

Кинетика и катализ, 2020, T. 61, № 6, стр. 873-887
Анализ гетерогенно-гомогенной модели окислительной конденсации метана с использованием процедуры редуцирования кинетических схем
В. И. Ломоносов a, *, М. Ю. Синев a
a ФГБУН Федеральный исследовательский центр химической физики им. Н.Н. Семенова РАН (ФИЦ ХФ РАН)
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: vlomonosov@mail.ru
Поступила в редакцию 23.03.2020
После доработки 10.04.2020
Принята к публикации 10.04.2020
Аннотация
Проанализирована возможность использования редуцирования кинетических схем большого объема для сравнения различных кинетических моделей и параметров отдельных реакций, представленных в литературе. Рассмотрены особенности формирования блока газофазных реакций, входящего в гетерогенно-гомогенную кинетическую модель процесса окислительной конденсации метана (ОКМ) и проанализированы подходы к выбору кинетических констант элементарных стадий. Показано, что кинетические модели, полученные при строгом соблюдении принципа “независимости кинетических параметров”, могут обладать низкой предсказательной способностью ввиду существующих неопределенностей в величинах параметров, приведенных в широко известных обзорных работах и базах данных. Отдельно рассмотрен вопрос о том, как учет блока гетерогенных реакций и варьирование условий протекания процесса ОКМ влияют на результаты редуцирования детальной кинетической схемы процесса. Показано, что использование процедуры редуцирования для анализа механизма сложного процесса ограничено ввиду высокой степени сопряжения между его отдельными стадиями, а также сильной зависимости кинетических констант от физических параметров.
Процессы парциального окисления и окислительного дегидрирования легких алканов – основных компонентов природного и попутных нефтяных газов – рассматриваются как перспективная альтернатива существующим технологиям получения основных полупродуктов органического и нефтехимического синтеза из нефтяного сырья [1]. Особое место среди них занимает окислительная конденсация метана (ОКМ) с получением высших углеводородов, включая этилен [2–6]. С одной стороны, метан является наиболее распространенным и дешевым углеводородным сырьем. С другой стороны, этилен – один из наиболее востребованных и дефицитных полупродуктов нефтехимии.
Характерная особенность реакции ОКМ состоит в том, что активация метана происходит с участием активных центров поверхности катализатора и сопровождается выходом в объем газовой фазы свободных радикалов [7–9]. При этом молекулярные продукты образуются в сложной цепочке превращений промежуточных частиц радикальной природы как в газовой фазе, так и с участием активных центров поверхности [10, 11]. По этой причине кинетические закономерности процесса определяются наличием сопряжения гетерогенной и гомогенной составляющих. Экспериментально наблюдаемые величины скорости реакции являются сложной комбинацией гетерогенных и гомогенных процессов, причем активные центры катализатора играют двойную роль, участвуя как в генерации первичных радикалов, так и в обрыве гомогенных цепей [10, 12]. С учетом того, что катализаторы ОКМ весьма эффективны также в процессах высокотемпературного окислительного дегидрирования и крекинга алканов C2+, которые протекают по аналогичному механизму [10, 13], реакция ОКМ может рассматриваться в качестве удобной модели при изучении широкого класса процессов окисления углеводородов. Для анализа их кинетики приходится использовать сложные многостадийные схемы, которые позволяют проследить основные закономерности развития процесса во времени, выявить маршруты образования продуктов, спрогнозировать влияние различных факторов на параметры процесса. Такие модели являются эффективным инструментом для изучения детального механизма реакции, а также позволяют объяснять и предсказывать сложные кинетические явления, сопровождающие процессы рассматриваемого типа.
Вскоре после открытия процесса ОКМ были предприняты первые попытки объяснить некоторые его закономерности участием свободных радикалов и кинетикой их превращений [14–16], а также построить кинетическое описание процесса в целом с помощью гетерогенно-гомогенных схем [17–19]. В настоящее время наиболее распространен подход, согласно которому кинетическая схема гетерогенно-гомогенного процесса формируется на основании двух блоков – гомогенного и гетерогенного (см., например, [11, 12, 20]). Первый представляет собой детальное описание процессов, протекающих при окислении углеводородов в газовой фазе. Все элементарные стадии с участием молекулярных и радикальных частиц, образующихся при гомогенном окислении, протекают и в ходе каталитических процессов, сопровождающихся выходом свободных радикалов в газовую фазу. Второй блок необходим для описания взаимодействия этих частиц с центрами поверхности катализаторов. Разработка обоих блоков требует соблюдения ряда принципов, включающих полноту и открытость описания, его термодинамическую согласованность, а также независимость кинетических параметров [11].
В настоящей работе рассмотрены особенности формирования гомогенного блока кинетической модели; основное внимание при этом уделено такому важному аспекту, как выбор кинетических параметров элементарных стадий. В доступных источниках представлено большое число кинетических моделей газофазного окисления углеводородов, отличающихся как по количеству частиц и стадий, так и по принципу их наполнения кинетическими параметрами. Наиболее детально проработанной кинетической схемой, использующейся для анализа превращений легких алканов, сегодня, по-видимому, следует считать модель окисления С1–С5-углеводородов, разработанную в Научном центре химии процессов горения университета г. Галвэй (Северная Ирландия) [21]. Она построена по иерархическому принципу и включает блоки окисления водорода, СО, углеводородов С1–С5, а также различных кислородсодержащих соединений. Разработка модели была начата в 2010 г. и продолжается до сих пор. Последняя ее версия Aramco 3.0 (далее AramcoMech), включающая 3037 реакций 581 частицы, была опубликована в 2018 г. [22]. За время разработки было внесено большое число уточнений и поправок как в стадийную схему, так и в константы скорости отдельных элементарных стадий [23–30]. Более того, в ряде случаев уточнялись также термодинамические параметры некоторых веществ, участвующих в описываемых процессах (см., например, [30]). Важным достоинством этого семейства моделей, на наш взгляд, является то, что их разработчики избегают “подгонки” параметров отдельных стадий для повышения точности описания. При выборе кинетических констант используются наиболее свежие и, по мнению авторов модели, наиболее надежные литературные источники. Дальнейшая оптимизация включает детальный анализ всей схемы и ее отдельных блоков (под-механизмов), который проводится следующим образом. Например, рассматривался процесс окисления метана в реакторах различного типа (в проточном реакторе постоянного давления, адиабатическом реакторе, реакторе с реактивным перемешиванием, эксперименты в ударных трубах и др.), и анализируется чувствительность по отдельным элементарным стадиям [30]. Далее кинетические параметры выбранных стадий выверяются на основании сопоставления результатов расчетов и независимых экспериментальных данных. При этом рассматриваются только кинетические данные, опубликованные в открытых источниках (включая как экспериментальные, так и теоретические оценки). Другими словами, разработчики стремятся максимально исключить механическую (или лучше сказать математическую) подгонку кинетических параметров и одновременное варьирование параметров значительного числа элементарных стадий. Верификация итоговой модели проводилась на большом массиве независимых экспериментальных данных в широком интервале начальных условий. При этом результаты расчетов сравнивались также с результатами, полученными при использовании других кинетических схем [31–36].
Во многих случаях модель AramcoMech позволяла получить наиболее точное описание имеющихся экспериментальных данных. Результаты сравнительного анализа находятся в открытом доступе [21]. Как было отмечено выше, эту схему можно считать наиболее полной и согласованной из представленных на сегодня в литературе. Однако принятый ее разработчиками подход к выбору кинетических параметров отдельных стадий вызывает некоторые вопросы. В частности, в [30] утверждается, что наполнение модели кинетическими параметрами не должно осуществляться без привязки к экспериментальным данным и их сопоставления с результатами моделирования. Другими словами, по мнению авторов, построение модели, обладающей высокой предсказательной силой и вычислительной точностью невозможно в рамках “априорного” выполнения принципа “независимости кинетических параметров” – использования при построении кинетических моделей значений параметров, представленных в наиболее авторитетных обзорах и базах данных. То есть необходима проверка того, в какой степени разные данные по кинетическим параметрам, представленные в литературе, позволяют в лучшей степени описать имеющиеся экспериментальные данные. На наш взгляд, представляет несомненный интерес провести независимую оценку справедливости данного утверждения. Для этого в настоящей работе сравниваются результаты расчетов по моделям, параметры которых получены в соответствии с двумя различающимися подходами: при строгом следовании принципу использования кинетических параметров, определенных из независимых экспериментов [11], и при более “мягком” к нему отношении, позволяющем варьирование величин параметров в неких пределах, допускаемых имеющимся в литературе разбросом данных [22].
Одной из наиболее известных кинетических схем, при разработке которой был соблюден принцип независимости кинетических параметров, по-видимому, является модель окисления метана, разработанная в начале 2000-х гг. сотрудниками университета г. Лидс (Великобритания) [34]. Модель также содержит блоки окисления водорода, монооксида углерода, этана и этилена. При ее разработке авторы полностью отказались от возможности “подгонки” кинетических параметров для достижения точности описания, однако в отличие от работы [22] наполнение модели осуществлялось без привязки к каким-либо экспериментальным данным. При выборе значений кинетических констант использовались открытые базы данных [37, 38], при этом был предложен алгоритм, в котором источники были расставлены в порядке наибольшей достоверности предоставляемых сведений [39]. При расчете констант скорости обратных реакций использовались термодинамические данные пакета программ CHEMKIN [40], что делало схему термодинамически согласованной. Было показано, что полученная кинетическая модель позволяет довольно точно описать экспериментальные данные о скорости распространения ламинарного пламени, а также задержек воспламенения при окислении метана. Как было отмечено выше, верификацию модели [22] проводили на гораздо большем массиве независимых экспериментальных данных и в более широком интервале начальных условий. При этом сравнительный анализ, проведенный ее разработчиками, показал, что в некоторых случаях разница в результатах расчетов по схемам [22] и [34] оказывается весьма значительной. Однако нельзя однозначно утверждать, связано ли это с разницей подходах к выбору кинетических параметров или нет. Строго говоря, прямое сравнение результатов расчетов по схемам [22] и [34] не вполне корректно, поскольку рассматриваемые модели изначально создавались для решения различных задач и существенно различаются по набору элементарных стадий. Наполнение же стадийной схемы [22] кинетическими параметрами на основе “независимого” подхода, использованного в [34], потребует значительного времени. По этой причине в качестве альтернативного подхода для анализа различных описаний и наборов параметров нами предпринято сравнение кинетических схем, полученных редуцированием “полных” моделей с использованием стандартных подходов. В данной статье описаны результаты такого анализа, в котором в качестве базовой “полной” схемы была выбрана модель [22]. Кроме того, изучено влияние начальных условий и учета блока гетерогенных реакций на результаты редуцирования “полной” схемы с различными наборами кинетических параметров.
Редуцирование больших кинетических моделей имеет самостоятельное значение. Возрастающая роль вычислительных методов в исследовании различных процессов, разработке технологий и аппаратуры (в том числе химических производств) требует создания достаточно компактных кинетических моделей, которые могут включаться в расчеты гидродинамики, тепло- и массопереноса. С этой точки зрения представляет несомненный интерес то, в какой степени возможно снижение объема “полных” кинетических схем (по числу включенных в модель компонентов и реакций между ними) с сохранением заданной точности описания. Учитывая высокую степень сопряженности кинетики гетерогенно-гомогенных процессов и сильную зависимость констант скорости отдельных стадий от физических параметров (в первую очередь от температуры), мы рассмотрели также вопрос о том, как учет блока гетерогенных реакций и варьирование условий протекания процесса ОКМ влияют на результаты редуцирования “полной” схемы.
МЕТОДИКА ВЫЧИСЛЕНИЙ
В данной работе использовали программный пакет Chemical WorkBench, разработанный в компании КинтехЛаб (Россия) [41]. Он позволяет проводить расчеты сложных многостадийных процессов, протекающих в различных условиях (режимах, типах реакторов). Описание методов редуцирования многостадийных кинетических схем, а также их основные особенности подробно изложены в работе [42]. Редуцирование кинетической схемы [22] осуществляли путем последовательного использования трех алгоритмов:
– DRG – метод анализа графа прямых связей,
– CSP – граф вычислительных сингулярных возмущений,
– DSA – метод прямого анализа чувствительности.
Все расчеты проводили для режима непроточного изотермического реактора (при температурах 900, 1000, 1100 и 1200 К), работающего при постоянном давлении (1–10 атм), для состава исходной смеси (в мол. %), равного 79CH4, 20O2 и 1N2. Критерием оценки приближения редуцированного механизма к исходному (“полному”) было выбрано следующее условие: максимальное относительное отклонение концентраций реагентов (CH4, O2) и основных продуктов (CO2, CO, C2H6, C2H4, H2) при расчетах по “полной” и редуцированной схемам в заданный момент времени не должно превышать заранее заданной величины. В англоязычной литературе этот параметр – допустимое отклонение – принято обозначать термином “relative tolerance” (RT). Чтобы оценить, в какой степени этот параметр влияет на результат редуцирования, на начальном этапе были заданы два его значения – 0.05 и 0.1. При дальнейших вычислениях принималась величина RT, равная 0.1.
Для наполнения редуцированной модели кинетическими параметрами в рамках “независимого” подхода использовали базу данных Национального Института Стандартов и Технологий “NIST Chemical Kinetics Database” [37]. В качестве приоритетных источников были выбраны работы [43–46]. В случае отсутствия в них сведений по какой-то из стадий кинетические параметры не заменяли, т.е. использовали параметры, приведенные в работе [22].
Для сравнительного анализа редуцированных схем с разным набором кинетическим параметров использовали метод прямого анализа чувствительности, который заключается в определении изменения концентрации компонента i при варьировании константы скорости стадии j в заданный момент времени. Этот метод, с одной стороны, позволяет выделить стадии, вносящие основной вклад в превращение отдельных компонентов реакционной смеси, а с другой – оценить степень влияния погрешности в определении кинетических параметров отдельных стадий на результаты расчета.
При расчетах гетерогенно-гомогенной схемы использовали ранее описанный подход [12]: к “полной” схеме AramcoMech добавляли блок гетерогенных реакций, состав которого и величины констант скорости были определены в соответствии с ранее разработанной методикой (см. [11, 47–49]). Принималось, что частицы, моделирующие каталитические активные центры, распределяются гомогенно. Их концентрацию варьировали от 0 до величины, соответствующей усредненной концентрации поверхностных центров в единице объема гранул твердого вещества, имеющего насыпную плотность 1 г/см3 и удельную поверхность 10 м2/г, т.е. ~2 × 1019 см–3. Это практически перекрывает диапазон усредненных концентраций поверхностных центров всех наиболее эффективных катализаторов ОКМ в пересчете на единицу объема реакционной зоны.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние величины допустимого отклонения на результаты редуцирования
На начальном этапе была проведена проверка влияния заданной величины параметра RT на результаты редуцирования кинетической схемы AramcoMech. Расчет проводили для изотермического реактора в интервале температур 900–1000 К и давлений 1–10 атм. В данном случае для упрощения анализа учитывали реакции только частиц, имеющих в своем составе не более трех атомов углерода. В результате получено два описания, основные параметры которых приведены в табл. 1. Из данных таблицы видно, что при допущении более грубого приближения удается существенно сократить редуцированную кинетическую схему.
Менее ожидаемым был другой результат. Оказалось, что в существенно более “компактной” схеме, полученной при RT = 0.1, присутствует несколько реакций, которые не вошли в редуцированную схему при RT = 0.05. Очевидно, исчезновение из описания даже реакций, не вносящих большого вклада в суммарное превращение/образование определенных компонентов реакционной смеси, приводит к существенному перераспределению вклада различных маршрутов реакции. Это является важным указанием на то, что использованная процедура, будучи сугубо формальной, может применяться для решения задач оптимизации, но не пригодна для анализа механизма сложного процесса.
Результаты редуцирования схемы AramcoMech с разными наборами кинетических параметров
Недостатком метода прямого анализа чувствительности является то, что определяемая величина, носящая название “чувствительность” – производная dCi/d(lnkj) – зависит в том числе и от того, какие кинетические параметры изначально заложены в модель. То же самое можно сказать и о других методах редуцирования, использованных в настоящей работе.
Сравнение кинетических схем, полученных редуцированием различных моделей, не может выявить влияния величин параметров на результат редуцирования ввиду структурных различий исходных описаний. Для того, чтобы оценить это влияние напрямую, был выбран следующий подход. Было проведено редуцирование исходной модели AramcoMech при 1000 К и атмосферном давлении. После этого кинетические параметры стадий, вошедших в редуцированную схему, были заменены в исходной модели на приведенные в работах [31–34], а затем проведено редуцирование такой модифицированной кинетической модели. Для упрощения в дальнейшем схема, полученная редуцированием с кинетическими параметрами, принятыми в работе [22], будет называться “исходной редуцированной”, а с параметрами, внесенными из базы данных [37] – “модифицированной”. Стадийная схема, полученная при редуцировании, два набора кинетических параметров, а также значения констант скоростей, рассчитанные для температуры 1000 К, приведены в табл. 2.
Таблица 2.
Стадийная схема, полученная при редуцировании модели AramcoMech 3.0 (1000 К, 1 атм)*
| Реакция | AramcoMech 3.0 [22] | База данных NIST [37]*** | k (NIST)/ k (Aramco) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A** | N | Ea, ккал/моль |
k (1000 К) | A** | N | Ea, ккал/моль |
k (1000 К) | лит. ссылка |
|||
| 1. | O2 + H ⇄ O + OH | 1.73 × 10–10 | 0 | 15.29 | 7.88 × 10–14 | 1.62 × 10–10 | 0 | 14.84 | 9.25 × 10–14 | [46] | 1.17 |
| 2. | H2O2 + M ⇄ OH + OH + M | Аппроксимация Troe | 8.99 × 10–18 | 2.01 × 10–7 | 0 | 45.51 | 2.27 × 10–17 | [46] | 2.53 | ||
| 3. | HO2 + HO2 ⇄ H2O2 + O2 | 1.66 × 10–10 | 0 | 11.04 | 6.41 × 10–13 | 7.01 ×1 0–10 | 0 | 11.98 | 1.69 × 10–12 | [46] | 2.63 |
| 4. | HO2 + HO2 ⇄ H2O2 + O2 | 3.16 × 10–13 | 0 | –1.41 | 6.41 × 10–13 | Данные отсутствуют | – | ||||
| 5. | H + O2 + M ⇄ HO2 + M | Аппроксимация Troe | 9.23 × 10–33 | 3.89 × 10–30 | –0.80 | 0.00 | 1.55 × 10–32 | [46] | 1.68 | ||
| 6. | CO + OH ⇄ CO2 + H | 1.16 × 10–19 | 2.05 | –0.36 | 2.01 × 10–13 | 1.05 × 10–17 | 1.50 | –2.08 | 9.46 × 10–13 | [45] | 4.71 |
| 7. | CO + OH ⇄ CO2 + H | 9.56 × 10–12 | –0.66 | 0.33 | 8.24 × 10–14 | Данные отсутствуют | – | ||||
| 8. | CO + HO2 ⇄ CO2 + OH | 2.61 × 10–19 | 2.18 | 17.94 | 1.08 × 10–16 | 2.51 × 10–10 | 0 | 23.65 | 1.70 × 10–15 | [43] | 15.69 |
| 9. | CH3 + H + M ⇄ CH4 + M | Аппроксимация Troe | 5.07 × 10–30 | 1.71 × 10–24 | –1.80 | 0.00 | 6.80 × 10–30 | [45] | 1.34 | ||
| 10. | CH4 + H ⇄ CH3 + H2 | 1.02 × 10–18 | 2.50 | 9.59 | 2.59 × 10–13 | 2.20 × 10–20 | 3.00 | 8.03 | 3.87 × 10–13 | [45] | 1.50 |
| 11. | CH4 + O ⇄ CH3 + OH | 1.69 × 10–15 | 1.50 | 8.60 | 7.07 × 10–13 | 1.15 × 10–15 | 1.56 | 8.49 | 7.67 × 10–13 | [45] | 1.09 |
| 12. | CH4 + OH ⇄ CH3 + H2O | 9.68 × 10–20 | 2.60 | 2.19 | 2.03 × 10–12 | 2.60 × 10–17 | 1.83 | 2.78 | 1.98 × 10–13 | [45] | 0.98 |
| 13. | CH4 + HO2 ⇄ CH3 + H2O2 | 1.88 × 10–23 | 3.74 | 21.01 | 7.97 × 10–17 | 1.50 × 10–11 | 0 | 24.64 | 6.17 × 10–17 | [45] | 0.78 |
| 14. | CH4 + CH3O2 ⇄ CH3 + CH3O2H | 1.59 × 10–24 | 3.77 | 17.81 | 4.17 × 10–17 | 3.01 × 10–13 | 0 | 18.48 | 2.75 × 10–17 | [43] | 0.66 |
| 15. | CH3 + HO2 ⇄ CH4 + O2 | 1.93 × 10–19 | 2.23 | –3.02 | 4.32 × 10–12 | 5.99 × 10–12 | 0 | 0.00 | 5.99 × 10–12 | [43] | 1.39 |
| 16. | CH3 + O2 + M ⇄ CH3O2 + M | Аппроксимация Troe | 1.11 × 10–32 | 1.59 × 10–22 | –3.30 | 0.00 | 2.01 × 10–32 | [45] | 1.80 | ||
| 17. | CH3 + HO2 ⇄ CH3O + OH | 1.66 × 10–12 | 0.27 | –0.69 | 1.50 × 10–11 | 3.01 × 10–11 | 0 | 0.00 | 3.01 × 10–11 | [45] | 2.00 |
| 18. | CH3 + O2 ⇄ CH2O + OH | 4.39 × 10–24 | 3.28 | 8.11 | 5.24 × 10–16 | 5.50 × 10–13 | 0 | 8.94 | 6.11 × 10–15 | [45] | 11.66 |
| 19. | CH3O2 + CH3 ⇄ CH3O + CH3O | 8.44 × 10–12 | 0 | –1.41 | 1.72 × 10–11 | 4.00 × 10–11 | 0 | 0.00 | 4.00 × 10–11 | [43] | 2.33 |
| 20. | CH3O2H ⇄ CH3O + OH | 6.31 × 1014 | 0 | 42.30 | 3.59 × 105 | 6.00 × 1014 | 0 | 42.33 | 3.36 × 105 | [46] | 0.94 |
| 21. | CH3O + O2 ⇄ CH2O + HO2 | 7.27 × 10–43 | 9.50 | –5.50 | 3.66 ×10–13 | 3.60 × 10–14 | 0 | 1.75 | 1.49 × 10–14 | [46] | 0.04 |
| 22. | CH2O + O2 ⇄ HCO + HO2 | 1.34 × 10–8 | 0 | 53.42 | 2.83 × 10–20 | 1.00 × 10–10 | 0 | 40.74 | 1.25 × 10–19 | [45] | 4.41 |
| 23. | CH2O + H ⇄ HCO + H2 | 9.53 × 10–17 | 1.90 | 2.74 | 1.20 × 10–11 | 2.10 × 10–16 | 1.62 | 2.17 | 5.10 × 10–12 | [46] | 0.42 |
| 24. | CH2O + HO2 ⇄ HCO + H2O2 | 3.12 × 10–20 | 2.70 | 11.52 | 1.19 × 10–14 | 5.00 × 10–12 | 0 | 13.08 | 6.92 × 10–15 | [45] | 0.58 |
| 25. | CH2O + CH3 ⇄ HCO + CH4 | 6.36 × 10–23 | 3.36 | 4.31 | 8.73 × 10–14 | 1.29 × 10–31 | 6.10 | 1.97 | 9.57 × 10–14 | [46] | 1.10 |
| 26. | CH2O + CH3O2 ⇄ HCO + CH3O2H | 3.30 × 10–12 | 0 | 11.66 | 9.35 × 10–15 | 3.30 × 10–12 | 0 | 11.66 | 9.33 × 10–15 | [43] | 1.00 |
| 27. | HCO + M ⇄ H + CO + M | 9.47 × 10–13 | 0.66 | 14.87 | 5.08 × 10–14 | Данные отсутствуют | – | ||||
| 28. | HCO + O2 ⇄ CO + HO2 | 1.26 × 10–11 | 0 | 0.41 | 1.02 × 10–11 | 8.50 × 10–11 | 0 | 1.69 | 3.63 × 10–11 | [43] | 3.55 |
| 29. | CH3O + M ⇄ CH2O + H + M | Аппроксимация Troe | 1.20 × 10–13 | Данные отсутствуют | – | ||||||
| 30. | CH3 + CH3 + M ⇄ C2H6 + M | Аппроксимация Troe | 2.57 × 10–30 | 3.51 × 10–7 | –7.00 | 2.76 | 8.74 × 10–29 | [46] | 34.00 | ||
| 31. | C2H6 + H ⇄ C2H5 + H2 | 1.91 × 10–16 | 1.90 | 7.53 | 2.16 × 10–12 | 2.39 × 10–15 | 1.50 | 7.41 | 1.82 × 10–12 | [45] | 0.84 |
| 32. | C2H6 + OH ⇄ C2H5 + H2O | 2.46 × 10–17 | 1.90 | 0.95 | 7.64 × 10–12 | 1.19 × 10–17 | 2.00 | 0.86 | 7.74 × 10–12 | [45] | 1.01 |
| 33. | C2H6 + CH3 ⇄ C2H5 + CH4 | 9.22 × 10–28 | 4.72 | 3.23 | 2.62 × 10–14 | 2.48 × 10–31 | 6.00 | 6.04 | 1.19 × 10–14 | [45] | 0.45 |
| 34. | C2H4 + H + M ⇄ C2H5 + M | Аппроксимация Troe | 3.65 × 10–31 | 7.69 × 10–30 | 0 | 0.76 | 5.25 × 10–30 | [45] | 14.38 | ||
| 35. | C2H5 + HO2 ⇄ C2H5O + OH | 1.83 × 10–11 | 0 | 0.00 | 1.83 × 10–11 | Данные отсутствуют | – | ||||
| 36. | C2H5 + O2 ⇄ C2H4 + HO2 | Логарифм. интерполяция | 1.08 × 10–13 | 1.69 × 10–13 | 0 | –2.19 | 5.09 × 10–13 | [45] | 4.71 | ||
| 37. | C2H4O(1–2) ⇄ CH3 + HCO | 3.63 × 1013 | 0 | 57.20 | 11. 4 | 7.00 × 1015 | 0 | 81.67 | 9.88 × 10–3 | [45] | 8.65 × 10–4 |
| 38. | C2H4 + OH ⇄ C2H3 + H2O | 3.70 × 10–20 | 2.75 | 2.22 | 2.09 × 10–12 | 3.40 × 10–11 | 0 | 5.94 | 1.71 × 10–12 | [45] | 0.82 |
| 39. | C2H4 + CH3 ⇄ C2H3 + CH4 | 1.62 × 10–21 | 2.95 | 15.15 | 5.49 × 10–16 | 6.91 × 10–12 | 0 | 11.13 | 2.55 × 10–14 | [45] | 46.45 |
| 40. | C2H4 + HO2 ⇄ C2H4O(1–2) + OH | 5.55 × 10–12 | 0 | 17.19 | 9.72 × 10–16 | 1.00 × 10–14 | 0 | 7.95 | 1.83 × 10–16 | [43] | 0.19 |
| 41. | C2H3 + O2 ⇄ CH2O + H + CO | Логарифм. интерполяция | Данные отсутствуют | – | |||||||
| 42. | CH3 + CH2O ⇄ C2H5O | 4.98 × 10–13 | 0 | 6.34 | 2.05 × 10–14 | – | |||||
| 43. | C3H8 + M ⇄ CH3 + C2H5 + M | Аппроксимация Troe | 2.12 × 10–21 | 1.30 × 10–5 | 0 | 64.98 | 8.15 × 10–20 | [46] | 38.46 | ||
| 44. | C3H6 + O2 ⇄ CH2CHCH2 + HO2 | 9.90 × 10–5 | –1.67 | 46.19 | 7.75 × 10–20 | 3.20 × 10–12 | 0 | 39.15 | 8.88 × 10–21 | [46] | 0.11 |
| 45. | C3H6 + CH3 ⇄ CH2CHCH2 + CH4 | 3.67 × 10–24 | 3.50 | 5.68 | 6.67 × 10–15 | 3.68 × 10–24 | 3.50 | 5.68 | 6.67 × 10–15 | [43] | 1.00 |
| 46. | C3H6 + H ⇄ C2H4 + CH3 | Логарифм. интерполяция | Данные отсутствуют | ||||||||
* Кинетические параметры представлены для трехпараметрической формы уравнения Аррениуса: k = A(T)Ne(–Ea/RT). ** Размерности A – молек., cм3, с. *** Ссылки на конкретные работы, приведенные в базе данных [37] (см. в тексте).
Как видно из табл. 3, в модели, полученной при редуцировании модифицированной схемы, отсутствует ряд важных стадий с участием метильного радикала. Одна из них – образование и распад радикала CH3O2. Именно этот процесс определяет положение нижней границы “окна ОКМ” и резкое падение селективности по С2-углеводородам при низких конверсиях в области температур <900–950 К [11, 16]. Полученный результат, вероятно, обусловлен тем, что при использовании кинетических параметров [43–46] существенно возрастает скорость стадий (18), (30) и (39) (см. табл. 2). Это приводит к снижению вклада других реакций с участием метильного радикала в протекание процесса. Увеличение скорости стадии (39) в модифицированной схеме также приводит к тому, что схема 2 содержит большее число стадий с участием радикала С2Н3 по сравнению со схемой 1 . Таким образом, данный пример очень наглядно демонстрирует влияние неопределенностей в величинах кинетических параметров отдельных стадий на вид редуцированных кинетических схем.
Таблица 3.
Различия в стадийных схемах, полученных редуцированием модели AramcoMech 3.0 (1000 К, 1 атм), в зависимости от начальных значений кинетических параметров*
| Номер стадии | Реакция |
|---|---|
| Есть в схеме 1 , нет в схеме 2 | |
| 1 | H + O2 + M ⇄ HO2 + M |
| 2 | CH3 + H + M ⇄ CH4 + M |
| 3 | CH4 + CH3O2 ⇄ CH3 + CH3O2H |
| 4 | CH3 + O2 + M ⇄ CH3O2 + M |
| 5 | CH3O2 + CH3 ⇄ CH3O + CH3O |
| 6 | CH3O2H ⇄ CH3O + OH |
| 7 | CH3O + O2 ⇄ CH2O + HO2 |
| 8 | CH2O + CH3O2 ⇄ HCO + CH3O2H |
| 9 | HCO + M ⇄ H + CO + M |
| 10 | C2H4 + H + M ⇄ C2H5 + M |
| 11 | C2H5 + HO2 ⇄ C2H5O + OH |
| 12 | C2H4O(1–2 ) ⇄ CH3 + HCO |
| 13 | C2H4 + HO2 ⇄ C2H4O(1–2) + OH |
| Есть в схеме 2 , нет в схеме 1 | |
| 14 | CH2O + OH ⇄ HCO + H2O |
| 15 | C2H3 + O2 ⇄ CH2CHO + O |
| 16 | C2H3 + O2 ⇄ CHOCHO + H |
| 17 | C2H3 + O2 ⇄ CH2O + HCO |
| 18 | C2H4 + O ⇄ CH2CHO + H |
| 19 | C2H6 + HO2 ⇄ C2H5 + H2O2 |
| 20 | CH2CHO + M ⇄ CH3 + CO + M |
| 21 | CH2CHO + O2 ⇄ CH2O + CO + OH |
Представленные в табл. 2 данные показывают, что для некоторых стадий различие в величинах констант скорости составляет более одного порядка. Подобное расхождение в параметрах ожидаемо отражается на результатах расчета. В частности, если в качестве критерия для оценки суммарной скорости протекания процесса использовать концентрацию кислорода (лимитирующего реагента при стандартной организации процесса ОКМ) в заданный момент времени, то в случае модифицированной схемы реакция протекает с несколько большей скоростью (рис. 1). Если сравнивать распределение продуктов окисления метана, то разница для двух схем оказывается еще более существенной (см. рис. 2а–2г). Анализ чувствительности показывает, что в случае модифицированной кинетической схемы текущая концентрация кислорода наиболее чувствительна к стадиям (13), (17), (18) и (30). Согласно результатам анализа, первые три реакции оказывают “положительное” влияние на скорость превращения кислорода (т.е. при увеличении константы скорости растет скорость процесса), тогда как реакция (30) – наоборот, “отрицательное”.
Рис. 1.
Зависимость текущей концентрации кислорода от времени реакции. Кривые 1–5 получены при замене кинетических параметров отдельных стадий модифицированной схемы на параметры AramcoMech: 1 – замена параметров стадии (13), 2 – стадии (17), 3 – стадии (18), 4 – стадии (30), 5 – стадий (13), (17), (18) и (30).
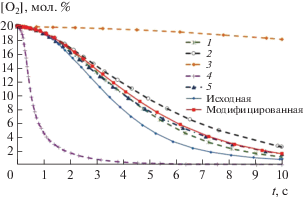
Рис. 2.
Зависимость текущих концентраций этана (а), этилена (б), CO (в) и CO2 (г) от времени реакции. Кривые 1–3 получены при замене кинетических параметров отдельных стадий модифицированной схемы на параметры AramcoMech: 1 – замена параметров стадий (13), (17), (18) и (30); 2 – стадий (13), (15), (17), (18) и (30); 3 – стадий (13), (15), (17), (18), (30) и (39).
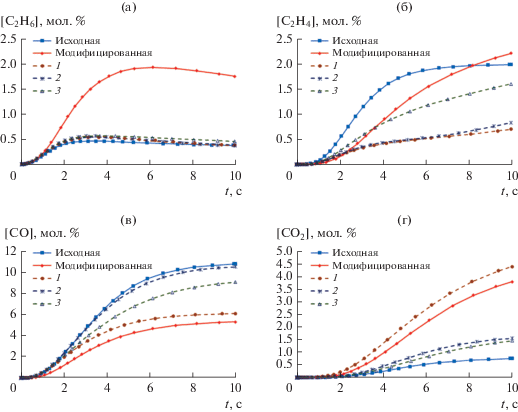
На рис. 1 приведены результаты расчетов, которые показывают, как попеременная замена кинетических параметров стадий (13), (17), (18) и (30) из данных работ [45, 46] на данные [22] влияет на скорость превращения кислорода. В случае стадий (13) и (17) замена оказывает на нее незначительное влияние, что, по-видимому, обусловлено относительно небольшой разницей в значениях констант скорости для этих реакций (см. табл. 1). В случае реакций (18) и (30) значения констант скорости при 1000 К, рассчитанные по данным [45, 46], соответственно в 12 и 34 раза выше по сравнению с данными [22]. При переходе от данных [45, 46] к [22] происходит снижение скорости стадии разветвления (18) и обрыва (30). Соответственно, в первом случае мы наблюдаем резкое снижение скорости процесса, а во втором – наоборот, увеличение (рис. 1, кривые 3 и 4). Интересно отметить, что эффекты от замены кинетических параметров разных стадий компенсируют друг друга: при одновременной замене параметров для стадий (13), (17), (18) и (30) изменение зависимости степени превращения кислорода от времени становится незначительным (рис. 1, кривая 5). При этом, однако, существенно меняется распределение продуктов реакции: количество С2-углеводородов снижается при одновременном увеличении доли оксидов углерода (рис. 2а–2г). Такое перераспределение продуктов в сторону глубокого окисления выглядит закономерным, поскольку принятая в модели AramcoMech константа скорости рекомбинации метильных радикалов существенно ниже. В ходе дальнейшего анализа было установлено, что количество образующегося этилена также заметно зависит от скорости стадии взаимодействия молекулы этилена с метильным радикалом (39), для которой константа скорости, рассчитанная по данным [45], в 46 раз выше, чем принятая в модели [22]. Соответственно, при варьировании константы скорости для этой стадии доля этилена в продуктах реакции существенно меняется (рис. 2б). Что касается оксидов углерода, то анализ кинетической схемы показал, что их концентрации наиболее чувствительны к изменению параметров стадии (15). Ее константа скорости, рассчитанная по данным [43], в 16 раз выше по сравнению с использованной в [22]. Прогнозируемо замена параметров этой стадии приводит к существенному изменению соотношения между концентрациями оксидов углерода (рис. 2в, 2г). При этом следует отметить, что величина константы скорости реакции (15) слабо влияет на количество образующихся углеводородов С2 (рис. 2а, 2б).
Последующий анализ показал, что концентрации продуктов и реагентов имеют существенно более низкую чувствительность к параметрам всех других стадий в отдельности, а наблюдаемые на рис. 1 и 2 расхождения в ходе соответствующих кривых обусловлены накоплением различий в значениях кинетических параметров (табл. 1).
Таким образом, проведенный анализ показывает, что наиболее существенный вклад в расхождение результатов расчетов по данным [22] и [45] вносят стадии (15), (18), (30) и (39), кинетические параметры которых отличаются более чем на порядок. Ниже мы подробнее рассмотрим каждую из этих реакций.
Окисление CO гидроперекисным радикалом. Реакция
(15)
${\text{CO}} + {\text{H}}{{{\text{O}}}_{2}} \rightleftarrows {\text{C}}{{{\text{O}}}_{2}} + {\text{OH}}$В модели [22] использованы данные, предложенные в относительно недавней работе [50], в которой кинетические параметры стадии (15) были определены на основании квантово-химических расчетов. Ввиду отсутствия в работах [45, 46] данных о кинетических параметрах стадии (15) рассмотрим параметры, предложенные в [43, 44]. Как видно из табл. 4, значения констант скорости, полученные по данным работ [43] и [44], практически совпадают, тогда как их разница по сравнению с [50] составляет более порядка. Кинетические параметры из [43] и [44] были использованы в моделях [31] и [33] соответственно. Что касается схем [35, 36], то принятые в них значения параметров дают промежуточные величины констант скорости стадии (15). Таким образом, проведенный анализ показывает, что кинетические параметры для стадии (15), используемые разработчиками модели [22], также могут требовать уточнения.
Таблица 4.
Зависимость величин констант скорости реакции CO + HO2 ⇄ CO2 + OH от температуры по данным различных источников
| Реакция | T, К | k × 1017, см3 молек.–1 с–1 | ||||
|---|---|---|---|---|---|---|
| Aramco 3.0 [22] | Tsang и др. [43], Konnov [33] | Warnatz [44], Gri-mech 3.0 [31] |
San-diego [35] |
Milan [36] | ||
| CO + HO2 ⇄ CO2 + OH | 800 | 0.7 | 8.7 | 8.9 | 1.8 | 2.6 |
| 900 | 3.2 | 45.3 | 46.2 | 9.1 | 12.9 | |
| 1000 | 10.8 | 170.0 | 173.0 | 32.8 | 46.8 | |
| 1100 | 30.3 | 501.7 | 509.4 | 93.6 | 134.1 | |
| 1200 | 72.6 | 1236.1 | 1252.7 | 224.0 | 322.2 | |
| 1300 | 154.3 | 2651.1 | 2682.2 | 468.9 | 676.7 | |
Окисление метильных радикалов. Еще более сложно обстоит дело с анализом данных по константам скорости одной из реакций взаимодействия метильного радикала с кислородом:
(18)
${\text{C}}{{{\text{H}}}_{3}} + {{{\text{O}}}_{2}} \rightleftarrows {\text{C}}{{{\text{H}}}_{2}}{\text{O}} + {\text{OH}}.$Именно эта реакция играет ключевую роль в резком падении селективности в процессе ОКМ при температурах выше 1100–1200 К (определяя верхнюю границу “окна ОКМ” [11]) и в образовании синтез-газа при гомогенном парциальном окислении метана.
В качестве первоисточника параметров для этой стадии разработчики модели [22] указали: “Klippenstein S.J. Personal communication, 2009”. Можно предположить, что авторы использовали параметры, предложенные Klippenstein S.J. в частной беседе. При этом не известно, являются ли эти данные опубликованными. В базе [37] для реакции (18) указано небольшое число источников, большая часть данных получена в высокотемпературной области (>1500 К). Помимо работы [45], в табл. 5 также приведены значения констант скорости, рассчитанные по данным относительно недавних экспериментальных работ [51, 52]. Что касается других кинетических схем, то в модели [31] были использованы параметры, близкие по значениям к [52], тогда как в схемах [33] и [35] – параметры из [45]. В модели [36] реакция (18) отсутствует.
Таблица 5.
Зависимость величин констант скорости реакции CH3 + O2 ⇄ CH2O + OH от температуры по данным различных источников
| Реакция | T, К | k × 1017, см3 молек.–1 с–1 | |||||
|---|---|---|---|---|---|---|---|
| Aramco 3.0 [22] | Baulch и др. [45] |
Gri-mech 3.0 [31] | Konnov [33], San-diego [35] |
Srinivasan и др. [51] | Yu и др. [52] |
||
| CH3 + O2 ⇄ CH2O + OH | 800 | 9.1 | 198.5 | 1.1 | 198.5 | 21.6 | 0.9 |
| 900 | 23.6 | 370.9 | 4.5 | 370.9 | 55.5 | 3.7 | |
| 1000 | 52.4 | 611.5 | 13.9 | 611.5 | 118.2 | 11.4 | |
| 1100 | 103.9 | 920.5 | 35.2 | 920.5 | 219.2 | 28.8 | |
| 1200 | 188.2 | 1294.3 | 76.3 | 1294.3 | 366.9 | 62.4 | |
| 1300 | 318.0 | 1727.0 | 147.1 | 1727.0 | 567.4 | 120.0 | |
Таким образом, для стадии (18) также наблюдается серьезное противоречие между параметрами, приведенными в [45] и используемыми разработчиками кинетических схем [22, 32, 33, 35, 36].
Рекомбинация метильных радикалов. Реакция
(30)
${\text{C}}{{{\text{H}}}_{3}} + {\text{C}}{{{\text{H}}}_{3}} + {\text{M}} \rightleftarrows {{{\text{C}}}_{2}}{{{\text{H}}}_{6}} + {\text{M}}$В табл. 6 представлена зависимость величин констант скорости реакции (30) от температуры в диапазоне 800–1300 К, рассчитанная по данным [22] и [46]. В модели [22] в качестве источника были использованы результаты экспериментальной работы [53]. Как видно из представленных результатов, константы скорости, рассчитанные по данным работ [22] и [46], различаются более чем на порядок. Выше было показано, что такая разница оказывает критическое влияние на результаты расчетов. Для дальнейшего анализа в качестве дополнения были рассмотрены описания скорости реакции (30), используемые в наиболее известных на сегодня кинетических моделях окисления легких алканов [31, 33, 35, 36]. Как можно видеть, значения констант скорости, рассчитанные по этим данным, являются величинами одного порядка. При этом данные [53] практически полностью совпадают с [33]. Таким образом, наблюдаемое расхождение в результатах расчета по данным широко известных и хорошо зарекомендовавших себя моделей с результатами, полученными с использованием данных обзора [46], заставляют усомниться в достоверности приведенных в нем параметров.
Таблица 6.
Зависимость величин констант скорости реакции CH3 + CH3 + M ⇄ C2H6 + M от температуры по данным различных источников
| Реакция | T, К | k × 1030, см6 молек.–2 с–1 | |||||
|---|---|---|---|---|---|---|---|
| Aramco 3.0 [22] |
Baulch и др. [46] |
Gri-mech 3.0 [31] |
Konnov [33] |
San-diego [35] |
Milan [36] |
||
| CH3 + CH3 + M ⇄ C2H6 + M | 800 | 2.9 | 294.5 | 2.5 | 3.3 | 2.4 | 2.6 |
| 900 | 2.8 | 156.6 | 2.3 | 3.0 | 2.3 | 2.3 | |
| 1000 | 2.6 | 87.4 | 2.0 | 2.7 | 2.1 | 1.9 | |
| 1100 | 2.3 | 50.9 | 1.7 | 2.4 | 1.7 | 1.5 | |
| 1200 | 2.1 | 30.8 | 1.4 | 2.0 | 1.4 | 1.2 | |
| 1300 | 1.8 | 19.2 | 1.2 | 1.7 | 1.1 | 0.9 | |
Реакция этилена с метильным радикалом. Наверное, наиболее неоднозначная ситуация наблюдается в случае рассмотрения взаимодействия этилена с метильным радикалом. При этом стадия
(39)
${{{\text{C}}}_{2}}{{{\text{H}}}_{4}} + {\text{C}}{{{\text{H}}}_{3}} \rightleftarrows {{{\text{C}}}_{2}}{{{\text{H}}}_{3}} + {\text{C}}{{{\text{H}}}_{4}}$В описании модели [22] сведения об источнике кинетических параметров для этой стадии отсутствуют. При этом константа скорости, рассчитанная по использованным в [22] параметрам, отличается от полученной по данным [46] более чем в 40 раз (см табл. 7). Следует отметить, что величины, рассчитанные по данным других работ, включая как широко известные обзоры [43, 44], так и кинетические модели [31, 36] также различаются между собой и являются промежуточными между данными [22] и [46]. В модели [35] стадия (39) отсутствует, а в схеме [33] использованы параметры из работы [46].
Таблица 7.
Зависимость величин констант скорости реакции C2H4 + CH3 ⇄ C2H3 + CH4 от температуры по данным различных источников
| Реакция | T, К | k × 1015, см3 молек.–1 с–1 | |||||
|---|---|---|---|---|---|---|---|
| Aramco 3.0 [22] | Baulch и др. [46], Konnov [33] | Tsang и др. [43] | Warnatz [44] | Gri-mech 3.0 [31] |
Milan [36] |
||
| C2H4 + CH3 ⇄ C2H3 + CH4 | 800 | 0.04 | 6.3 | 1.5 | 0.6 | 0.7 | 0.3 |
| 900 | 0.2 | 13.7 | 4.6 | 1.4 | 1.8 | 0.7 | |
| 1000 | 0.5 | 25.5 | 11.6 | 2.6 | 3.7 | 1.7 | |
| 1100 | 1.5 | 42.5 | 25.5 | 4.3 | 6.8 | 3.4 | |
| 1200 | 3.3 | 64.9 | 50.6 | 6.6 | 11.5 | 6.3 | |
| 1300 | 6.9 | 92.9 | 92.4 | 9.4 | 18.1 | 10.6 | |
Влияние учета гетерогенных реакций на результаты редуцирования
Как уже упоминалось выше, процесс ОКМ является одновременно свободно-радикальным и гетерогенно-каталитическим, т.е. его суммарная скорость и состав образующихся продуктов определяются совокупностью большого числа стадий, протекающих как в газовой фазе, так и с участием поверхностных центров катализатора. Выше было показано, что выбор тех или иных величин кинетических параметров гомогенных стадий в значительной мере влияет на результаты моделирования газофазного процесса окисления метана и редуцирования “полной” схемы гомогенных превращений. Можно ожидать, что введение в рассмотрение блока гетерогенных реакций окажет существенное влияние не только на результаты расчетов кинетики окисления метана на базе схемы AramcoMech (что уже было отмечено ранее [12]), но и на результаты редуцирования гомогенной части объединенной модели.
Ранее было показано [11, 12, 49], что введение в модель блока гетерогенных реакций приводит к нескольким основным изменениям. Так, по мере увеличения концентрации каталитически активных центров (КАЦ) постепенно исчезает участок роста скорости реакции, при их наиболее высоких концентрациях процесс становится линейным, т.е. начинается с максимальной скорости реакции, которая постепенно снижается по мере израсходования реагентов. Хотя максимальная скорость реакции может достигаться в чисто гомогенном процессе (за счет развития цепного ускорения), время достижения определенной степени превращения сокращается с ростом концентрации КАЦ. Селективность по целевым продуктам реакции ОКМ (C2-углеводородам) проходит через максимум: на начальном участке зависимости селективности от концентрации КАЦ наблюдается ее рост за счет высокой скорости генерации метильных радикалов и возрастания эффективности их рекомбинации, который сменяется некоторым снижением за счет увеличения вклада гетерогенного глубокого окисления в суммарное превращение метана.
Что касается результатов редуцирования, то они также ожидаемо зависят от введения в систему КАЦ. Однако характер этой зависимости оказался немонотонным. При относительно низкой концентрации КАЦ (~7 × 1017 см–3) происходит увеличение общего числа стадий в гомогенной части редуцированной схемы. За счет того, что при резком возрастании скорости генерации метильных радикалов и концентрации углеводородов C2 растет вклад реакций с участием последних (дополнительно в схеме появляются 5 таких реакций), а также концентрация углеводородов C3 и, соответственно, вклад реакций с участием частиц, содержащих 3 атома углерода (еще дополнительно 11 реакций). При этом из редуцированной схемы исчезают 11 реакций частиц с числом атомов углерода ≤2, которые присутствовали в ней до введения КАЦ.
При дальнейшем увеличении концентрации КАЦ число гомогенных реакций в редуцированной схеме последовательно снижается. При достижении их концентрации ~1 × 1019 см–3 в ней остается всего 25 обратимых реакций. При этом варьирование концентрации КАЦ не приводит к изменению состава гетерогенного блока в редуцированной схеме.
Таким образом, результаты как моделирования, так и редуцирования гетерогенно-гомогенной схемы окисления метана указывают на существенное влияние гетерогенного фактора на протекание процесса, обусловленное сильным кинетическим сопряжением гомогенных и гетерогенных процессов. В частности, добавление гетерогенного блока приводит к изменению вида всех кинетических зависимостей, а также относительного вклада в них отдельных стадий. В практическом отношении присутствие поверхностных активных центров существенно меняет селективность по целевым продуктам.
Влияние температуры на результаты редуцирования
Высокая чувствительность результата редуцирования к величинам кинетических параметров неизбежно должна приводить к тому, что набор стадий, входящих в редуцированную кинетическую схему, будет меняться при изменении условий протекания реакции и начальных величин физических параметров. Действительно, например, ввиду сильной температурной зависимости констант скорости соотношения между вкладами различных стадий в суммарную кинетику также сильно меняются при изменении температуры. Значит, относительная значимость разных стадий будет изменяться при варьировании температуры при изотермическом протекании процесса и тем более при неизотермическом (например, адиабатическом или автотермическом) режиме.
В данной работе влияние температуры (900–1200 К с шагом 100 К) на результаты редуцирования изучены для исходной кинетической схемы, включающей гомогенный и гетерогенный блоки (концентрация КАЦ 7 × 1018 см–3). Как и в случае варьирования концентрации КАЦ, оказалось, что состав гетерогенного блока в редуцированной схеме меняется мало при варьировании температуры. Однако на гомогенную часть схемы температура оказывает сильное влияние. В целом, по мере ее повышения снижается вклад реакций с участием спиртов и альдегидов (веществ, образующихся в основном через радикалы CH3O2) и растет вклад реакций с участием частиц C1–C2 с высокой степенью окисления, которые являются промежуточными в образовании оксидов углерода (в первую очередь CO) через стадии типа (18) (см. выше). В итоге число обратимых гомогенных реакций в редуцированной схеме возрастает с 25 при 1000 К до 45 при 1200 К.
Важно отметить, что ввиду высокой экзотермичности реакций образования оксидов углерода использование “коротких” схем, полученных редуцированием при относительно низких температурах (например, при 1000 К) и не учитывающих высокотемпературные стадии, может приводить к значительным ошибкам при расчете адиабатического или автотермического (с частичной потерей выделяющегося тепла) протекания процесса. Аналогично, при относительно низких температурах для более точного воспроизведения данных по селективности следует включать стадии, которые не попадают в редуцированные схемы, полученные при высоких температурах.
Нами были рассмотрены два подхода к получению “укороченных” схем, позволяющих работать в широком диапазоне температур. Один из них – проведение редуцирования в ходе расчета адиабатического протекания процесса. Второй – объединение схем, полученных в разных температурных диапазонах, путем добавления веществ и реакций из высокотемпературных схем в низкотемпературные (или наоборот). Хотя второй подход приводит к получению описаний большего объема, точность расчетов с их использованием, как правило, гораздо выше (результаты ближе к получаемым по “полным” схемам), чем при использовании редуцирования первого типа.
ЗАКЛЮЧЕНИЕ
Выполнение принципа “независимости кинетических параметров”, т.е. полный отказ от “подгонки” значений кинетических констант для согласования результатов расчетов с экспериментальными данными, является одним из основных требований при создании согласованного и непротиворечивого описания процесса [11]. В противном случае параметры внутри сложной схемы становятся сильно связанными друг с другом и с результатами “базовых” экспериментов. Также такая подгонка накладывает ограничения на дальнейшее развитие модели и делает неопределенными результаты расчетов в областях параметров за пределами “базовых” экспериментов. В идеале наполнение кинетической модели параметрами должно осуществляться с использованием открытых баз данных без какой-либо привязки к экспериментальным результатам. Однако проведенный в настоящей работе анализ показывает, что модели, полученные в рамках такого подхода, могут обладать низкой предсказательной способностью ввиду существующих неопределенностей в величинах параметров, приведенных в широко известных обзорных работах и базах данных [43–46].
В качестве альтернативного “промежуточного” решения можно рассматривать подход, предложенный авторами модели [22]. Он также предполагает отказ от подгонки значений кинетический параметров. При этом основным критерием при их выборе является достижение максимального приближения описания к наблюдаемым экспериментальным зависимостям для широкого круга реакционных смесей и условий протекания реакций. Авторы [22] утверждают, что кинетические параметры, приведенные в независимых источниках (например, в [43–46]), не учитывают взаимное влияние всех элементарных реакций и частиц, возникающее при их включении в общее кинетическое описание. Поэтому простое наполнение кинетическое схемы такими параметрами не будет давать достаточно точное описание процесса. Само по себе такое утверждение противоречит одному из основных положений химической кинетики – принципу независимости скоростей элементарных реакций. По этой причине мнение авторов семейства моделей AramcoMech, приведенное в [30], по-видимому, можно понимать только в следующем смысле: некоторые значения кинетических параметров, приводимые в литературе, получены не в прямом эксперименте, а на основании анализа закономерностей сложных процессов и с использованием некоторых кинетических моделей. По этой причине определенные значения искомых параметров становятся “модельно зависимыми”. Иными словами, если бы при их определении на основании имеющихся данных о закономерностях брутто-процесса были использованы другие модели с другим набором стадий и/или их параметров, численные значения могли бы быть иными. При такой интерпретации подход, предлагаемый разработчиками модели [22], является разумным компромиссом между строгим выполнением принципа “независимости” в совокупности с априорным предпочтением одних литературных данных другим и произвольной “подгонкой” значений кинетических параметров для достижения максимальной точности описания “базовых” экспериментов.
Итак, нами показано, что редуцирование кинетических схем большого объема является удобным и надежным инструментом, позволяющим сравнивать различные кинетические описания и наборы параметров, имеющиеся в литературе. При этом, однако, использование полученных сокращенных описаний (как для анализа механизма процессов, так и для моделирования реальных процессов и аппаратов) требует большой осторожности. В первом случае возможна неправильная трактовка результатов сравнения расчетных данных с экспериментальными из-за многомаршрутности сложных реакций и высокой степени сопряжения различных маршрутов внутри них. Во втором случае возможно возникновение значительных ошибок расчета из-за высокой параметрической зависимости кинетических констант и скоростей процесса в целом (от температуры, давления, концентраций участников реакции). Реальные сочетания параметров могут существенно отличаться от тех, при которых производится редуцирование. В итоге выпадающие из описания реакции и даже компоненты реакционной смеси могут становиться весьма существенными на тех или иных этапах протекания процесса в моделируемых условиях.
Список литературы
Крылов О.В., Арутюнов В.С. Окислительные превращения метана. М.: Наука, 1998. 350 с.
Mitchell H.L., Waghorne R.H. Pat. US, 1980. № 4205194.
Fang T., Yeh C. // J. Catal. 1981. V. 69. № 2. P. 227.
Keller G.E., Bhasin M.M. // J. Catal. 1982. V. 73. № 1. P. 9.
Hinsen W., Bytyn W., Baerns M. // 8th International Congress on Catalysis: Proceedings. Berlin: Verlag Chemie, 1984. V. 3. P. 581.
Ito T., Lunsford J.H. // Nature. 1985. V. 314. № 6013. P. 721.
Driscoll D.J., Martir W., Wang J.-X., Lunsford J.H. // J. Am. Chem. Soc. 1985. V. 107. № 1. P. 58.
Синев М.Ю., Корчак В.Н., Крылов О.В., Григорян Р.Р., Гарибян Т.А. // Кинетика и катализ. 1988. Т. 29. № 5. С. 1105.
Feng Y., Niiranen J., Gutman D. // J. Phys. Chem. 1991. V. 95. № 17. P. 6558.
Лoмoнoсoв B.И., Синeв М.Ю. // Кинетика и катализ. 2016. Т. 57. № 5. С. 652.
Sinev M., Arutyunov V., Romanets A. // Adv. Chem. Eng. 2007. V. 32. P. 167.
Lomonosov V., Gordienko Yu., Ponomareva E., Sinev M. // Chem. Eng. J. 2019. V. 370. P. 1210.
Sinev M.Yu. // J. Catal. 2003. V. 216. № 1–2. P. 468.
Синев М.Ю., Воробьева Г.А., Корчак В.Н. // Кинетика и катализ. 1986. Т. 27. № 5. С. 1164.
Синев М.Ю., Корчак В.Н., Крылов О.В. // Кинетика и катализ. 1987. Т. 28. № 6. С. 1376.
Labinger J.A., Ott K.C. // J. Phys. Chem. 1987. V. 91. № 11. P. 2682.
Zanthoff H., Baerns M. // Ind. Eng. Chem. Res. 1990. V. 29. № 1. P. 2.
McCarty J.G., McEwen A.B., Quinlan M.A. // Stud. Surf. Sci. Catal. 1990. V. 55. P. 405.
Tjatjopoulos G.J., Vasalos I.A. // Catal. Today. 1992. V. 13. № 2–3. P. 361.
Sun J., Thybaut J.W., Marin G.B. // Catal. Today. 2008. V. 137. № 1. P. 90.
http://www.nuigalway.ie/combustionchemistrycentre
AramcoMech Version 3.0, 2018. Available at http://www.nuigalway.ie/combustionchemistrycentre/mechanismdownloads/
Zhou C.-W., Li Y., Burke U., Banyon C., Somers K.P., Ding S., Khan S., Hargis J.W., Sikes T., Mathieu O., Petersen E.L., AlAbbad M., Farooq A., Pan Y., Zhang Y., Huang Z., Lopez J., Loparo Z., Vasu S.S., Curran H.J. // Combust. Flame. 2018. V. 197. P. 423.
Li Y., Zhou C.-W., Somers K.P., Zhang K., Curran H.J. // Proc. Combust. Inst. 2017. V. 36. № 1. P. 403.
Zhou C.-W., Li Y., O’Connor E., Somers K.P., Thion S., Keesee C., Mathieu J., Petersen E.L., DeVerter T.A., Oehlschlaeger M.A., Kukkadapu G., Sung C.-J., Alrefae M., Khaled F., Farooq A., Dirrenberger P., Glaude P.-A., Battin-Leclerc F., Santner J., Ju Y., Held T., Haas F.M., Dryer F.L., Curran H.J. // Combust. Flame. 2016. V. 167. P. 353.
Burke U., Metcalfe W.K., Burke S.M., Heufer K.A., Dagaut P., Curran H.J. // Combust. Flame. 2016. V. 165. P. 125.
Burke S.M., Burke U., Mc Donagh R., Mathieu O., Osorio I., Keesee C., Morones A., Petersen E.L, Wang W., DeVerter T.A., Oehlschlaeger M.A., Rhodes B., Hanson R.K., Davidson D.F., Weber B.W., Sung C.-J., Santner J., Ju Y., Haas F.M., Dryer F.L., Volkov E.N., Nilsson E.J.K., Konnov A.A., Alrefae M., Khaled F., Farooq A., Dirrenberger P., Glaude P.-A., Battin-Leclerc F., Curran H.J. // Combust. Flame. 2015. V. 162. P. 296.
Burke S.M., Metcalfe W.K., Herbinet O., Battin-Leclerc F., Haas F.M., Santner J., Dryer F.L., Curran H.J. // Combust. Flame. 2014. V. 161. P. 2765.
Kéromnès A., Metcalfe W.K., Heufer K.A., Donohoe N., Das A.K., Sung C.-J., Herzler J., Naumann C., Griebel P., Mathieu O., Krejci M.C., Petersen E.L., Pitz W.J., Curran H.J. // Combust. Flame. 2013. V. 160. P. 995.
Metcalfe W.K., Burke S.M., Ahmed S.S., Curran H.J. // Int. J. Chem. Kinet. 2013. V. 45. P. 638.
Smith G.P., Golden D.M., Frenklach M., Moriarty N.W., Eiteneer B., Goldenberg M., Bowman C.T., Hanson R.K., Song S., Gardiner W.C., Jr., Lissianski V., Qin Z. Available at http://www.me.berkeley.edu/gri_mech/
Wang H., You X., Joshi A.V., Davis S.G., Laskin A., Egolfopoulos F., Law C.K. USC Mech Version II. High-Temperature Combustion Reaction Model of H2/CO/C1-C4 Compounds. Available at http://ignis.usc.edu/Mechanisms/USC-Mech%20II/USC_Mech%20II.htm
Konnov A.A. Detailed reaction mechanism for small hydrocarbons combustion. Release 0.5, (2000), available as Electronic Supplementary Material to: Coppens F.H.V., de Ruyck J. Konnov A.A. // Combust. Flame. 2007. V. 149. P. 409.
Hughes K.J., Turanyi T., Clague A.R., Pilling M.J. The Leeds methane oxidation mechanism, Version 1.5. 2001. Available at http://garfield.chem.elte.hu/Combustion/mechanisms/metan15.dat
Chemical-Kinetic Mechanisms for Combustion Applications, San Diego Mechanism web page, Mechanical and Aerospace Engineering (Combustion Research), University of California at San Diego (http://combustion.ucsd.edu).
C1–C3 mechanism, Version 1412, December 2014. Available at http://creckmodeling.chem.polimi.it/
База данных Национального Института Стандартов и Технологий “NIST Chemical Kinetics Database” https://kinetics.nist.gov/kinetics/
Baulch D.L. Reaction Kinetics Database. School of Chemistry. The University of Leeds.
Hughes K.J., Turányi T., Clague A.R., Pilling M.J. // Int. J. Chem. Kinet. 2001. V. 33. P. 513.
http://www.reactiondesign.com/products/chemkin/chemkin-2/
http://www.kintechlab.com/ru/produkty/chemical-workbench/
Lebedev A.V., Okun M.V., Chorkov V.A., Tokar P.M., Strelkova M. // J. Math. Chem. 2013. V 51. P. 73.
Tsang W., Hampson R.F. // J. Phys. Chem. Ref. Data. 1986. V. 15. P. 1087.
Warnatz J. Combustion Chemistry / Ed. Gardiner W.C., Jr. N.Y.: Springer-Verlag, 1984. P. 197.
Baulch D.L., Cobos C.J., Cox R.A., Esser C., Frank P., Just T., Kerr J.A., Pilling M.J., Troe J., Walker R.W., Warnatz J. // J. Phys. Chem. Ref. Data. 1992. V. 21. P. 411.
Baulch D.L., Bowman C.T., Cobos C.J., Cox R.A., Just T., Kerr J.A., Pilling M.J., Stocker D., Troe J., Tsang W., Walker R.W., Warnatz J. // J. Phys. Chem. Ref. 2005. V. 34. P. 757.
Sinev M.Yu. // Catal. Today. 1992. V. 13. № 4. P. 561.
Sinev M.Yu. // Catal. Today. 1995. V. 24. № 3. P. 389.
Sinev M.Yu. // Russ. J. Phys. Chem. B. 2007. V. 1. № 4. P. 329.
You X.Q., Wang H., Goos E., Sung C.J., Klippenstein S.J. // J. Phys. Chem. A. 2007. V. 111. P. 4031.
Srinivasan N.K., Su M.C., Michael J.V. // J. Phys. Chem. A. 2007. V. 111. P. 11589.
Yu C.L., Wang C., Frenklach M. // J. Phys. Chem. 1995. V. 99. P. 14377.
Wang B.S., Hou H., Yoder L.M., Muckerman J.T., Fockenberg C. // J. Phys. Chem. A. 2003. V. 107 P. 11414.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ