

Кинетика и катализ, 2020, T. 61, № 5, стр. 654-662
Молекулярная организация реагентов в кинетике и катализе жидкофазных реакций. XIII. Циклические переходные состояния с участием молекул растворителя в механизме аминолиза циклокарбонатов в спиртовой среде
М. В. Забалов a, *, М. А. Левина a, Р. П. Тигер a
a ФГБУН Федеральный исследовательский центр химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: zabalov@chph.ras.ru
Поступила в редакцию 02.12.2019
После доработки 10.03.2020
Принята к публикации 14.04.2020
Аннотация
Исследованы закономерности влияния протонодонорного растворителя в реакции аминолиза этиленкарбоната, протекающей по концертному и стадийному механизмам. В основу рассмотрения положены кинетические данные по модельным реакциям в спиртовых растворах и квантово-химические расчеты методом DFT путей реакций с участием Н-связанных переходных состояний и интермедиатов, содержащих различное число молекул растворителя в циклах переноса протона и циклах стабилизации. Расчетные данные по энергиям активации реакции, включающей циклы разного размера, свидетельствуют о предпочтительности стадийного механизма по сравнению с концертным при протекании реакции в метаноле. Барьер стадийного пути реакции ниже на 2.9 ккал/моль по сравнению с концертным. Для протекания реакции необходим только цикл переноса протона, наличие цикла стабилизации не приводит к снижению энергии активации.
ВВЕДЕНИЕ
Реакции циклокарбонатов с аминами относятся к малоизученным процессам органической химии. Они были открыты в середине прошлого века, но интерес к их механизму возник лишь в последние годы в связи с появлением нового направления зеленой химии полимеров – процессов синтеза полиуретанов без использования высокотоксичных изоцианатов, получаемых к тому же, с применением фосгена [1–9]. Эти реакции протекают путем нуклеофильной атаки циклокарбоната молекулой амина с раскрытием циклокарбонатного цикла и, что важно, без выделения побочных продуктов, также как и классические реакции уретанообразования (схема 1 ).
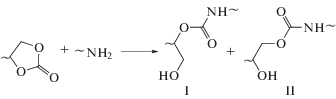
Схема 1 . Получение полиуретанов из циклокарбонатов и аминов.
Полиуретаны, образующиеся в этой реакции, содержат гидроксильные группы – первичные I и вторичные II в равных или несколько отличающихся соотношениях в зависимости от строения реагентов и условий проведения процесса, которые при необходимости могут быть использованы для модификации полимеров. Важно, что в процессах синтеза новых уретанов не принимают участия высокотоксичные изоцианаты, а исходные олигомеры с циклокарбонатными группами для них можно получать из эпоксидсодержащих предшественников за счет реакции эпоксидных групп с диоксидом углерода, безупречной в экологическом отношении, в том числе из возобновляемого сырья – растительных масел.
Количественные сведения о механизме и кинетических закономерностях образования гидроксиуретанов из циклокарбонатов и аминов появились лишь в последние годы. В работе [10] была отмечена возможность протекания реакции по переменному порядку по амину – первому или второму в зависимости от его строения, концентрации и природы растворителя. При этом второй порядок интерпретировался как результат смены лимитирующей стадии и участия дополнительной молекулы амина в депротонировании тетраэдрического интермедиата, образующегося в первой стадии реакции. Проведенные нами [11, 12] в рамках теории функционала плотности (DFT) квантово-химические расчеты взаимодействия этиленкарбоната и пропиленкарбоната с метиламином показали, что реакция действительно может протекать параллельно по двум каналам с участием одной (схема 2 ) и двух (схема 3 ) молекул амина по одностадийному (концертному) пути и по более сложному многостадийному механизму, включающему образование аминоспирта в качестве интермедиата.
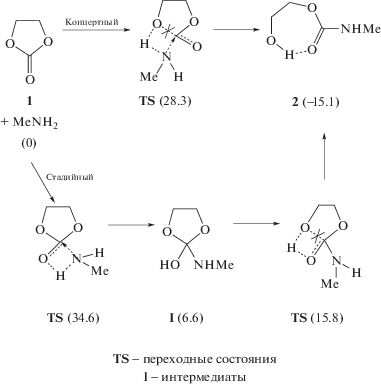
Схема 2 . Механизм реакции этиленкарбоната с одной молекулой амина в газовой фазе. Конечный продукт 2 – наиболее устойчивая структура гидроксиуретана с внутримолекулярной водородной связью.
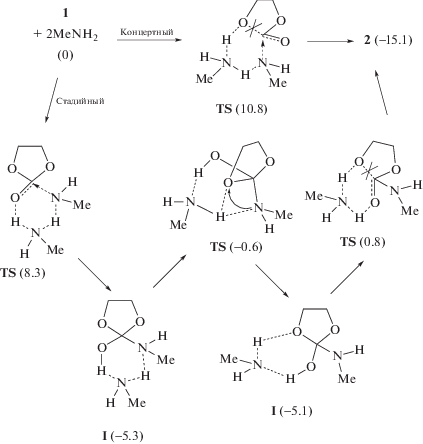
Схема 3 . Механизм реакции этиленкарбоната с двумя молекулами амина в газовой фазе.
На схемах 2 и 3 в скобках указаны минимальные энергии изомеров (ккал/моль), рассчитанные относительно суммы энергий исходных реагентов, разделенных на бесконечность (в газофазном приближении) [11, 12]. В этих работах, а также в наших последующих теоретических и экспериментальных исследованиях [13–19] было установлено, что вторая молекула амина, встраиваясь в циклическую структуру переходного состояния, осуществляет каталитическое содействие акту переноса протона к молекуле циклокарбоната. Результатом этого является троекратное снижение эффективной энергии активации по сравнению с энергией активации реакции с участием одной молекулы амина. Такого рода содействие имеет место также в реакциях, катализируемых карбоновыми кислотами, другими соединениями, для которых характерен бифункциональный катализ переноса атомов и групп [15, 18, 19]. Аналогичную роль могут играть гидроксилсодержащие соединения, в частности алифатические спирты, выступающие в качестве среды или компонентов смешанного растворителя, образуя полимолекулярные структуры за счет водородных связей. Образование подобных структур было изучено нами на примере взаимодействия этиленкарбоната с одной молекулой метиламина по концертному пути в метаноле с помощью квантово-химических расчетов в приближении супермолекулы [17].
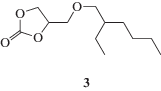
При исследовании различных путей реакций, проходящих с участием молекул спирта как растворителя и катализатора, были введены представления о так называемых циклах переноса протона и циклах стабилизации в переходных состояниях [17]. При этом подробно в приближении супермолекулы были изучены лишь закономерности концертного механизма аминолиза. Настоящая работа направлена на детализацию роли протонодонорного растворителя при протекании реакции аминолиза по стадийному механизму. В основу положены экспериментальные данные по модельным реакциям в спиртовых растворах и квантово-химические расчеты путей реакций с участием Н-связанных переходных состояний и интермедиатов, содержащих различное число молекул растворителя в циклах переноса протона и стабилизации. Обсуждение ведется с использованием полученных ранее результатов по реакционной способности этиленкарбоната 1 при взаимодействии с н-бутиламином [13] и новых данных по аминолизу 4-(2-этилгексилоксиметил)-1,3-диоксолан-2-она – модельного монофункционального олигомера 3, содержащего циклокарбонатную группу на одном из концов цепи:
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и методика эксперимента
Циклокарбонат 3, производства компании “Макромер” (г. Владимир), содержал 33.2 мас. % циклокарбонатных групп (при 1.0 мас. % непрореагировавших при карбонизации эпоксидных групп) и не подвергался дополнительной очистке, как и н-бутиламин, н-бутанол и диоксан (все “Sigma-Aldrich”).
Кинетические измерения проводили методом ИК-Фурье-спектроскопии на приборе FTIR Tensor 27 (“Bruker”, Германия) по изменению во времени оптических плотностей на частотах валентных колебаний карбонильных групп исходного циклокарбоната 3 (1795 см–1 в н-бутаноле и 1820 см–1 в диоксане). Методика эксперимента подробно описана в [13] на примере аминолиза 1. Измерения осуществляли при Т = 55 ± 0.1°С непосредственно в кюветном отделении прибора в стандартных кюветах постоянной толщины 0.1 или 0.4 мм с окошками из CaF2, оборудованных термостатирующей насадкой. При такой постановке эксперимента не требовалось использования реперной полосы, интенсивность которой не менялась бы в ходе реакции. При концентрациях 3 равной (1.5–5.0) × 10–2 моль/л в избытке амина в интервале 0.1–0.75 моль/л реакция протекала по псевдопервому порядку до 95–100% превращения, как и изученная ранее реакция аминолиза 1. В этих условиях наблюдаемая константа скорости первого порядка не зависела от исходного содержания циклокарбоната в растворе. Реакция с участием 3 была существенно медленней, чем с участием 1. В связи с этим в отличие от экспериментов по аминолизу 1, исследованному в интервале температур 20–55°С, температурные зависимости констант скорости аминолиза 3 не измерялись.
Методика квантово-химических расчетов
Квантово-химические расчеты проводили в рамках теории функционала плотности (DFT) с использованием неэмпирически обобщенного градиентного приближения и функционала PBE [20, 21] в базисе TZ2P по программе PRIRODA [22, 23]. Для всех исходных реагентов и переходных состояний осуществляли оптимизацию геометрии. Характер найденных стационарных точек (минимум или седловая точка на поверхности потенциальной энергии) определяли вычислением собственных значений матрицы вторых производных энергии по координатам ядер. Принадлежность переходных состояний данному превращению устанавливали с помощью расчетов координаты реакции (IRC). Величины относительных энергий исследуемых систем отсчитывались от энергий сольватированных реагентов (см. ниже).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Типичные зависимости наблюдаемой константы скорости псевдопервого порядка kнабл реакции циклокарбонатов 1 и 3 с н-бутиламином от концентрации амина [BuNH2] в н-бутаноле и диоксане показаны на рис. 1, из которого видно, что циклокарбонат 1 в реакции аминолиза почти вдвое активнее, чем 3. Кривые, приведенные на рисунке, с хорошей точностью (среднеквадратичное отклонение R2 = 0.98–0.99) описываются двухпараметрическим уравнением:
Рис. 1.
Зависимости наблюдаемой константы скорости аминолиза 1 (1) и 3 (2) от концентрации н-бутиламина в н-бутаноле (а) и диоксане (б).
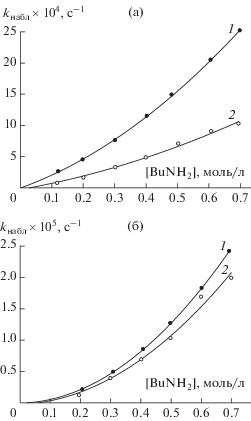
Таблица 1.
Константы скорости k1 и k2 реакций н-бутиламинолиза циклокарбонатов 1 и 3 в н-бутаноле и диоксане при различных температурах
| Циклокарбонат | Т, °С | k1 × 104, л моль–1 с–1 |
k2 × 104, л2 моль–2 с–1 |
k1 × 107, л моль–1 с–1 |
k2 × 105, л2 моль–2 с–1 |
|---|---|---|---|---|---|
| н-бутанол | диоксан | ||||
| 1 | 20 | 4.9 | 16.4 | – | – |
| 28 | 7.5 | 18.8 | 0.84 | 2.98 | |
| 40 | 11.0 | 21.5 | 2.13 | 3.92 | |
| 55 | 17.9 | 26.2 | 5.60 | 4.96 | |
| 3 | 55 | 7.4 | 11.4 | 3.9 | 4.2 |
Используя полученные данные, можно оценить соотношение скоростей реакций, протекающих по разным каналам при постоянной исходной концентрации циклокарбоната: W2/W1 = = k2[BuNH2]/k1. При низких концентрациях амина ([BuNH2] = 0.1 моль/л) основной вклад в скорость реакции в спиртовой среде вносит мономерный канал (W2/W1 < 1); при высоких ([BuNH2] = = 1.0 моль/л) – соотношение W2/W1 во всем диапазоне исследованных температур возрастает на порядок, и вклад канала с участием двух молекул амина в общую скорость процесса начинает преобладать (W2/W1$ \gg $ 1). В диоксане константы скорости k1 и k2 ниже, чем в спирте, примерно на 2 порядка, причем канал с участием двух молекул амина является основным путем реакции независимо от температуры и [BuNH2], т.е. W2/W1$ \gg $ 1.
Как было установлено ранее [13], энергии активации аминолиза 1, рассчитанные из температурных зависимостей k1 и k2, соответственно равны 14.0 ± 0.3 и 3.7 ± 0.2 ккал/моль в диоксане и 7.0 ± 0.2 и 2.5 ± 0.1 ккал/моль в н-бутаноле. Для реакций с участием одной и двух молекул амина характерны высокие отрицательные значения энтропий активации: –46.2 и –68.8 кал моль–1 град–1 в диоксане и –51.4 и –64.6 кал моль–1 град–1 в н-бутаноле соответственно, что является типичным для процессов, протекающих в молекулярно-организованных системах через циклические переходные состояния [24].
Обращает на себя внимание почти двукратное снижение экспериментальной величины активационного барьера реакций при переходе от диоксана к спирту как протонодонорному растворителю. При этом энергии активации каналов с участием одной и двух молекул амина различаются примерно втрое, как и расчетные значения в газовой фазе, составляющие 28.3 и 10.8 ккал/моль для реакций с одной или с двумя молекулами амина соответственно (отсчитаны от суммы энергий реагентов, разделенных друг от друга на бесконечность) [11, 12].
Причиной расхождения теории и эксперимента является сольватация исходных реагентов и переходных состояний в растворе, а в случае протонодонорного растворителя – непосредственное участие его в реакции путем встраивания в структуру активированного комплекса и облегчения тем самым переноса протона от амина к циклокарбонату. При расчете концертного пути в метаноле этой реакции с одной молекулой амина величина минимального барьера реакции составила 15.1 ккал/моль [17], что существенно ниже барьера реакции в газовой фазе [11, 12], но все равно его значение остается высоким по сравнению с экспериментально найденной кажущейся энергией активации для канала реакции с одной молекулой амина. В связи с этим в настоящей работе были выполнены модельные квантово-химические расчеты влияния гидроксилсодержащего растворителя на стадийный путь реакции с одной молекулой амина, который потенциально может протекать с меньшим барьером по сравнению с концертным. По аналогии с исследованием концертного пути реакции рассматривались структуры с вовлечением различного числа молекул метанола.
При проведении квантово-химических расчетов в качестве уровня отсчета относительных энергий обычно выбирают или сумму энергий реагентов, разделенных на бесконечность, или энергию предреакционного комплекса (RC). У таких способов расчета есть свои недостатки. Сумма энергий исходных реагентов вносит погрешность, связанную с неполнотой базиса, которая в случае сильного межмолекулярного взаимодействия, как в нашем случае из-за образования водородных связей, может приводить к заниженным энергиям активации или даже к отрицательным их значениям [15, 17]. При вычислении энергии активации от RC ее величина оказывается наоборот завышенной, так как в нее входит энергия образования самого RC. Для реакций в газовой фазе с этим приходится мириться. Для реакций в растворе нами предложена новая структура в качестве уровня отсчета – сольватированные реагенты (SR). В SR реагенты разделены в пространстве молекулами растворителя и между собой не взаимодействуют. Таким образом, в энергию SR вносят вклад только энергии взаимодействия реагентов с растворителем, и расчет величин энергии активации, когда за точку отсчета принимаются такие структуры, получается более точным. Поскольку те же молекулы растворителя присутствуют и в переходном состоянии, то энергия взаимодействия исследуемых молекул с растворителем взаимно вычитается при вычислении энергии активации, если молекулы растворителя не принимают участия в элементарном акте превращения [15, 17]. Комплексы SR рассчитываются с тем же набором базисных функций, как и TS, и RC, что позволяет избежать проблем с неполнотой базиса [15].
Как было отмечено выше, реакция аминолиза может протекать по двум механизмам, концертному и стадийному. При концертном пути перенос протона происходит одновременно с присоединением амина и раскрытием этиленкарбонатного цикла. При стадийном механизме вначале протон переносится на карбонильный атом кислорода, а затем на стадии раскрытия цикла происходит второй перенос протона с карбонильного атома кислорода на алкоксильный атом O с образованием гидроксильной группы (схемы 2, 3 ). Для реакции с участием одной молекулы амина в газовой фазе, как показали расчеты, концертный механизм выгоднее стадийного: минимальные барьеры, отсчитанные от суммы энергий исходных реагентов, разделенных на бесконечность, составляют 28.3 и 34.6 ккал/моль соответственно [11, 12]. Однако ситуация принципиально меняется при катализе реакции второй молекулой амина. В этом случае более выгодным оказывается стадийный механизм: для концертного и стадийного путей энергетические барьеры соответственно равны 10.8 и 8.3 ккал/моль [11, 12].
В работе [17] путем расчетов методом DFT в приближении супермолекулы нами были изучены основные закономерности концертного механизма аминолиза этиленкарбоната с участием одной молекулы метиламина в метаноле. Найдены возможности стабилизации переходных состояний и снижения активационных барьеров за счет образования двух видов циклов из водородных связей с участием молекул метанола. Расширение цикла переноса протона до 8- или 10-членного способствует более легкому переносу протона через менее напряженные структуры, нежели через 4-членный цикл, как в газовой фазе. Второй цикл из водородных связей, также 8- или 10-членный, способствует экранированию избыточных зарядов на атомах кислорода этиленкарбоната (в основном на карбонильном атоме O) при присоединении молекулы амина к карбонильной группе. При стабилизирующем эффекте двух циклов из водородных связей минимальный барьер протекания реакции по концертному пути снижается до 15.1 ккал/моль [17]. Кроме того, в работе [17] удалось решить ряд методологических вопросов применения метода супермолекулы для расчета механизмов реакций в протонодонорном растворителе. Однако остался нерешенным вопрос о барьерах реакции аминолиза этиленкарбоната в метаноле при ее протекании по стадийному механизму.
Поскольку для концертного механизма был проведен большой объем вычислений, то для стадийного механизма выбраны только характерные модели переходных состояний, а именно структуры с 6-, 8- и 10-членными циклами переноса протона (число молекул спирта, входящих в их состав, соответственно равно 1, 2 и 3) и структуры, включающие два цикла – 8-членный цикл переноса протона и 8-членный цикл стабилизации.
Стадийный механизм реакции в протонодонорной среде (МеОН) представлен на схеме 4 , где c(1) – цикл переноса протона, c(2) – цикл стабилизации. Пунктиром обозначены цепочки из Н‑связанных молекул метанола. Вначале из сольватированных реагентов SR, как видно из схемы 4 , образуется предреакционный комплекс RC. Далее в переходном состоянии TS1 метиламин присоединяется к карбонильной группе с образованием интермедиата I1. В TS2 происходит перенос протона между молекулой амина и карбонильным атомом O с участием цепочки из молекул метанола. Образование TS2 на потенциальной поверхности возможно только из-за стабилизирующего действия молекул метанола. В газовой фазе, как показывают расчеты, такое переходное состояние не образуется из-за нестабильности I1.
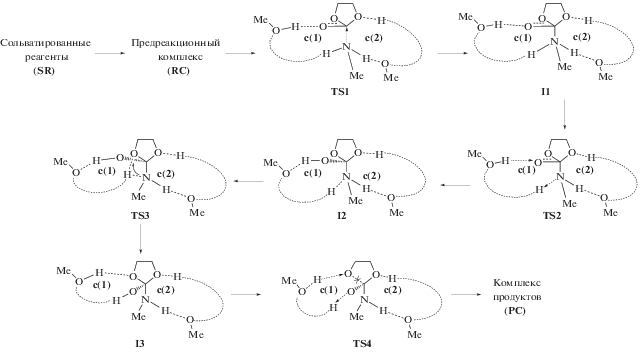
Схема 4 . Стадийный механизм реакции аминолиза в среде метанола.
В дальнейшем в переходном состоянии TS3 происходит поворот цикла c(1) с переносом протона с атома N на алкоксильный атом O. В TS4 раскрывается этиленкарбонатный цикл и образуется сольватированный комплекс продукта (РС). Относительные энергии переходных состояний и интермедиатов, отсчитанные от SR, а также другие параметры расчета представлены в табл. 2 в сравнении с энергетическими барьерами концертного пути реакции.
Таблица 2.
Результаты расчета стадийного механизма реакции этиленкарбоната с одной молекулой амина в метаноле в сравнении с концертным механизмом в метаноле и реакцией в газовой фазе*
| Путь реакции | I | II | III | IV | V |
|---|---|---|---|---|---|
| Размер цикла c(1) | 4 | 6 | 8 | 10 | 8 |
| Размер цикла c(2) | – | – | – | – | 8 |
| Минимальный барьер стадийного пути | 34.6a | 15.5 | 12.2 | 13.3 | 13.7 |
| Минимальный барьер концертного пути | 28.3a | 19.2b | 18.5b | 18.4b | 16.9b |
| Число изомеров | 2 | 4 | 8 | 16 | 30 |
| Число учтенных молекул MeOH | 0 | 1 | 2 | 3 | 4 |
| RC | –4.7 (–4.5)b, c | –4.4 (–4.2) | –3.8 (–3.0) | –3.3 (–2.3) | –4.6 (5.8) |
| TS1 | 34.6 (34.7)a | – | – | 6.0 (8.9) | 6.4 (10.4) |
| I1 | 6.6a | – | – | 6.0 (8.8) | 1.5 (10.1) |
| TS2 | – | 15.5 (16.6) | 11.9 (14.2) | 12.8 (16.0) | 9.3 (18.2) |
| I2 | – | 3.3 (4.9) | 1.9 (3.8) | 2.8 (5.1) | 3.2 (9.3) |
| TS3 | – | 5.6 (6.6) | 4.9 (6.3) | 5.9 (7.7) | 7.1 (12.2) |
| I3 | – | 6.6 (8.0) | 5.7 (8.1) | 5.1 (9.0) | 6.5 (14.1) |
| TS4 | 15.8a | 12.4 (13.1) | 10.5 (13.3) | 11.2 (16.2) | 13.7 (18.4) |
| PC | –15.1a | –15.5 (–13.9) | –14.8(–12.6) | –13.9 (–11.3) | –17.7 (–6.9) |
* Энергии рассчитаны в ккал/моль относительно SR, если не указано другое. Примечание. Для третьей строки таблицы прочерки означают, что цикл такого типа в структуре отсутствует, для 9–14 строк – что структуры такого типа не образуются на данной поверхности потенциальной энергии (ППЭ). a По данным [11, 12]. За уровень отсчета принята сумма энергий реагентов, разделенных на бесконечность. b По данным [17]. c Минимальные энергии изомеров (ккал/моль), в скобках максимальные энергии.
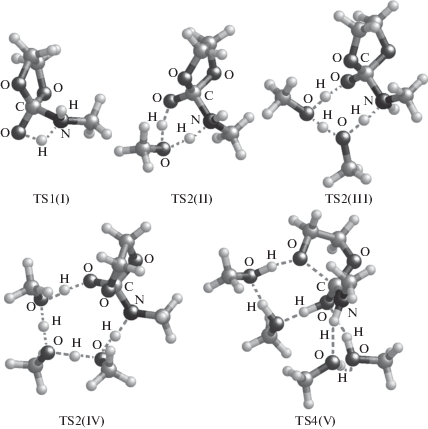
Характерные структуры, определяющие энергетические барьеры реакции, представлены на рис. 2. Следует отметить, что для путей реакции без образования цикла стабилизации (пути II–IV в табл. 2) барьер всей реакции определяется стадией переноса протона между амином и карбонильным атомом O (TS2), а для пути реакции с двумя циклами (путь V в табл. 2) – стадией раскрытия этиленкарбонатного цикла (TS4).
Рис. 2.
Структуры переходных состояний, определяющих энергетические барьеры реакций I–V (см. табл. 2).
Во всех случаях возможна стереоизомерия в циклах c(1) и c(2) и изомерия присоединения молекулы метиламина (рис. 3). Интерес представляют пути с наименьшей затрачиваемой энергией. Поскольку невозможно предугадать наиболее стабильный изомер, а разница в энергиях изомеров может достигать значений, соизмеримых с энергией активации [17], приходится проводить расчет для всех возможных изомеров. В табл. 2 приведены данные для минимальных и максимальных (в скобках) значений энергий изомеров для реакции по двум механизмам, а также минимальный энергетический барьер для протекания реакции, который определяется переходным состоянием с наибольшей энергией в каждой конкретной изомерной цепочке превращений.
Рис. 3.
Возможные типы стереоизомерии на примере TS1: A – изомерия присоединения метиламина к карбонильной группе; B – изомерия метильных групп метанола в циклах c(1) и c(2).
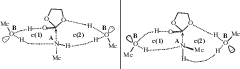
Таким образом, барьер стадийного пути реакции аминолиза этиленкарбоната в метаноле ниже на 2.9 ккал/моль по сравнению с концертным путем. При этом наиболее выгодно протекание реакции по концертному механизму через TS с 8-членным циклом переноса протона и 10-членным циклом стабилизации, а для протекания реакции по стадийному механизму необходим только 8-членный цикл переноса протона в TS. Цикл стабилизации c(2) для стадийного пути не нужен. Это связано с тем, что в стадийном пути возникает значительно меньшая избыточная электронная плотность на атомах O этиленкарбоната при присоединении амина к карбонильной группе, так как в интермедиатах I2 и I3 образуется гидроксильная группа.
ЗАКЛЮЧЕНИЕ
Реакция аминолиза циклокарбонатов может протекать по концертному и стадийному путям. В газофазном приближении более выгодным оказывается концертный путь, а в среде метанола – стадийный. Отличие связано с возможностью стабилизации переходных состояний и интермедиатов за счет образования комплексов с молекулами метанола связанных водородными связями, в том числе за счет встраивания молекул метанола в циклы переноса протона в переходных состояниях. Это подтверждает необходимость учета среды при квантово-химическом моделировании химических реакций для более точного и количественного описания механизма их протекания.
Список литературы
Guan J., Song Y., Lin Y., Yin X., Zuo M., Zhao Y., Tao X., Zheng Q. // Ind. Eng. Chem. Res. 2011. V. 50. № 11. P. 6517.
Figovsky O., Shapovalov L., Leykin A., Birukova O., Potashnikova R. // PU Magazine. 2013. V. 10. № 4. P. 1.
Nohra B., Candy L., Blanco J.-F., Guerin C., Raoul Y., Mouloungui Z. // Macromolecules. 2013. V. 46. № 10. P. 3771.
Blattmann H., Fleischer M., Bähr M., Mülhaupt R. // Macromol. Rapid Comm. 2014. V. 35. № 14. P. 1238.
Rokicki G., Parzuchowski P.G., Mazurek M. // Polymer Adv. Technol. 2015. V. 26. № 7. P. 707.
Maisonneuve L., Lamarzelle O., Rix E., Grau E., Cramail H. // Chem. Rev. 2015. V. 115. P. 12 407.
Cornille A., Auvergne R., Figovsky O., Boutevin B., Caillol S. // Eur. Polym. J. 2017. V. 87. P. 535.
Błażek K., Datta J. // Crit. Rev. Environ. Sci. Technol. 2019. V. 49. № 3. P. 173.
Carré C., Ecochard Y., Caillol S., Avérous L. // ChemSusChem. 2019. V. 12. № 15. P. 3410.
Гарипов Р.М., Сысоев В.А., Михеев В.В., Загидуллин А.И., Дебердеев Р.Я., Иржак В.И., Берлин А.А. // Докл. АН. 2003. Т. 393. № 1. С. 61.
Забалов М.В., Тигер Р.П., Берлин А.А. // Докл. АН. 2011. Т. 441. № 4. С. 480.
Забалов М.В., Тигер Р.П., Берлин А.А. // Изв. АН. Сер. хим. 2012. № 3. С. 518.
Левина М.А., Крашенинников В.Г., Забалов М.В., Тигер Р.П. // Высокомол. соед. 2014. Т. 56-Б. № 2. С. 152.
Забалов М.В., Левина М.А., Крашенинников В.Г., Тигер Р.П. // Изв. АН. Сер. хим. 2014. № 8. С. 1740.
Забалов М.В., Тигер Р.П. // Изв. АН. Сер. хим. 2016. № 3. С. 631.
Левина М.А., Забалов М.В., Крашенинников В.Г., Тигер Р.П. // Высокомол. соед. 2017. Т. 59-Б. № 5. С. 317.
Zabalov M.V., Tiger R.P. // Theor. Chem. Acc. 2017. V. 136. Article 95.
Левина М.А., Забалов М.В., Крашенинников В.Г., Тигер Р.П. // Высокомол. соед. 2018. Т. 60-Б. С. 372.
Zabalov M.V., Levina M.A., Krasheninnikov V.G., Tiger R.P. // Reac. Kinet. Mech. Cat. 2020. V. 129. № 1. P. 65.
Perdew J.P., Burke K., Ernzerhoff M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865.
Ernzerhoff M., Scuseria G.E. // J. Chem. Phys. 1999. V. 110. № 11. P. 5029.
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. P. 151.
Лайков Д.Н., Устынюк Ю.А. // Изв. АН. Сер. хим. 2005. № 3. С. 804.
Энтелис С.Г., Тигер Р.П. Кинетика реакций в жидкой фазе: количественный учет влияния среды. М.: Химия, 1973. 416 с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ