активный катализатор et-W обладает высокой стабильностью в окислении тиофена на протяжении пяти последовательных циклов реакции. Дробная загрузка окислителя способствует росту конверсии тиофена и ОДС дизельного топлива. При использовании катализаторов SILP получено дизельное топливо с содержанием серы менее 10 ppm.
Список литературы
da Silva M.J., de Oliveira C.M. // Curr. Catal. 2018. V. 7. P. 26. https://doi.org/10.2174/2211544707666171219161414
Ghubayra R., Nuttall C., Hodgkiss S., Craven M., Kozhevnikova E.F., Kozhevnikov I.V. // Appl. Catal. B: Environ. 2019. V. 253. P. 309. https://doi.org/10.1016/j.apcatb.2019.04.063
Julião D., Valença R., Ribeiro J.C., de Castro B., Balula S.S. // Appl. Catal. A: Gen. 2017. V. 537. P. 93. https://doi.org/10.1016/j.apcata.2017.02.021
Bryzhin A.A., Gantman M.G., Buryak A.K., Tarkhanova I.G. // Appl. Catal. B: Environ. 2019. V. 257. P. 117 938. https://doi.org/10.1016/j.apcatb.2019.117938
Bhadra B.N., Jhung S.H. // Appl. Catal. B: Environ. 2019. V. 259. P. 118 021. https://doi.org/10.1016/j.apcatb.2019.118021
Javadli R., de Klerk A. // Appl. Petrochem. Res. 2012. V. 1. P. 3. https://doi.org/10.1007/s13203-012-0006-6
Rezakazemi M., Zhang Z. Desulfurization Materials / Compr. Energy Syst. Elsevier. 2018. 944 p. https://doi.org/10.1016/B978-0-12-809597-3.00263-7
Dehghan R., Anbia M. // Fuel Process. Technol. 2017. V. 167. P. 99. https://doi.org/10.1016/j.fuproc.2017.06.015
Crandall B.S., Zhang J., Stavila V., Allendorf M.D., Li Z. // Ind. Eng. Chem. Res. 2019. V. 58. P. 19 322. https://doi.org/10.1021/acs.iecr.9b03183
Chandran D., Khalid M., Walvekar R., Mubarak N.M., Dharaskar S., Wong W.Y., Gupta T.C.S.M. // J. Mol. Liq. 2019. V. 275. P. 312. https://doi.org/10.1016/j.molliq.2018.11.051
Gao J., Zhu S., Dai Y., Xiong C., Li C., Yang W., Jiang X. // Fuel. 2018. V. 233. P. 704. https://doi.org/10.1016/j.fuel.2018.06.101
Abdul-Kadhim W., Deraman M.A., Abdullah S.B., Tajuddin S.N., Yusoff M.M., Taufiq-Yap Y.H., Rahim M.H.A. // J. Environ. Chem. Eng. 2017. V. 5. P. 1645. https://doi.org/10.1016/j.jece.2017.03.001
Zhang Y., Li G., Kong L., Lu H. // Fuel. 2018. V. 219. P. 103. https://doi.org/10.1016/j.fuel.2018.01.050
Wei S., He H., Cheng Y., Yang C., Zeng G., Kang L., Qian H., Zhu C. // Fuel. 2017. V. 200. P. 11. https://doi.org/10.1016/j.fuel.2017.03.052
Bazyari A., Khodadadi A.A., Haghighat Mamaghani A., Beheshtian J., Thompson L.T., Mortazavi Y. // Appl. Catal. B: Environ. 2016. V. 180. P. 65. https://doi.org/10.1016/j.apcatb.2015.06.011
Craven M., Xiao D., Kunstmann-Olsen C., Kozhevnikova E.F., Blanc F., Steiner A., Kozhevnikov I.V. // Appl. Catal. B: Environ. 2018. V. 231. P. 82. https://doi.org/10.1016/j.apcatb.2018.03.005
Kozhevnikov I.V. // Chem. Rev. 1998. V. 98. P. 171. https://doi.org/10.1021/cr960400y
García-Gutiérrez J.L., Fuentes G.A., Hernández-Terán M.E., García P., Murrieta-Guevara F., Jiménez-Cruz F. // Appl. Catal. A: Gen. 2008. V. 334. P. 366. https://doi.org/10.1016/j.apcata.2007.10.024
Marcì G., García-López E., Bellardita M., Parisi F., Colbeau-Justin C., Sorgues S., Liotta L.F., Palmisano L. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 13 329. https://doi.org/10.1039/c3cp51142a
Gao S., Li J., Chen X., Abdeltawab A.A., Yakout S.M., Yu G. // Fuel. 2018. V. 224. P. 545. https://doi.org/10.1016/j.fuel.2018.03.108
Wang J., Zhang L., Sun Y., Jiang B., Chen Y., Gao X., Yang H. // Fuel Process. Technol. 2018. V. 177. P. 81. https://doi.org/10.1016/j.fuproc.2018.04.013
Raj J.J., Magaret S., Pranesh M., Lethesh K.C., Devi W.C., Mutalib M.I.A. // J. Clean. Prod. 2019. V. 213. P. 989. https://doi.org/10.1016/j.jclepro.2018.12.207
Yasuda T., Uchiage E., Fujitani T., Tominaga K., Nishida M. // Appl. Catal. B: Environ. 2018. V. 232. P. 299. https://doi.org/10.1016/j.apcatb.2018.03.057
Zhao J., Yu Y., Xu X., Di S., Wang B., Xu H., Ni J., Guo L., Pan Z., Li X. // Appl. Catal. B Environ. 2017. V. 206. P. 175. https://doi.org/10.1016/j.apcatb.2017.01.026
Plechkova N.V., Seddon K.R., eds. Ionic Liquids Completely UnCOILed. John Wiley & Sons. Inc. Hoboken. N.J. 2015. 548 p. https://doi.org/10.1002/9781118840061
Steinrück H.-P., Wasserscheid P. // Catal. Lett. 2015. V. 145. P. 380. https://doi.org/10.1007/s10562-014-1435-x
More S., Jadhav S., Salunkhe R., Kumbhar A. // J. Mol. Catal. 2017. V. 442. P. 126. https://doi.org/10.1016/j.mcat.2017.08.023
Fehér C., Tomasek S., Hancsók J., Skoda-Földes R. // Appl. Catal. B: Environ. 2018. V. 239. P. 52. https://doi.org/10.1016/j.apcatb.2018.08.013
Rufete-Beneite M., Haumann M., Román-Martínez M.C. // J. Mol. Catal. 2018. V. 453. P. 31. https://doi.org/10.1016/j.mcat.2018.04.031
Ismagilov Z., Yashnik S., Kerzhentsev M., Parmon V., Bourane A., Al-Shahrani F.M., Hajji A.A., Koseoglu O.R. // Catal. Rev. 2011. V. 53. P. 199. https://doi.org/10.1080/01614940.2011.596426
Luo S.-C., Sun S., Deorukhkar A.R., Lu J.T., Bhattacharyya A., Lin I.J.B. // J. Mater. Chem. 2011. V. 21. P. 1866. https://doi.org/10.1039/C0JM02875D
Li M., Zhang M., Wei A., Zhu W., Xun S., Li Y., Li H., Li H. // J. Mol. Catal. A Chem. 2015. V. 406. P. 23. https://doi.org/10.1016/j.molcata.2015.05.007
Treiber A. // J. Org. Chem. 2002. V. 67. P. 7261. https://doi.org/10.1021/jo0202177
Kong L., Li G., Wang X. // Catal. Today. 2004. V. 93–95. P. 341–345. https://doi.org/10.1016/j.cattod.2004.06.016
Chen L., Li F. // Energy & Fuels. 2010. V. 24. P. 3443.https://doi.org/10.1021/ef1002205
Kreja L. // Monatshefte Fur Chemie / Chem. Mon. 1987. V. 118. P. 717. https://doi.org/10.1007/BF00809221
Kreja L., Plewka A. // Angew. Makromol. Chem. 1982. V. 102. P. 45. https://doi.org/10.1002/apmc.1982.051020106
Pignatello J.J. // Environ. Sci. Technol. 1992. V. 26. P. 944. https://doi.org/10.1021/es00029a012
Zhang H., Fei C., Zhang D., Tang F. // J. Hazard. Mater. 2007. V. 145. P. 227. https://doi.org/10.1016/j.jhazmat.2006.11.016
Брыжин А.А., Тарханова И.Г, Маслаков К.И., Николаев С.А., Гуревич С.А., Кожевин В.М., Явсин Д.А., Гантман М.Г., Ростовщикова Т.Н. // Журн. физ. хим. 2019. Т. 93. № 10. С. 1575. https://doi.org/10.1134/S0036024419100029
Tarkhanova I.G., Bryzhin A.A., Gantman M.G., Yarovaya T.P., Lukiyanchuk I.V., Nedozorov P.M., Rudnev V.S. // Surf. Coatings Technol. 2019. V. 362. P. 132. https://doi.org/10.1016/j.surfcoat.2019.01.101
Тарханова И.Г., Анисимов А.В., Буряк А.К., Брыжин А.А., Али-Заде А.Г., Акопян А.В., Зеликман В.М. // Журн. нефтехим. 2017. Т. 51. № 5. С. 536. https://doi.org/10.1134/S0965544117100164
Elwell W.T., Wood D.F. Qualitative Detection / Anal. Chem. Molybdenum Tungsten. Elsevier. 1971. P. 15. https://doi.org/10.1016/B978-0-08-016673-5.50008-9
Zhu W., Huang W., Li H., Zhang M., Jiang W., Chen G., Han C. // Fuel Process. Technol. 2011. V. 92. P. 1842. https://doi.org/10.1016/j.fuproc.2011.04.030
Ranga Rao G., Rajkumar T., Varghese B. // Solid State Sci. 2009. V. 11. P. 36. https://doi.org/10.1016/j.solidstatesciences.2008.05.017
Sopa M., Wącław-Held A., Grossy M., Pijanka J., Nowińska K. // Appl. Catal. A: Gen. 2005. V. 285. P. 119. https://doi.org/10.1016/j.apcata.2005.02.013
Izumi Y. // J. Catal. 1983. V. 84. P. 402. https://doi.org/10.1016/0021-9517(83)90011-8
Wang S.-S., Yang G.-Y. // Chem. Rev. 2015. V. 115. P. 4893. https://doi.org/10.1021/cr500390v
Zheng H.-Q., Zeng Y.-N., Chen J., Lin R.-G., Zhuang W.-E., Cao R., Lin Z.-J. // Inorg. Chem. 2019. V. 58. P. 6983. https://doi.org/10.1021/acs.inorgchem.9b00604
Lopes A.R., de P. Scheer A., Silva G.V., Yamamoto C.I. // Matéria (Rio Janeiro). 2016. V. 21. P. 407. https://doi.org/10.1590/S1517-707620160002.0038
Chen L., Guo S., Zhao D. // Chin. J. Chem. Eng. 2007. V. 15. P. 520. https://doi.org/10.1016/S1004-9541(07)60118-9
Al-Shahrani F., Xiao T., Llewellyn S.A., Barri S., Jiang Z., Shi H., Martinie G., Green M.L.H. // Appl. Catal. B: Environ. 2007. V. 73. P. 311. https://doi.org/10.1016/j.apcatb.2006.12.016
Zhang B., Jiang Z., Li J., Zhang Y., Lin F., Liu Y., Li C. // J. Catal. 2012. V. 287. P. 5. https://doi.org/10.1016/j.jcat.2011.11.003
Yahya R., Craven M., Kozhevnikova E.F., Steiner A., Samunual P., Kozhevnikov I. V., Bergbreiter D.E. // Catal. Sci. Technol. 2015. V. 5. P. 818. https://doi.org/10.1039/C4CY01394H
Oh H.S., Kim J.-J., Kim Y.-H. // J. Chem. Eng. 2016. V. 33. P. 885. https://doi.org/10.1007/s11814-015-0204-x
Mirante F., Alves A.C., Julião D., Almeida P.L., Gago S., Valença R., Ribeiro J.C., de Castro B., Granadeiro C.M., Balula S.S. // Fuel. 2020. V. 259. P. 116 213. https://doi.org/10.1016/j.fuel.2019.116213
Banisharif F., Dehghani M.R., Capel-Sanchez M.C., Campos-Martin J.M. // Catal. Today. 2019. V. 333. P. 219. https://doi.org/10.1016/j.cattod.2018.07.009
Andevary H.H., Akbari A., Omidkhah M. // Fuel Process. Technol. 2019. V. 185. P. 8. https://doi.org/10.1016/j.fuproc.2018.11.014
Julião D., Mirante F., Ribeiro S.O., Gomes A.C., Valença R., Ribeiro J.C., Pillinger M., de Castro B., Gonçalves I.S., Balula S.S. // Fuel. 2019. V. 241. P. 616. https://doi.org/10.1016/j.fuel.2018.11.095
Ribeiro S.O., Granadeiro C.M., Almeida P.L., Pires J., Capel-Sanchez M.C., Campos-Martin J.M., Gago S., de Castro B., Balula S.S. // Catal. Today. 2019. V. 333. P. 226. https://doi.org/10.1016/j.cattod.2018.10.046
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ