

Кинетика и катализ, 2020, T. 61, № 6, стр. 789-796
Механизм антирадикального действия природных фенилпропаноидов в полярных неионизирующих средах
Н. И. Белая a, *, А. В. Белый a, А. А. Давыдова b
a ГОУ ВПО Донецкий национальный университет
283001 Донецк, ул. Университетская, 24, Украина
b ГУ Институт физико-органической химии и углехимии им. Л.М. Литвиненко
283114 Донецк, ул. Р. Люксембург, 70, Украина
* E-mail: nat.iv.belaya@gmail.com
Поступила в редакцию 29.01.2020
После доработки 22.03.2020
Принята к публикации 10.04.2020
Аннотация
Исследованы кинетика и механизмы реакции некоторых природных фенилпропаноидов группы гидроксикоричных кислот со стабильным радикалом 2,2'-дифенил-1-пикрилгидразилом в полярных неионизирующих средах путем комбинирования кинетического и спектрофотометрического методов исследования. Определены кинетические и стехиометрические параметры изученной реакции. По величине дейтериевого изотопного эффекта показано, что в полярных растворителях с низкой ионизирующей способностью (диметилсульфоксид) реакция протекает по механизму переноса электрона с последующей передачей протона. Аналогичный механизм антирадикального действия гидроксикоричных кислот может реализовываться в сильнокислых водных средах.
Фенилпропаноиды относятся к классу растительных органических соединений ароматического ряда, характерным структурным фрагментом которых является бензольное кольцо с присоединенной к нему неразветвленной трехуглеродной цепью [1]. Они широко распространены в растительном мире, однако лишь в последнее время интенсивно изучаются в качестве перспективных биологически активных веществ с выраженными адаптогенными, иммуностимулирующими, антиоксидантными и гепатопротекторными свойствами [2, 3].
Особого внимания заслуживают фенилпропаноиды, которые относятся к группе гидроксикоричных кислот, дезактивирующих активные формы кислорода (${\text{О}}_{2}^{{\centerdot {\kern 1pt} - }}$, НОО•, ROO• и т.д.), способные повреждать клеточные структуры в организме человека [2]. Эти соединения применяются как биологически активные добавки с выраженной антиоксидантной активностью в составе многочисленных лекарственных растительных препаратов, но при этом механизм их взаимодействия со свободными радикалами либо считается подобным механизму большинства растительных фенолов (флавоноидов, гидроксибензойных кислот), либо изучается только теоретически [4, 5].
Рассматривая гидроксикоричные кислоты как антиоксиданты (АО) с фенольной функциональной группой, можно выделить несколько механизмов их действия. В неполярных (жироподобных) средах с низкой ионизирующей способностью предполагается механизм отрыва атома водорода НАТ (Hydrogen Atom Transfer) [6, 7]. В полярных средах происходит сопряженный перенос электрона и протона. Если потеря протона предшествует лимитирующей стадии переноса электрона, то механизм именуется как SPLET (Sequential Proton Loss–Electron Transfer), и в реакции непосредственно участвует ионизированная форма антиоксиданта [8, 9]. Это характерно для щелочных сред c рН ≥ 7. Если потеря протона следует за медленной стадией переноса электрона, то осуществляется механизм ET–PT (Electron Transfer–Proton Transfer, также он упоминается как SET–PT или SEPT) [4, 10, 11]. Тогда в реакции участвует молекулярная форма антиоксиданта, что характерно для водных сред с низким значением рН ≤ 3.
Механизмы НАТ и SPLET исследуются более детально, поскольку они связаны с двумя крайними случаями – либо с неполярными неионизирующими растворителями (углеводородами, липидами), либо с полярными ионизирующими средами (водой, спиртами). Механизм ET–PT, как правило, изучается редко и в основном квантово-химическим методом без экспериментального подтверждения [4, 5, 11], что обусловлено определенными сложностями при установлении этого механизма (часто его принимают за НАТ, поскольку в реакции участвует молекулярная форма АО).
Цель данной работы _ спектрально-кинетическое исследование возможных механизмов реакции гидроксикоричных кислот (Cinnam–OH) с N-центрированным радикалом 2,2'-дифенил-1-пикрилгидразилом (DPPH•) в полярных неионизирующих средах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
Объектами исследования служили некоторые представители класса растительных фенилпропаноидов – гидроксикоричные кислоты и их эфиры следующего строения:
 |
I – о-кумаровая кислота | R1,3,4,5,6 = H, R2 = ОH; |
| II – м-кумаровая кислота | R1,2,4,5,6 = H, R3 = ОH; | |
| III – п-кумаровая кислота | R1,2,3,5,6 = H, R4 = ОH; | |
| IV – кофейная кислота | R1,2,5,6 = H, R3,4 = ОH; | |
| V – феруловая кислота | R1,2,5,6 = H, R3 = ОCH3, R4 = ОH; |
|
| VI – метилферулат | R2,5,6 = H, R3 = ОCH3, R4 = ОH, R1 = CH3; |
|
| VII – этилферулат | R2,5,6 = H, R3 = ОCH3, R4 = ОH, R1 = C2H5; |
|
| VIII – синаповая кислота | R1,2,6 = H, R3,5 = ОCH3, R4 = ОH. |
Гидроксикоричные (I–V, VIII) кислоты и их эфиры (VI, VII) (“Fluka”, Швейцария; “Merck”, Германия; “Panreac”, Испания) дополнительно не очищали.
Антирадикальную активность соединений I–VIII изучали в реакции с DPPH• (“Merck”), который является одним из наиболее удобных и часто используемых стабильных радикалов при оценке эффективности антиоксидантов и установления механизмов их действия [12, 13]. Раствор радикала в диметилсульфоксиде (ДМСО) и бензоле (“Merck”) имел интенсивную фиолетовую окраску с максимумом поглощения при 520 нм. В условиях его хранения в темноте интенсивность спектра в максимуме поглощения оставалась неизменной на протяжении 72 ч.
ДМСО очищали по методике [14]. рН водного раствора задавали с помощью солянокислой (рН 2) буферной системы, приготовленной как указано в работе [15]. Значение точного рН буферного раствора контролировали с помощью иономера И-160МИ (“ООО “Измерительная техника”, Россия).
Кинетические исследования
Реакцию Cinnam–OH с DPPH• проводили в растворителях, из которых предварительно удаляли кислород путем барботирования аргона в течение 15–20 мин, что позволило исключить возможные реакции фенолокислот и продуктов их превращения с участием кислорода. Кинетику реакции исследовали при λmax = 520 нм на спектрофотометре Specord S300 UV-VIS (“Carl Zeiss Jena”, Германия) при температуре Т = 293 ± 2 К в интервале начальных концентраций реагирующих веществ 10–3–10–5 моль/л. Реагенты смешивали, затем измеряли оптическое поглощение смеси и с помощью молярного коэффициента светопоглощения (εДМСО = 1.8 × 103, εбензол = 1.1 × 103 л моль–1 мм–1) по закону Бугера–Ламберта–Бера рассчитывали концентрацию радикала DPPH•.
Синтез дейтеропроизводных гидроксикоричных кислот
Для изучения дейтериевого изотопного эффекта (ДИЭ) получали дейтеропроизводные гидроксикоричных кислот (Cinnam–OD) путем изотопного обмена [16] между Cinnam–OH (С = 0.025 моль/л) и диоксидом дейтерия (D2О) (“Merck”), объемную долю которого в реакционной системе варьировали от 20 до 30 об. %. Для этого их смешивали в ДМСО-d6 (“Panreac”) при постоянном барботировании аргоном в течение 10 мин. Методом ЯМР-спектроскопии [17] было установлено, что за указанный промежуток времени Cinnam-OH превращаются на 97–98% в соответствующие производные – Cinnam–OD. Это подтверждалось практически полным исчезновением сигналов, относящихся к протонам фенольных (в интервале от 7.5 до 9.5 м. д.) и карбоксильных (в интервале от 11 до 12 м. д.) ОН-групп.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В ДМСО как в полярном растворителе с крайне низкой ионизирующей способностью DPPH• расходуется за счет его реакции с молекулярной формой исследуемых Cinnam–OH (I–VIII) (рис. 1). Общий порядок (n) и константу скорости (kДМСО) реакции как характеристику антирадикальной активности (АРА) вещества определяли при эквимольном соотношении фенолокислота–радикал. Для этого кинетические данные обрабатывали нелинейным методом обобщенного приведенного градиента (ОПГ) [18], реализованного в Solver MS Excel. В качестве критерия выбора порядка реакции использовали параметр S, отражающий относительный разброс вычисленного ряда констант.
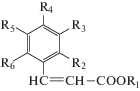
Рис. 1.
а – Кинетические данные расходования DРРН• в реакции с метилферулатом (С = 9 × 10–5 моль/л) при Т = = 293 ± 2 К в разных растворителях: 1 – ДМСО (смоделировано в Solver MS Exсel нелинейным методом ОПГ), 2 – ДМСО (эксперимент), 3 – смесь ДМСО (20 об. %) и бензола (80 об. %), 4 – бензол. б – Зависимость в координатах уравнения (1) для реакции радикала DPPH• с кофейной (1) и синаповой (2) кислотами в смеси бензола с ДМСО.
Порядок реакции соответствует значению, при котором относительный разброс S константы скорости реакции, вычисленной из исходных данных (концентрации и времени) по этому порядку, является наименьшим. Кинетические кривые, смоделированные на основе констант реакции второго порядка, рассчитанных в Solver MS Exсel, хорошо согласуются с экспериментальными данными (рис. 1). Отклонения наблюдаются при степени превращения радикала более 80%, что может быть вызвано влиянием вторичных продуктов превращения гидроксикоричных кислот – димерных фенолов [19, 20]. В связи с этим расчет порядков реакции и констант скорости проводили до момента времени, соответствующего 60–70% расходования DPPH•.
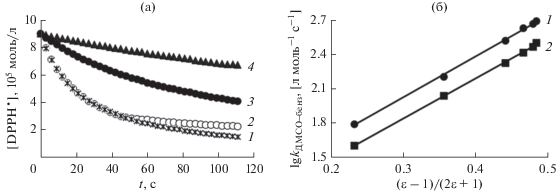
В результате были установлены второй общий порядок реакции (табл. 1) и первый псевдопорядок по DPPH• (${{n}_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}}$). Тогда порядок по Cinnam–OH (${{n}_{{{\text{Cinnam}} - {\text{OH}}}}}$), рассчитанный как разность между общим порядком и частным порядком по радикалу, также будет первым. При определении ${{n}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}}}}$ кинетику реакции исследовали в условиях большого недостатка радикала. На начальных участках кривых (рис. 2а) путем численного дифференцирования определяли начальные скорости w0 исследуемой реакции. Затем, используя метод изолирования Оствальда [21], по величине углового параметра линейной зависимости (рис. 2б), построенной в координатах уравнения (1), был установлен порядок ${{n}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}}}}$, равный единице.
(1)
$\begin{gathered} {{w}_{0}} = k{\kern 1pt} '[{\text{DPP}}{{{\text{H}}}^{\centerdot }}]_{0}^{{{{n}_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}}}}{{[{\text{Cinnam}}{\kern 1pt} - {\kern 1pt} {\text{OH}}]}_{0}}^{{{{n}_{{{\text{Cinnam}}{\kern 1pt} - {\text{OH}}}}}}}, \\ k{\kern 1pt} '[{\text{Cinnam}}{\kern 1pt} - {\kern 1pt} {\text{OH}}]_{0}^{{{{n}_{{{\text{Cinnam}}{\kern 1pt} - {\text{OH}}}}}}} = k, \\ {{w}_{0}} = k[{\text{DPP}}{{{\text{H}}}^{\centerdot }}]_{0}^{{{{n}_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}}}}, \\ {\text{lg}}{\kern 1pt} ~{{w}_{0}} = {\text{lg}}~k + {{n}_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}}{\kern 1pt} {\text{lg\;}}{{[{\text{DPP}}{{{\text{H}}}^{\centerdot }}]}_{0}}. \\ \end{gathered} $Таблица 1.
Экспериментальные константы скорости k и общий порядок (n) реакции Cinnam–OH с DPPH• в разных средах
| Соединение | kДМСО, л моль–1 с–1 | n | kбенз, л моль–1 с–1 | kрН 2, л моль–1 с–1 |
|---|---|---|---|---|
| I | 6.4 ± 0.3 | 1.99 | <1 × 10–3 | 7.9 ± 0.3 |
| II | 5.24 ± 0.15 | 1.98 | <1 × 10–3 | 6.8 ± 0.3 |
| III | 8.3 ± 0.3 | 1.98 | <1 × 10–3 | 10.5 ± 0.5 |
| IV | (4.84 ± 0.15) × 102 | 2.01 | 62.4 ± 1.7 | (5.88 ± 0.16) × 102 |
| V | (4.03 ± 0.15) × 102 | 2 | 30.2 ± 1.4 | (4.53 ± 0.15) × 102 |
| VI | (4.39 ± 0.15) × 102 | 1.99 | 33.9 ± 1.4 | (5.21 ± 0.15) × 102 |
| VII | (4.62 ± 0.15) × 102 | 2.02 | 38.4 ± 1.4 | (5.79 ± 0.16) × 102 |
| VIII | (3.15 ± 0.14) × 102 | 1.97 | 29.2 ± 1.3 | (4.18 ± 0.15) × 102 |
Рис. 2.
Определение псевдопорядка реакции по радикалу DPPH• методом изоляции Оствальда на основе данных о начальных скоростях реакции (б), рассчитанных из кинетических кривых (а) расходования радикала при взаимодействии с кофейной кислотой (С = 5.6 × 10–5 моль/л) в ДМСО при T = 293 ± 2 К.
Среди исследованных веществ (табл. 1) наибольшую АРА (kДМСО) проявили кофейная кислота (IV) с двумя функциональными ОН-группами и ферулаты (VI, VII), в которых электроноакцепторная карбоксильная СООН-группа заменена на электронодонорную сложноэфирную группу. Медленно реагируют с радикалом кумаровые кислоты (I–III) с одной ОН-группой. Полученные закономерности по изменению АРА кислот в неионизирующих средах согласуются с литературными данными [11, 22]. Однако остается спорным вопрос о механизме антирадикального действия Cinnam–OH.
В полярных средах с низкой ионизирующей способностью гидроксикоричные кислоты могут дезактивировать радикалы по механизмам с участием молекулярных форм АО (перенос атома водорода НАТ или механизм переноса электрона ЕТ–РТ [6, 7, 10, 11]). При взаимодействии Cinnam–OH с DPPH• в ДМСО исследовали возможность протекания реакции по этим направлениям. Перенос электрона с участием ионных форм антиоксидантов (механизм SPLET) мы не рассматривали, считая его маловероятным, поскольку доля образовавшихся в ДМСО фенолят-ионов изученных фенилпропаноидов будет ничтожно мала (по сравнению с молекулярной формой), чтобы влиять на скорость лимитирующей стадии.
Одним из способов установления механизмов антирадикального действия вещества является чувствительность реакционной способности АО к изменению природы растворителя [23, 24]. Так, механизм НАТ (реакция (I)) протекает в неполярных средах с низкой протоноакцепторной способностью (в гексане, бензоле, углеводородах, жирах), поскольку в этом случае не образуются водородные связи между атомом Н фенольной группы антиоксиданта и молекулой растворителя, а значит, ОН-группа остается “свободной” и более активной по отношению к радикалу.
(I)
$\begin{gathered} ~{\text{Cinnam--OH}} + {\text{DPP}}{{{\text{H}}}^{\centerdot }} \to \\ \to {\text{Cinnam--}}{{{\text{O}}}^{\centerdot }} + {\text{DPPH--H}}. \\ \end{gathered} $Для изучения влияния растворителя на реакционную способность АО по реакции (I) используют ${\beta }_{2}^{{\text{Н}}}$-шкалу основности водородных связей вещества [6], предложенную в работе [25]. Согласно ей, в неполярных средах чем меньше параметр ${\beta }_{2}^{{\text{Н}}}$, тем выше реакционная способность фенольных соединений.
Механизм ЕТ–РТ (реакция (II)) реализуется в полярных растворителях, способствующих формированию переходного состояния реакции с разделением заряда [6, 26]. Это приводит к образованию катион-радикала, который обладает бóльшими кислотными свойствами, чем исходная частица и способен быстро диссоциировать с образованием соответствующего радикала.
(II)
$\begin{gathered} {\text{Cinnam}}{\kern 1pt} - {\kern 1pt} {\text{OH}} + {\text{DPP}}{{{\text{H}}}^{\centerdot }}{\text{\;}}\xrightarrow{{{\text{slow}}}} \\ \to {\text{Cinnam}}{\kern 1pt} - {\kern 1pt} {\text{O}}{{{\text{H}}}^{{\centerdot {\kern 1pt} + }}} + {\text{DPP}}{{{\text{H}}}^{--}}, \\ {\text{Cinnam}}{\kern 1pt} - {\kern 1pt} {\text{O}}{{{\text{H}}}^{{\centerdot {\kern 1pt} + }}} \rightleftarrows {\text{\;Cinnam}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}^{\centerdot }} + {{{\text{H}}}^{ + }}. \\ \end{gathered} $Величина кинетического эффекта растворителя будет определяться его диэлектрической проницаемостью (ε). Механизм переноса электрона считается традиционно более быстрым, чем перенос атома [6, 26].
Таким, образом, чтобы оценить возможность протекания реакций (I) или (II) с участием молекулярных форм Cinnam–OH, необходимо подбирать растворитель, руководствуясь параметрами ε и ${\beta }_{2}^{{\text{Н}}}$. Так, если к ДМСО с ε = 78.3 и ${\beta }_{2}^{{\text{Н}}}$ = 0.78 добавлять бензол с ε = 2.3 и ${\beta }_{2}^{{\text{Н}}}$ = 0.14 [15, 25], то стоит ожидать снижения реакционной способности (k) гидроксикоричных кислот за счет замены механизма ЕТ–РТ на более медленный механизм НАТ.
При добавлении в исследуемую систему бензола реакционная способность кислот по отношению к DPPH• уменьшается (табл. 2), а в чистом бензоле в заданных условиях некоторые соединения (I–III) фактически не реагируют с гидразильным радикалом (kбенз < 1 × 10–3 л моль–1 с–1, табл. 1). По величине кинетического эффекта растворителя (КЭР, ${\mathbf{КЭР}} = \frac{{{{k}_{{{\text{ДМСО}}}}}}}{{{{k}_{{{\text{бенз}}}}}}}~$) видно, что скорость исследуемой реакции в ДМСО превышает таковую в бензоле в среднем на порядок (табл. 2). Это может быть связано как с влиянием специфической сольватации – образованием водородных связей радикала с молекулами растворителя, так и с неспецифической сольватацией – увеличением диэлектрической проницаемости среды. Установленная линейная зависимость в координатах уравнения Лейдлера–Эйринга [21] показывает (рис. 1б), что скорость исследуемой реакции в бинарной смеси сильно зависит от ε растворителя.
(2)
$\begin{gathered} {\text{lg\;}}{\kern 1pt} {{k}_{{{\text{ДМСО}} - {\text{бенз}}}}} = {\text{lg\;}}{\kern 1pt} {{k}_{0}} - \frac{{2.3}}{{RT}} \times \\ \times \,\,\left( {\frac{{{\mu }_{{{\text{Cinnam}} - {\text{OH}}}}^{2}}}{{r_{{{\text{Cinnam}}{\kern 1pt} - {\text{OH}}}}^{3}}} + \frac{{{\mu }_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}^{2}}}{{r_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}^{3}}} - \frac{{{\mu }_{ \ne }^{2}}}{{r_{ \ne }^{3}}}} \right)\frac{{\left( {{\varepsilon }~ - ~1} \right)}}{{\left( {2{\varepsilon }~ + ~1} \right)}}, \\ \end{gathered} $Таблица 2.
Зависимость экспериментальных величин констант скоростей (kДМСО–бенз) реакции Cinnam–OH с DPPH• от содержания бензола в смеси с ДМСО
| Соединение | kДМСО–бенз, л моль–1 с–1 | КЭР | ДИЭ | ||||
|---|---|---|---|---|---|---|---|
| содержание бензола в смеси с ДМСО, об. % | $\frac{{{{k}_{{{\text{ДМСО}}}}}}}{{{{k}_{{{\text{бенз}}}}}}}$ | $\frac{{{{k}_{{{\text{ДМСО}}}}}}}{{{{k}_{{{\text{ДМСО(Cinnam}} - {\text{OD}})}}}}}$ | $\frac{{{{k}_{{{\text{бенз}}}}}}}{{{{k}_{{{\text{бенз(Cinnam}} - {\text{OD}})}}}}}$ | ||||
| 80 | 50 | 30 | 10 | ||||
| IV | (1.59 ± 0.05) × 102 | (3.31 ± 0.12) × 102 | (4.22 ± 0.14) × 102 | (4.59 ± 0.13) × 102 | 8 | 0.96 | 1.94 |
| V | (1.12 ± 0.05) × 102 | (2.49 ± 0.12) × 102 | (3.32 ± 0.12) × 102 | (3.78 ± 0.13) × 102 | 13 | 1.01 | 2.32 |
| VI | (1.19 ± 0.05) × 102 | (2.82 ± 0.12) × 102 | (3.59 ± 0.12) × 102 | (4.11 ± 0.13) × 102 | 13 | 1.01 | 1.92 |
| VII | (1.33 ± 0.05) × 102 | (2.89 ± 0.12) × 102 | (3.82 ± 0.13) × 102 | (4.32 ± 0.13) × 102 | 12 | 0.98 | 2.13 |
| VIII | (1.09 ± 0.05) × 102 | (2.11 ± 0.11) × 102 | (2.62 ± 0.12) × 102 | (2.89 ± 0.12) × 102 | 8 | 0.96 | 2.45 |
С увеличением скорости реакции при переходе от менее полярной к более полярной смеси растворителей наблюдается прямая пропорциональная зависимость и величина углового параметра $\frac{{2.3}}{{RT}}\left( {\frac{{{\mu }_{{{\text{Cinnam}} - {\text{OH}}}}^{2}}}{{r_{{{\text{Cinnam}} - {\text{OH}}}}^{3}}} + \frac{{\mu _{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}^{2}}}{{r_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}}}^{3}}} - \frac{{\mu _{ \ne }^{2}}}{{r_{ \ne }^{3}}}} \right){\text{\;}}~$линейной регрессии (2) остается положительной (рис. 1б). Это указывает на то, что активный комплекс реакции является более полярным и легче сольватируется, чем исходные реагенты, что ускоряет протекание реакции. В связи с этим можно полагать, что в полярных средах с низкой ионизирующей способностью доминирующее влияние на антирадикальную активность Cinnam–OH оказывает не специфическая сольватация, а именно рост диэлектрической проницаемости среды, способной изменять механизм реакции. По-видимому, увеличение ε приводит к существенному разделению заряда в переходном состоянии реакции (II) и способствует дальнейшему распаду на ионы активного комплекса с переносом заряда.
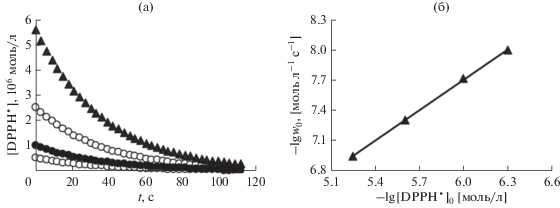
Высокая чувствительность скорости реакции к полярности среды характерна для реакций с лимитирующей стадией переноса электрона, что подтверждается разными значениями дейтериевого изотопного эффекта${\text{\;ДИЭ}} = \frac{{{{k}_{{{\text{ДМСО}}}}}}}{{{{k}_{{{\text{ДМСО}}\left( {{\text{Cinnam}} - {\text{OD}}} \right)}}}}}$ в среде ДМСО и бензола (табл. 2). Константы скорости реакции в диметилсульфоксидном растворе DPPH• в присутствии легкой кислоты (${{k}_{{{\text{ДМСО}}}}}$) и ее дейтеропроизводного (${{k}_{{{\text{ДМСО}}\left( {{\text{Cinnam}} - {\text{OD}}} \right)}}}$) фактически не отличаются (рис. 3). Величина ДИЭ, равная 1, подтверждает отсутствие переноса протона в реакции (II), а значит, возможна реализация реакции DPPH• с Cinnam–OH в ДМСО по пути ЕТ–РТ. Наличие же выраженного эффекта в бензоле (рис. 2, табл. 2) свидетельствует о реализации механизма НАТ в неполярных средах, что согласуется с литературными значениями ДИЭ для природных фенолов в реакции с DPPH• в циклогексане [7].
Рис. 3.
Кинетические данные расходования DРРН• в реакции с синаповой кислотой (⚪) (С = 9 × 10–5 моль/л) и с ее дейтеропроизводным (⚫) (С = 9 × 10–5 моль/л) в ДМСО (1) и бензоле (2) при Т = 293 ± 2 К.
На основании полученных данных можно предположить, что механизм переноса электрона от молекулярной формы фенолокислоты будет реализовываться и в водных средах с низким рН, при котором подавляется процесс ионизации Cinnam–OH. Установление механизма действия фенилпропаноидов в кислых средах крайне важно при оценке их реакционной способности как ингибиторов радикальных процессов в биологических системах.
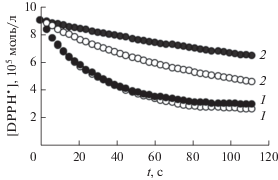
Поскольку гидразильный радикал не растворим в воде, то определение АРА (kрН 2) гидроксикоричных кислот проводили в смеси ДМСО с солянокислым буфером (рН 2) в соотношении 10 : 90 (об. %, рис. 4), при котором диэлектрическая проницаемость смеси (ε = 75.9) близка к таковой в воде (ε = 78.3 [15]).
Рис. 4.
Кинетические кривые расходования DРРН• в реакции с Cinnam–OH (С = 8 × 10–5 моль/л): 1 – кофейная кислота, 2 – синаповая кислота, 3 – п-кумаровая кислота. Растворитель – ДМСО–солянокислый буфер с рН 2 (10 : 90, об. %), Т = 293 ± 2 К.
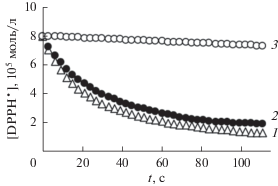
По определенным величинам kрН 2 видно (табл. 1), что в буфере все изученные АО проявляют несколько более высокую активность, чем в ДМСО, что связано, вероятно, с ускорением механизма переноса электрона за счет увеличения ε среды при переходе от ДМСО к водному раствору. В кислой среде наиболее активны соединения, относящиеся к дигидроксикоричным кислотам. Уменьшение числа функциональных фенольных ОН-групп, как и в ДМСО, приводит к резкому падению реакционной способности соединений. Так, величины kрН 2 для кумаровых кислот (I–III) малы и достигают порядка 10 л моль–1 с–1. При этом наблюдается симбатная зависимость антирадикальной активности в апротонном полярном растворителе (ln kДМСО) с данными в кислом буфере (ln kрН 2).
(3)
$\begin{gathered} \ln {{k}_{{{\text{ДМСО\;}}}}} = --\left( {0.26 \pm 0.05} \right) + \left( {1.01 \pm 0.01} \right)\ln {{k}_{{{\text{рН}}{\kern 1pt} {\kern 1pt} 2}}}, \\ n = 8,\,\,\,\,r = 0.996,\,\,\,\,{{r}^{2}} = 0.992, \\ F = 1122,\,\,\,p < 0.00001, \\ \end{gathered} $Полученная линейная зависимость (уравнение (3)) (с высоким коэффициентом корреляции r = 0.996) опосредованно подтверждает наши предположения о реализации механизма ЕТ–РТ при протекании реакции Cinnam–OH с DPPH• в сильнокислых средах.
ЗАКЛЮЧЕНИЕ
Природные фенилпропаноиды группы гидроксикоричных кислот реагируют с радикалом DPPH• в полярном апротонном растворителе по механизму переноса электрона от молекулярной формы кислоты к радикалу с последующим быстрым переносом протона от образующегося катион-радикала, что подтверждается величинами кинетического эффекта растворителя и дейтериевого изотопного эффекта при сравнении с реакцией в неполярной среде. Подобный механизм может реализовываться в водных средах с повышенной кислотностью. Установление механизма антирадикального действия гидроксикоричных кислот в модельных реакциях позволит корректировать скорость протекания радикальных процессов и в реальных системах, что является определяющим фактором при подборе природных фенилпропаноидов как эффективных антиоксидантов.
Список литературы
Vermerris W., Nicolson R. Phenolic Compound Biochemistry. Dodrecht: Springer, 2006. 276 p.
Li J., He D., Wang B., Zhang L., Li K., Xie Q., Zheng L. // Acta Pharm. Sin. B. 2017. V. 7. № 1. P. 106.
Alam M.A., Subhan N., Hossain H., Hossain M., Reza H.M., Rahman M.M., Ullah M.O. // Nutr. Metab. 2016. V. 13. № 1. P. 13.
Galano A., Mazzone G., Alvarez-Diduk R., Marino T., Alvarez-Idaboy J.R., Russo N. // Annu. Rev. Food Sci. T. 2016. V. 7. P. 335.
Amić A., Marković Z., Klein E., Dimitrić Marković J.M., Milenkoviće D. // Food Chem. 2018. V. 246. № 25. P. 481.
Litwinienko G., Ingold K.U. // Acc. Chem. Res. 2007. V. 40. № 3. P. 222.
Foti M.C., Daquino C., Mackie I.D., DiLabio G.A., Ingold K.U. // J. Org. Chem. 2008. V. 73. P. 9270.
Mazzone G., Russo N., Toscano M. // Comput. Theoret. Chem. 2016. V. 1077. P. 39.
Galano A., Alvarez-Idaboy J.R. // J. Comput. Chem. 2013. V. 34. P. 2430.
Milenković D., Yorović J., Jeremić S., Marković J.M.D., Avdović E.H., Marković Z. // J. Chem. 2017. V. 2017. P. 1.
Galano A., Alvarez-Idaboy J.R. // Int. J. Quantum. Chem. 2019. V. 119. № 2. P. 1.
Yeo J.D., Shahidi F. // J. Agric. Food Chem. 2019. V. 67. № 26. P. 7526.
Hamlaoui I., Bencheraiet R., Bensegueni R., Bencharif M. // J. Mol. Struct. 2018. V. 1156. № 15. P. 385.
Armarego W.L.F., Chai C.L.L. Purification of Laboratory Chemicals. Burlington: Elsevier Science, 2003. 608 p.
Рабинович В.А., Хавин З.Я. Краткий химический справочник. Л.: Химия, 1991. 432 с.
Блументаль Г., Энгельс 3., Фиц И., Хабердитцль В., Хекнер К.Х., Хенрион Г., Ландсберг Р., Шмидт В., Шольц Г., Штарке П., Вильке И., Вильке К.Т. Анорганикум. Т. 1. М.: Мир, 1984. 672 с.
Дероум Э. Современные методы ЯМP для химических исследований. М.: Мир, 1992. 403 с.
Сухарев А.Г., Тимохов А.В., Федоров В.В. Курс методов оптимизации. М.: Физматлит, 2005. 368 с.
Белая Н.И., Белый А.В., Заречная О.М., Щербаков И.Н., Помещенко А.И., Горбань О.А. // Кинетика и катализ. 2019. Т. 60. № 1. С. 33.
Amić A., Marković Z., Dimitrić Marković J.M., Milenković D., Stepanić V. // Phytochemistry. 2020. V. 170. art. 112218.
Денисов Е.Т. Кинетика гомогенных химических реакций. М.: Высш. шк., 1978. 367 с.
Kikuzaki H., Hisamoto M., Hirose K., Akiyama K., Taniguchi H. // J. Agric. Food Chem. 2002. V. 50. № 7. P. 2161.
Litwinienko G., Ingold K.U. // J. Org. Chem. 2005. V. 70. № 68. P. 8982.
Shang Y.-J., Liu B.-Y., Zhao M.-M. // Czech J. Food Sci. 2015. V. 33. P. 210.
Abraham M.H., Grellier P.L., Prior D.V, Morris J.J., Taylor P.J. // J. Chem. Soc., Perkin Trans. 2. 1990. № 4. P. 521.
Ингольд К., Робертс Б. Реакции свободнорадикального замещения. М.: Мир, 1974. 255 с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ