

Кинетика и катализ, 2020, T. 61, № 5, стр. 741-748
Механохимический синтез – альтернативный эффективный метод приготовления композитных катализаторов
О. С. Морозова a, *, А. А. Фирсова a, Ю. П. Тюленин a, Г. А. Воробьева a, А. В. Леонов b
a ФИЦ химической физики им. Н.Н. Семенова РАН
117977 Москва, ул. Косыгина, 4, Россия
b МГУ им. М.В. Ломоносова, Химический факультет
119911 Москва, Ленинские горы, 1/3, Россия
* E-mail: om@chph.ras.ru
Поступила в редакцию 18.12.2019
После доработки 20.03.2020
Принята к публикации 23.05.2020
Аннотация
Механохимический синтез в шаровой мельнице применен для приготовления нанокомпозитных катализаторов Cu(CuO)–CeO2 из смесей СеО2 и допантов: металлической меди и оксидов меди различной морфологии и состава (CuO и CuO с добавками Cu2O в количестве 4 и 16.5 мас. %). Полученные материалы изучены методами рентгенофазового анализа, сканирующей электронной микроскопии, температурно-программированного восстановления в СО, Н2 и С2Н6 (ТПВ-СО, ТПВ-Н2 и ТПВ-С2Н6) и испытаны в качестве катализаторов реакций селективного окисления СО в избытке Н2 (CO-PROX) и глубокого окисления С2Н6. Методом ТПВ установлено, что в синтезированных образцах появляются новые формы кислорода с большей реакционной способностью в области низких температур по отношению к СО, Н2 и С2Н6. Показано, что конверсия СО мало зависит от природы исходных медьсодержащих компонентов в составе смеси с СеО2, однако величина конверсии С2Н6 при низких температурах чувствительна к составу допантов. Максимальная конверсия С2Н6 при 400°С (91.4%) наблюдается на Cu–CeO2-образце, минимальная (54.2%) – на CuO–CeO2. Продемонстрировано, что механохимический синтез является универсальным методом приготовления оксидных медно-цериевых катализаторов.
ВВЕДЕНИЕ
В настоящее время оксидные медно-цериевые катализаторы широко используются в различных промышленных и экологически важных процессах, таких, например, как окисление летучих органических соединений и сажи, глубокое окисление углеводородов, реакция конверсии водяного пара, низкотемпературное окисление СО в избытке водорода (реакция CO-PROX) и многих других [1–7]. Активность этих катализаторов связывают с образованием межфазной границы CuOх–CeO2, на которой формируются центры Cu–O–Ce, и с высокой подвижностью кислорода в решетке СеО2, обусловленной большим количеством кислородных вакансий. Введение меди в диоксид церия способствует возникновению таких вакансий. Считается [8–10], что именно центры на межфазной границе стимулируют появление в катализаторе подвижного кислорода с высокой реакционной способностью. Максимальная каталитическая активность достигается на образцах, содержащих 4–5 мас. % CuO [13, 14].
Предложены различные способы синтеза оксидных медно-цериевых систем, направленные на создание катализаторов, активных в области низких температур (120–180°С), обладающих протяженной межфазной границой CuOх–CeO2 [11, 12]. В основном эти способы энергозатратны, длительны и сопровождаются образованием экологически вредных сточных вод.
В работе [16] был предложен экологически чистый (безотходный) и одноступенчатый метод приготовления оксидных медно-цериевых катализаторов с помощью прямого механохимического синтеза (МС) из смесей индивидуальных компонентов – оксидов СеО2, CuO и металлической Cu. Благодаря механической активации (интенсивному перемешиванию и локальным пластическим деформациям под высоким давлением) происходило модифицирование поверхности частиц СеО2 ионами меди, аналогичное тому, которое получали другими методами синтеза оксидных медно-цериевых систем. С помощью МС удалось создать систему CuOх–CeO2 с протяженной межфазной границей, физико-химические и каталитические свойства которой в реакции CO-PROX были практически идентичны тем, что описаны в литературе для образцов, синтезированных иными способами [17, 18].
Дальнейшие исследования позволили расширить состав медьсодержащих компонентов для получения катализаторов методом МС и испытать их не только в окислении СО (CO-PROX), но и в глубоком окислении этана. Ранее [19] была показана важная роль оксидов CuO в катализаторе CuO–CeO2/Al2O3 для реакций глубокого окисления.
В настоящей работе в качестве модифицирующих добавок к СеО2 были использованы оксиды меди CuO, содержавшие в своем составе ионы одновалентной меди (4 и 16.5 мас. % Cu2O). Это дало возможность проверить предположение о важнейшей роли ионов Cu1+ в образовании активных центров для реакции CO-PROX. Кроме того, морфология таких оксидных систем (размер и форма частиц) сильно отличалась от морфологии уже изученных CuO и Cu. Это позволяло выявить влияние и фазового состава, и морфологии исходных оксидов меди на формирование и физико-химические свойства оксидных медно-цериевых катализаторов.
Полученные с помощью МС катализаторы были исследованы методами рентгенофазового анализ РФА, сканирующей электронной микроскопии СЭМ, и температурно-программированного восстановления ТПВ в СО, Н2 и С2Н6. Удельную поверхность определяли методом БЭТ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для синтеза катализаторов использовали СеО2, приготовленный пиролизом Ce(NO3)3 ⋅ 6H2O (“Aldrich”, 99%) на воздухе [17]. Диоксид церия с удельной поверхностью Sуд = 89 м2/г состоял из кубической фазы СеО2 (JCPDS 89-8434). В качестве медьсодержащих добавок применяли металлическую медь Cu – дендритную электролитическую (“GGP”, 99.9%), фаза (JCPDS 89-2838), и различные оксиды меди CuO(I–III). CuO(I) был получен пиролизом Cu(NO3)2 ⋅ 2.5 H2O (марка “ч. д. а.”) [18], состоял на 100% из моноклинной фазы CuO (JCPDS 5-661); Sуд = 1 м2/г. CuO(II) был приготовлен разложением Cu(NO3)2 ⋅ 2.5 H2O (“Aldrich”, 99%) в плазменном разряде и содержал 96 мас. % фазы CuO (JCPDS 5-661) и 4 мас. % фазы Cu2O (JCPDS 5-667); Sуд = 6 м2/г. CuO(III) (“Advanced Powder Technology LLC”, Томск, Россия) содержал 82 мас. % фазы CuO (JCPDS 5-661), 16.5 мас. % фазы Cu2O (JCPDS 5-667) и 1.5 мас. % фазы Cu; Sуд = 12 м2/г.
МС проводили в течение 30–90 мин в шаровой вибромельнице, состоящей из стального контейнера, прикрепленного к вибратору и заполненного стальными шарами диаметром 3–5 мм и весом 15.3 г. Энергонапряженность составляла 1 Вт/г, амплитуда колебаний – 7.25 мм, частота колебаний – 50 Гц [17, 18]. В мельницу помещали 1.8 г смеси 8 мас. % Cu и 92 мас. % СеО2 или 10 мас. % CuO и 90 мас. % СеО2. Медьсодержащие добавки были взяты в двойном избытке по сравнению с оптимальным количеством для того, чтобы следить за изменением фазового состава образцов на разных стадиях обработки. Воспроизводимость характеристик образцов, полученных методом МС, проверяли по результатам ТПВ и измерениям каталитической активности.
Удельную поверхность определяли методом БЭТ по низкотемпературной адсорбции аргона. Химический анализ образцов проводили с помощью атомно-абсорбционной спектроскопии (ААС), используя прибор ThermoScientific iCAP 6300 Duo (“Thermo Scientific”, США).
Морфологию образцов изучали методом сканирующей электронной микроскопии с использованием микроскопов SEM 515 (“Philips”, Нидерланды) и JSM 6390LV (“JEOL”, Япония). Перед измерениями образец крепили на двухсторонний проводящий скотч.
Фазовый состав исходных оксидов и образцов на разных стадиях МС исследовали на дифрактометре ДРОН-3 (“Буревестник”, Россия) в области углов 2θ = 20°–90° (CuKα-излучение, графитовый монохроматор), откалиброванном по линии (112) порошка SiO (α-кварц, межплоскостное расстояние – 1.818 Å). Количественный фазовый анализ проводили с помощью компьютерных программ [20]. Размер кристаллитов рассчитывали по формуле Шеррера или, в случае СеО2, по программе [20].
Температурно-программированное восстановление исходных оксидов и синтезированных образцов в СО, Н2 и С2Н6 (ТПВ-СО, ТПВ-Н2 и ТПВ-С2Н6 соответственно) осуществляли с использованием проточного дифференциального сканирующего калориметра NETZSCH STA 449C (“NETZSCH”, Германия), оснащенного масс-спектрометром AEOLOS-32. Эксперименты проводили в интервале 50–400оС со скоростью нагрева 10°С/мин. В качестве восстановителей применяли газовые смеси, содержащие 10 об. % Н2, СО или С2Н6 в аргоне. Газом-носителем был гелий. Все газы были чистоты 99.999%. Детали эксперимента приведены в [17].
Каталитическую активность в реакции окисления СО в избытке Н2 тестировали в интервале температур 20–400°С в проточном микрореакторе. Реакционную смесь состава 98 об. % H2, 1 об. % СО и 1 об. % O2 пропускали со скоростью 40 мл/мин через трубчатый кварцевый микрореактор (диаметр 3 мм), содержавший 20 мг катализатора. Детально эксперимент описан в [18]. Каталитическую активность в реакции глубокого окисления С2Н6 проверяли в интервале температур 100–500°С. Реакционную смесь состава 0.5 об. % C2H6, 5 об. % O2 и 94.5 об. % N2 пропускали со скоростью 16 мл/мин через трубчатый кварцевый микрореактор (диаметр 3 мм), содержавший 20 мг катализатора, разбавленного 100 мг SiO2. Анализ продуктов реакции осуществляли хроматографически на приборе Кристалл-2000 (Россия) с использованием двух колонок, наполненных молекулярными ситами 13 А и Porapak QS. В обоих случаях предварительную обработку образцов не проводили.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Морфологические и структурные особенности композитов, полученных с помощью МС
На рис. 1 представлены микрофотографии СЭМ исходного СеО2 (рис. 1д) и модифицирующих медьсодержащих добавок – металлической Сu (рис. 1а) и оксидов СuО(I) (рис. 1б), СuО(II) (рис. 1в) и СuО(III) (рис. 1г), а также образца CuO (III)–CeO2 после 60 мин МС (рис. 1е). Частицы исходного СеО2 были разнообразны по размеру и форме, маленькие частицы располагались на поверхности более крупных частиц. Механическая активация СеО2 сопровождалась уплотнением и укрупнением частиц порошка до 20–30 мкм, что приводило к уменьшению удельной поверхности с 89 до 46.5 м2/г после 60 мин МС. Порошок Cu (рис. 1a) представлял собой агломерированные плоские частицы длиной около 0.6 мкм и толщиной ~0.14 мкм, а оксид меди CuO(I) – хрупкие шарообразные агломераты плоских частиц длинной 1–3 мкм и толщиной ~0.1 мкм (рис. 1б), которые легко разрушались при помоле. Большие рыхлые частицы CuO(II) (рис. 1в) состояли из мелких кубических частиц. В образце CuO(III) (рис. 1г) обнаружены два типа округлых или овальных частиц: мелкие диаметром 100–150 нм и крупные размером в несколько десятков мкм. Морфология образца CuO(III)–CeO2 после 60 мин МС полностью соответствует изменению в морфологии СеО2, являющегося основным компонентом катализаторов (90–92 вес. %). Во всех случаях мелкие частицы СеО2 “слипались” в крупные компактные частицы композита с размером 20–30 мкм, удельная поверхность после 60 мин МС составляла от 36 до 44 м2/г. Природа добавки практически не влияла на морфологию катализаторов. Отдельные мелкие фрагменты металлической меди и оксидов меди в плотных частицах СеО2 не наблюдались.
Рис. 1.
Микрофотографии СЭМ: а – кристаллы Cu; б – CuO(I); в – CuO(II); г – CuO(III); д – оксид серия СеО2; е – порошок CuO(III)–CeO2 после МС в течение 60 мин.

На рис. 2 представлены рентгеновские дифрактограммы исходных медьсодержащих смесей (рис. 2а) и порошков, образовавшихся после 60 мин МС (рис. 2б). На дифрактограммах последних присутствовали линии, относящиеся к кубической фазе СеО2 (JCPDS 34394) с a = 5.414 Å, моноклинной фазе CuO и/или кубической фазе Cu. Линии, характеризующие кубическую фазу Cu2O, не обнаружены либо по причине низкой концентрации этого компонента, либо из-за окисления Cu2O в CuO во время МС.
Рис. 2.
Рентгеновские дифрактограммы исходных медьсодержащих смесей с СеО2 (а) и соответствующих нанокомпозитов после 60 мин МС (б): 1 – CuO(I)–СеО2; 2 – CuO(II)–СеО2; 3 – CuO(III)–СеО2; 4 – Cu–СеО2.
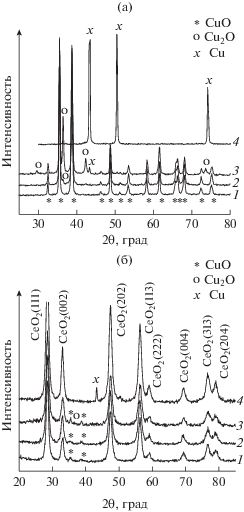
Анализ дифрактограмм, проведенный в соответствии с [20], показал, что размер кристаллитов СеО2 составлял 10–14 нм и практически не зависел от времени МС и присутствия того или иного модификатора. Концентрация микронапряжений в оксиде церия возрастала от 0.2–0.3 до 0.8–1.2% в процессе МС, однако прямой зависимости этой величины от времени МС не было выявлено. В рентгеновских дифрактограммах наблюдалось значительное уширение рентгеновских пиков CuO и Cu и уменьшение их интенсивности по мере увеличения времени МС вплоть до почти полного их исчезновения. Несмотря на снижение интенсивности соответствующих рентгеновских пиков, ААС-анализ показал неизменность концентрации меди в образцах после МС. Размер кристаллитов фаз CuO и Cu оценивали по формуле Шеррера: после 90 мин МС величина частиц CuO уменьшилась с 30 до 7–10 нм, а Cu – с 80 до ~16 нм. По-видимому, микроструктура синтезированных методом МС образцов представляет собой нанокомпозит, состоящий из матрицы СеО2 (нанофрагментов СеО2 размером 10–14 нм) и кристаллитов Cu или CuO, распределенных на поверхности или в межзеренных (межкристаллитных) границах СеО2. Часть этих кристаллитов является рентгеноаморфными, что согласуется с данными работ [6–14].
Окислительная способность кислорода решетки катализаторов CuO(Cu)–СеО2 (ТПВ-СО, ТПВ-Н2 и ТПВ-С2Н6) и фазовые превращения, сопровождающие процессы окисления
Механизм Марса–ван Кревелена считается наиболее вероятным для протекания окислительных реакций на оксидных медно-цериевых катализаторах [1–6], поэтому исследование окислительной способности кислорода решетки синтезированных композитов было необходимым шагом в изучении их каталитической активности. На рис. 3 приведены результаты масс-спектрального анализа продуктов окисления СО (рис. 3а), Н2 (рис. 3б) и С2Н6 (рис. 3в) в режиме температурно-программированного восстановления образцов, полученных МС в течение 60 мин. Кривые изменения интенсивности масс-спектральных сигналов на линиях m/е = 44 (СО2) и m/е = 18 (Н2О) записаны в ходе ТПВ-СО, ТПВ-Н2 и ТПВ-С2Н6 соответственно. Процессы окисления СО и Н2 кислородом решетки композитов, или, что тоже самое, восстановления всех синтезированных катализаторов в СО и в Н2, происходили в две стадии (рис. 3а и 3б) в низко- и высокотемпературных областях.
Рис. 3.
Изменение интенсивности масс-спектрального сигнала на линиях m/e = 44 (СО2) и m/e = 18 (Н2О) в процессе ТПВ для нанокомпозитов Cu(CuO)–CeO2 (60 мин МС): а – ТПВ-СО (СО2, m/e = 44): 1 – CuO(I)–CeO2; 2 – CuO(II)–CeO2; 3 – CuO(III)–CeO2; 4 – Cu–CeO2; б – ТПВ-Н2 (Н2О, m/e = 18): 1 – CuO(I)–CeO2; 2 – CuO(II)–CeO2; 3 – CuO(III)–CeO2; 4 – Cu–CeO2; 5 – CuO(III)–CeO2, смесь перетерта в ступке перед МС; в – ТПВ-С2Н6 (СО2, m/e = = 44): 1 – Cu–CeO2; 2 – Cu–CeO2, повторный опыт; 3 – Cu–CeO2 после предварительной обработки образца в смеси О2/Ar; 4 – CuO(I)–CeO2.
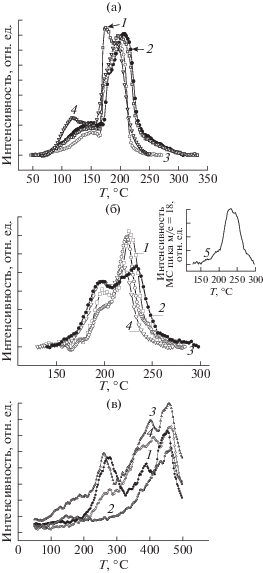
В табл. 1 приведены температуры низко- и высокотемпературных максимумов концентраций продуктов реакции для кривых, представленных на рис. 3а и 3б, и соотношения соответствующих площадей низко- и высокотемпературных пиков.
Таблица 1.
Некоторые характеристики ТПВ-Н2 и ТПВ-СО образцов Cu–CeO2 и CuO–CeO2
| № | Образец | ТПВ-СО | ТПВ-Н2 | ||||
|---|---|---|---|---|---|---|---|
| Т1, °С | Т2, °С | S1/S2 | Т1, °С | Т2, °С | S1/S2 | ||
| 1 | Cu–CeO2 | 120 | ~194.6 | 0.58 | 194 | 217.6 | 0.92 |
| 2 | CuO(I)–CeO2 | ~150 | 178–200 | 0.41 | ~198 | 225.5 | 0.42 |
| 3 | CuO(II)–CeO2 | ~150 | 185–209 | 0.42 | ~228 | ~259 | 0.92 |
| 4 | CuO(III)–CeO2 | ~159 | 199–219 | 0.18 | ~199 | ~223 | 0.25 |
Двухпиковая кривая восстановления является характерной особенностью именно композитных катализаторов. Для сравнения на вставке рис. 3б показано изменение интенсивности масс-спектрального сигнала m/е = 18 (Н2О) для смеси оксидов CuO(III) и CeO2 (кривая 5), просто перетертых в агатовой ступке. Здесь присутствует только один высокотемпературный пик (ср. с кривой 4). Следует отметить, что аналогичная “двухпиковая” форма кривых ТПВ отмечается во многих работах, посвященных катализаторам Cu/CuO–CeO2, приготовленным разными методами, например, в [14, 21–28]. Температурные области восстановления образцов в СО и Н2 различаются. В случае ТПВ-СО первая стадия восстановления протекает при 65–165°С, вторая – при 165–300°С, в случае ТПВ-Н2 – при 140–208 и 208–300°С соответственно. По данным РФА после второй стадии обнаружены только фазы металлической меди и СеО2, т.е. оксиды меди полностью восстанавливались. Напротив, после первой стадии ТПВ фаза металлической меди не наблюдалась, а интенсивность рентгеновских линий, относящихся к CuO, практически не изменялась. Эти результаты позволили предположить, что в решетке синтезированных композитов имелось, по крайней мере, два типа локализации активного кислорода. Были проведены специальные эксперименты ТПВ с попеременным восстановлением образца CuO(III)–CeO2 в СО и Н2: ТПВ-СО останавливали в момент окончания первой (низкотемпературной) стадии восстановления, образец остужали в потоке чистого Не и после этого проводили ТПВ-Н2. При этом на кривой Н2-ТПВ исчезал низкотемпературный пик в области 140–208°С. Таким образом было показано, что один и тот же “активный” кислород участвует в низкотемпературном окислении и СО, и Н2 [17]. Вероятно, именно этот кислород может быть локализован на границе фаз оксидов меди и церия, и именно этот “активный” кислород участвует в реакции CO-PROX.
Максимальная конверсия СО на изученных катализаторах достигалась при 150–160°С, что близко к температуре максимальной конверсии СО в ТПВ-экспериментах. Окисление водорода происходило при более высокой температуре. Именно это различие, отчетливо проявляющееся при ТПВ, позволяет осуществлять селективное окисление СО в присутствии Н2.
На рис. 3в приведены кривые ТПВ-С2Н6 образцов Сu–CeO2 (кривые 1–3) и СuO(I)–CeO2 (кривая 4), полученных МС в течение 60 мин. Из рис. 3в следует, что окисление С2Н6 кислородом решетки композитов также протекало в две стадии. Специальные эксперименты, проведенные для Сu–CeO2, показали, что в первой, низкотемпературной, стадии участвовал подвижный высокореакционный кислород решетки катализатора. Этот тип кислорода можно было полностью удалить из образца в первом цикле ТПВ-С2Н6 (ср. кривые 1 и 2 на рис. 3в). Обработка катализатора в смеси О2/Ar компенсировала удаленный кислород катализатора за счет кислорода газовой фазы, и кривая ТПВ-С2Н6 (рис. 3в, кривая 3) приобретала прежний вид. Сравнение кривых ТПВ-С2Н6 для образцов Сu–CeO2 и СuO(I) –CeO2 (кривые 3 и 4) показало, что они практически идентичны, следовательно низкотемпературное окисление этана на этих катализаторах протекает с участием одного и того же типа кислорода. Этот результат еще раз указывает на высокую подвижность “активного” кислорода в МС-катализаторах и возможность компенсации прореагировавшего кислорода за счет кислорода газовой фазы. Такая характерная особенность систем Cu/CuO–CeO2, отмечается во многих публикациях, например [1, 8, 14].
Высокотемпературные пики на кривых ТПВ были отнесены к восстановлению нанофрагментов CuO, распределенных в матрице СеО2. На это указывает обнаруженное методом РФА полное восстановление CuO в Cu после второй стадии ТПВ-СО, -Н2 и -С2Н6 для всех CuO-содержащих композитов. Ранее было установлено, что даже частичного восстановления оксида церия в этих условиях не происходило [22].
Каталитическая активность образцов CuO(Cu)–СеО2, полученных методом МС
В табл. 2 приведены данные по максимальной конверсии СО в СО2 в условиях селективного окисления СО в избытке Н2 на композитных катализаторах Cu–CeO2 и CuO–CeO2 при ~160°C. С увеличением длительности механической активации во время МС конверсия СО возрастала, проходя через максимум для образцов Cu–CeO2 и CuO(III)–CeO2 после 60 мин обработки. При механической активации смесей металлической меди или ее оксидов с диоксидом церия происходит модифицирование поверхности СеО2 ионами меди, которое по данным ТПВ составляет примерно <1, 1.5 и ~4.5% при МС в течение 30, 60 и 90 мин соответственно. Оказалось, что присутствие в CuO(III) “биографического” Cu2O, предполагаемой ”активной” составляющей катализаторов Cu/CuO–CeO2 [23, 13, 15, 29–31 ], не влияет на его активность. Возможно, что происходит окисление этой фазы до CuO кислородом СеО2 в процессе МС. Методом РФА показано, что катализаторы, извлеченные из реактора на максимуме их активности (160–170°С), содержали фазы Cu и CuO в практически равном количестве, а катализаторы, извлеченные после реакции (230–240°С) – только фазу металлической меди.
Таблица 2.
Конверсия СО при 160°С (реакция PROX) на образцах Cu–CeO2 и CuO–CeO2
| № | Образец | Конверсия СО, % | ||
|---|---|---|---|---|
| МС 30 мин | МС 60 мин | МС 90 мин | ||
| 1 | Cu–CeO2 | 72 | 95 | 89 |
| 2 | CuO(I)–CeO2 | 83 | 92 | 94 |
| 3 | CuO(II)–CeO2 | 85 | 83 | 85 |
| 4 | CuO(III)–CeO2 | 91 | 95 | 81 |
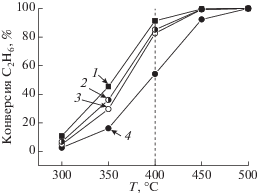
Реакция глубокого окисления С2Н6 оказалась более чувствительна к исходному составу медьсодержащих катализаторов. Сопоставление данных по конверсии С2Н6 (рис. 4) на исследованных системах позволяет сделать вывод о том, что лучшим катализатором оказался композит Cu–CeO2, а наименее активным – CuO(I)–CeO2: при 400°С конверсия С2Н6 составила 91.5 и 54% соответственно. Каталитические свойства двух других образцов были близки. Конверсия этана на всех катализаторах достигала 100% при 500°С.
Обсуждение результатов
Оксидные медно-цериевые катализаторы были приготовленные методом МС из основного компонента СеО2 и медьсодержащих добавок различного фазового состава и морфологии. Две из этих добавок – элементарная медь и чистый оксид меди – были подробно исследованы нами ранее [17, 21]. Было показано, что физико-химические свойства образцов Cu–СеО2 и CuO–СеО2, приготовленных МС, аналогичны свойствам оксидных систем, синтезированных другими способами, описанными в литературе, такими как соосаждение из солей соответствующих металлов или пропитка носителей растворами солей меди с последующим прокаливанием [3, 10, 14, 31], а также новыми методами – лазерной aбляцией и напылением [6, 9, 11, 32]. Все эти способы, так же как и МС, нацелены на создание протяженных межфазных границ, на которых и локализуются центры, активные в селективном окислении СО в избытке Н2 [7, 14, 15]. В случае применения МС сама методика способствует интенсивному перемешиванию материала. Близкий контакт между компонентами катализатора, распределение модифицирующей добавки в матрице основного компонента, СеО2, очень высокие локальные давления – все это способствует модифицированию поверхности диоксида церия ионами меди. Механическая активация как таковая в сочетании с модифицированием поверхности [28, 33] способствует возникновению кислородных вакансий на поверхности СеО2. Ранее в [17] мы показали методом РФЭС, что поверхность Cu–СеО2 уже после 30 мин МС содержит ионы Cu1+, Се4+ и Се3+, их концентрация зависит от времени МС. Одновременно в композитах фиксировалось появление нового поверхностного состояния кислорода с энергией связи 530.8 эВ (в дополнение к основному с Есв = 529.3 эВ), которое интерпретировано в литературе как: “ионы кислорода, находящиеся в необычной координации в CeOx (x < 2) [34]”; кислород поверхности [23, 30], способный к окислению СО; “сильно поляризованные ионы кислорода на поверхности (или в подповерхностном слое) нанокристаллитов, находящиеся в особо низкой координации” [24]. Такой же тип кислорода был обнаружен в катализаторах Cu/CuO–CeO2, приготовленных различными способами [23, 24, 30, 34, 35]. Возникновение “нового” состояния кислорода, участвующего в окислении СО, Н2 и С2Н6 при более низкой температуре, было зафиксировано также методами ТПВ-СО, ТПВ-Н2 и ТПВ-С2Н6 в настоящей работе. Не вызывает сомнений, что именно поверхность СеО2, модифицированная атомами или ионами меди, является источником такого реакционноспособного кислорода. Как показано на рис. 3в (кривая 5), простое перемешивание компонентов катализатора недостаточно для взаимодействия оксидов и не сопровождается появлением “активного кислорода”.
Важнейшим результатом представленной работы явилось то, что композиты, полученные из разных исходных медьсодержащих соединений, обладают очень сходными физико-химическими и, следовательно, каталитическими свойствами в реакции СО-PROX. Если на ранних стадиях МС каталитическая активность образцов несколько различалась, что можно объяснить разным размером частиц исходных компонентов и их фазовым составом, то в дальнейшем сам процесс МС приводит, вероятно, к “усреднению” поверхностного состава нанокомпозитов. В реакционной среде такая “усредненная” активная поверхность формируется окончательно. Доказательством такого предположения служит отсутствие каких-либо значительных различий в каталитических свойствах четырех изученных образцов: после 90 мин их каталитическая активность была практически одинаковой. Это указывает на своего рода “унификацию” поверхности нанокомпозитов после определенной обработки независимо от первоначального фазового состава и морфологии исходных материалов.
В [36] было показано, что глубокое окисление предельных углеводородов на оксидных медно-цериевых катализаторах также определяется подвижностью активного кислорода СеО2, которая, в свою очередь, связана с модифицированием поверхности СеО2 ионами меди. Два фактора могут существенно влиять на каталитические свойства композитов в этой реакции: состав поверхности образцов и подвижность кислорода. Согласно [37], центром активации этана на поверхности является ион меди Cu1+, с которым этан образует структуру типа сигма-комплекса. Ранее методом рентгеновской фотоэлектронной спектроскопии ионы меди Cu1+ были обнаружены на поверхности МС-катализатора Cu–CeO2 [17]. Также они присутствовали в исходных образцах CuO(II) и CuO(III). Исследование методом РФА катализаторов после разных стадий ТПВ-С2Н6 показало, что восстановление CuO в образце CuO(I)–СеО2 происходит при температуре выше 350°С. Следовательно, образование необходимых активных центров затруднено, и их мало. В результате ТПВ-С2Н6 было установлено, что количество активного кислорода в Cu–CeO2 более чем в 3 раза превышало таковое в CuO(I)–CeO2. Это способствовало большей конверсии С2Н6 на образце Cu–CeO2 в области относительно низких температур. При высокой температуре подвижность кислорода существенно возрастала, дополнительно в реакции начинал работать кислород решетки катализатора, и каталитическая активность всех исследованных образцов достигала максимума при 500°С.
ЗАКЛЮЧЕНИЕ
Была показана эффективность метода МС в приготовлении катализаторов Cu–CeO2 и CuO–CeO2 из смесей металлической меди и оксидов меди различной морфологии и фазового состава с диоксидом церия СеО2. Физико-химические свойства синтезированных систем (наличие протяженной межфазной границы, появление новой формы кислорода с высокой реакционной способностью при более низкой температуре, чем решеточный кислород оксидов) были аналогичны тем, которые были обнаружены для аналогичных катализаторов, приготовленных другими методами. Образцы катализаторов были испытаны в двух процессах: селективном окислении СО в избытке Н2 (CO-PROX) и глубоком окислении этана. Оказалось, что природа исходных медьсодержащих компонентов практически не важна для первого процесса. Каталитическая активность зависит только от времени МС (дозы энергии, подводимой к материалу), которая обеспечивает степень модифицирования поверхности СеО2 ионами меди. При достаточно длительном (60–90 мин) МС свойства катализаторов сближаются.
Напротив, реакция глубокого окисления этана чувствительна к составу добавок: наиболее активным оказался Cu–CeO2, а наименее активным – CuO(I)–CeO2, в составе которого был чистый CuO. Это, вероятнее всего, связано с тем, что для активации С2Н6 на поверхности необходимы ионы Cu1+, биографические или образующиеся во время МС во всех композитах, кроме CuO(I)–CeO2. Восстановление поверхности этого образца и появление ионов Cu1+ затруднено даже в атмосфере С2Н6/Ar. Поэтому увеличение конверсии этана на CuO(I)–CeO2 происходило при более высоких температурах, когда в реакцию мог вступать кислород решетки CuO. Однако причины видимого различия низкотемпературной конверсии С2Н6 на изученных композитах требуют дальнейшего исследования.
Список литературы
Liu W., Stephanopoulos M. // J. Catal. 1995. V. 153. P. 304.
Yang W., Li D., Xu D and Wang X. // J. Nat. Gas Chem. 2009. V. 18. P. 458.
Yao S.Y., Xu W.Q., Johnston-Peck A.C., Zhao F.Z., Liu Z.Y., Luo S., Senanayake S.D., Martinez-Arias A., Liu W.J., Rodriguez J.A. // Phys. Chem. Chem. Phys. 2014. V. 16. P. 17 183.
Chiu K.L., Kwong F.-I., Ng Dickon H.L. // Curr. Appl. Phys. 2012. V. 12. I. 4. P. 1195.
Yao X., Gao F., Yu Q., Qi L., Tang Ch., Dong L., Chen Y. // Catal. Sci. Technol. 2013. V. 3. P. 1355.
Kydd R., Teoh W.Y., Wong K., Wang Y., Scott J., Zeng Q.-H., Yu A.-B., Zou J., Amal R. // Adv. Func. Mater. 2009. V. 19. P. 369.
Chen A., Yu X., Zhou Y., Miao Sh., Li Y., Kuld S., Sehested J., Liu J., Aoki T., Hong S., Camellone M.F., Fabris S., Ning J., Jin Ch., Yang Ch., Nefedov A., Wöll Ch., Wang Y., Shen W.// Nat. Catal. 2019. V. 2. P. 334.
Yu W.-Zh., Wang W.-W., Fu X.-Pu, Wang X., Wu K., Si R., Ma Ch., Jia Ch.-J., Yan Ch.H. // Am. Chem. Soc. 2019. V. 141. I. 44. P. 17 548.
Skårman B., Nakayama T., Grandjean D., Benfild R.E., Olsson E., Niihara K., Wallenberg L.R. // Chem. Mater. 2002. V. 14. P. 3686.
Wongkaew A., Kongsi W., Limsuwan P. // Adv. Mater. Sci. Eng. 2013. ID 374080.
Gurbani A., Ayastuy J.L., González-Marcos M.P., Gutiérrez-Ortiz M.A. // J. Hydrogen Ener. 2010. V. 35. P. 11 582.
Prasad R., Rattan G. // Bull. Chem. React. Eng. Catal. 2010. V. 5. P. 7.
Liu Z., Zhou R., Zheng X. // J. Mol. Catal. 2007. V. 267. P. 137.
Martínez-Arias A., Gamarra D., Hungría A.B., Fernández-García M., Munuera G., Hornés A., Bera P., Conesa J.C., Cámara A.L. // Catal. 2013. V. 3. P. 378.
Martínez-Arias A., Gamarra D., Fernández-García M., Hornés A., Bera P., Koppány Zs., Schay Z. // Catal. Today. 2009. V. 143. P. 211.
Xu Ch., De S., Balu F.V., Ojeda M., Luque R. // Chem. Commun. 2015. V. 31. I. 51. P. 6698.
Borchers Ch., Martin M.L., Vorobjeva G.A., Morozova O.S., Firsova A.A., Leonov A.V., Kurmaev E.Z., Kukharenko A.I., Zhidkov I.S., Cholakh S.O. // J. Nanopart. Res. 2016. V. 18. P. 344.
Фирсова А.А., Морозова О.С., Леонов А.В., Стрелецкий А.Н., Корчак В.Н. // Кинетика и катализ. 2014. Т. 55. № 6. С. 783.
Galvita V., Filez M., Poelman H., Bliznuk V., Marin G.B. // Catal. Lett. 2014. V. 144. P. 43.
Shelekhov E.V., Sviridova T.A. // Met. Sci. Heat. Treat. 2000. V. 42. P. 309.
Il’ichev A.N., Firsova A.A., Korchak V.N. // Kinet. Catal. 2006. V. 47. P. 585.
Borchers Ch., Martin M.L., Vorobjeva G.A., Morozova O.S., Firsova A.A., Leonov A.V., Kurmaev E.Z., Kukharenko A.I., Zhidkov I.S., Cholakh S.O. // AIP Adv. 2019. V. 9. P. 065 115.
Zeng Sh., Zhang W., Śliwa M., Su H. // Int. J. Hydrogen Energ. 2013. V. 38. P. 3597.
Tang X., Zhang B., Li Y., Xu Y., Xin Q., Shen W. // Appl. Catal. A: General. 2005. V. 288. P. 116.
Xu J., Harmer J., Li G., Chapman T., Collier P., Longworth S., Tsang S.C. // Chem. Commun. 2010. V. 46. P. 1887.
Moretti E., Lenardaa M., Riello P., Storaro L., Talon A., Frattini R., Reyes-Carmona A., Jimenez-Lopez A., Rodriguez-Castellon E. // Appl. Catal. B: Environ. 2013. V. 129. P. 556.
Qi X., Flytzani-Stephanopoulos M. // Ind. Eng. Chem. Res. 2004. V. 43. P. 3055.
Polster Ch.P., Nair H., Baertsch C.D. // J. Catal. 2009. V. 266. P. 308.
Barbato P.S., Colussi S., Di Benedetto A., Landi G., Lisi L., Llorca J., Trovarelli A. // J. Phys. Chem. C. 2016. V. 120. P. 13 039.
Natile M.M., Galenda A., Glisenti A. // Surf. Sci. Spectra. 2009. V. 16. P. 13.
Di Benedetto A., Landi G., Lisi L. // Catalyst. 2018. V. 8. I. 5. P. 209.
Sundar R.S., Deevi S. // J. Nanopart. Res. 2006. V. 8. P. 497.
Zhan W., Yang S., Zhang P., Guo Y., Lu G., Chisholm M.F., Dai Sh. // Chem. Mater. 2017. V. 29. I. 17. P. 7323.
Holgado P., Manuera G., Espinos J.P., Gonzanez-Elipe A.R. // Appl. Surf. Sci. 2000. V. 158. P. 164.
Luo M.-F., Song Yu.-P., Lu Ji.-Q., Wang X.-Yu., Pu Zh.-Y. // J. Phys. Chem. C. 2007. V. 111. P. 12 686.
Luong N.N., Okumura H., Yamasue E., Ishihara K.N. // R. Soc. Open Sci. 2019. V. 6. P. 181 861.
Pidko E., Kazansky V. // PCCP. 2005. V. 7. P. 1939.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ