

Кинетика и катализ, 2021, T. 62, № 1, стр. 94-108
In situ исследование окисления CO на нанокомпозитных Cu–Fe–Al-оксидных катализаторах методами спектроскопии рентгеновского поглощения
А. М. Кремнева a, А. В. Федоров a, А. А. Сараев a, О. А. Булавченко a, В. А. Яковлев a, В. В. Каичев a, *
a ФГБУН Институт катализа СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: vvk@catalysis.ru
Поступила в редакцию 12.05.2020
После доработки 07.07.2020
Принята к публикации 20.07.2020
Аннотация
Нанокомпозитные Fe–Al- и Cu–Fe–Al-оксидные катализаторы исследованы в реакции окисления СО. Показано, что введение меди более чем на два порядка увеличивает скорость реакции. Наиболее активным катализатором является нанокомпозит, содержащий 5 мас. % CuO, 78 мас. % Fe2O3 и 17 мас. % Al2O3. По данным РФА катализаторы состоят из наночастиц гематита, α-Fe2O3, и аморфного оксида алюминия. Медь находится в высокодисперсном состоянии, образуя кластеры CuO и CuFeOx. Результаты in situ исследований методами спектроскопии рентгеновского поглощения свидетельствуют, что в потоке CO при температуре 200–250°C начинается восстановление меди с Cu2+ до Cu1+ и Cu0; при 400–600°C медь находится преимущественно в металлическом состоянии; восстановление Fe3O4 → FeO → Fe начинается при 400–500°С. В стехиометрической смеси СО и О2 восстановления Fe3+ не наблюдается, восстановление оксида меди до металла происходит при температурах выше 400°C. В избытке кислорода в диапазоне температур от 100 до 600°C изменений в химическом состоянии катализаторов не обнаружено. Предложен механизм окисления СО на Cu–Fe–Al-нанокомпозитах.
ВВЕДЕНИЕ
В последние годы наблюдается устойчивая тенденция развития и внедрения каталитических технологий сжигания топлива как для производства тепловой энергии, так и для утилизации различных промышленных отходов [1–6]. Использование катализаторов приводит к значительному уменьшению материалоемкости и габаритов тепловых установок, а удельная нагрузка на объем реактора возрастает более чем в 20 раз [5]. Катализаторы позволяют снизить температуру горения с 1200–1600 до 350–750°С и тем самым существенно сократить выбросы в атмосферу оксидов азота. При этом заметно увеличивается эффективность горения, что позволяет использовать низкокалорийное топливо (торф, бурый уголь, древесные отходы и т.п.). Наиболее перспективным считается сжигание топлива в кипящем слое катализатора. В этом случае на первой стадии происходит газификация твердого топлива с образованием CO-содержащего газа. Далее CO окисляется в CO2 на поверхности частиц катализатора. Активность в реакции окисления CO является ключевым фактором, определяющим качество катализаторов для данного процесса [7]. Кроме того, для работы в режиме кипящего слоя катализатор необходимо формовать в виде сферических частиц, обладающих высокой механической прочностью и стойкостью к истиранию. Катализаторы на основе благородных металлов нецелесообразно применять для данного процесса [8]. Это обусловлено их высокой стоимостью и жесткими условиями эксплуатации в режиме кипящего слоя, что приводит к необратимой потере благородных металлов вследствие истирания гранул катализатора [9]. Каталитические системы на основе смешанных хромсодержащих оксидов (CuCr2O4 и MgCr2O4) успешно используются как катализаторы для процесса сжигания топлива в кипящем слое [10]. Однако их применение приводит к выбросам токсичных хромсодержащих соединений. Поэтому разработка новых, экологически безопасных и дешевых каталитических систем для сжигания топлива в кипящем слое по-прежнему остается актуальной задачей.
Недавно было показано [11, 12], что нанокомпозиты на основе оксидов железа и алюминия обладают высокой каталитической активностью в окислении CO. Добавление оксида меди приводит к заметному ее увеличению. Наличие в составе катализаторов оксида алюминия позволяет их использовать для получения прочных сферических гранул методом капельного формования [6]. При этом добавка псевдобемита на стадии капельного формования способствует не только получению униформных высокопрочных гранул, но также позволяет стабилизировать фазовое состояние активного компонента. В настоящей работе нанокомпозитные Cu–Fe–Al-оксидные катализаторы, содержащие различное количество меди, были изучены in situ методами спектроскопии рентгеновского поглощения – Extended X-ray Absorption Fine Structure (EXAFS) и X-ray Absorption Near Edge Structure (XANES). Проведение экспериментов в режиме in situ позволило определить, как меняется состояние катионов железа и меди под воздействием реакционных условий. Сравнение полученных данных с результатами кинетических исследований дало возможность раскрыть природу каталитического действия нанокомпозитных Cu–Fe–Al-оксидных катализаторов в реакции окисления СО.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Катализаторы были приготовлены сплавлением нитратов Cu(NO3)2 ⋅ 3H2O, Fe(NO3)3 ⋅ 9H2O и Al(NO3)3 ⋅ 9H2O. В процессе синтеза соли механически смешивали в требуемых соотношениях, затем полученную смесь нагревали до получения однородного расплава гидратов солей и выдерживали при температуре 200°C до полного удаления воды. Полученный порошок последовательно прокаливали на воздухе при температуре 400°C в течение 1 ч и при 700°C в течение 1 ч. Катализаторы обозначены как CuxFeyAlz, где x, y и z соответствуют содержанию (мас. %) CuO, Fe2O3 и Al2O3 соответственно.
Исследование активности катализаторов в реакции окисления CO проводили с помощью двух установок: в импульсном режиме в проточной установке и в проточно-циркуляционной установке. Тестирование катализаторов в импульсном режиме проводили на приборе “Хемосорб” (ООО “СОЛО”, Россия), состоящем из реактора, системы подачи газов и системы автоматического управления и сбора данных. Для этого 0.113 мл катализатора (высота слоя – 10 мм) помещали в U-образный кварцевый реактор объемом 13.6 мл, через который непрерывно проходил стационарный поток газа-окислителя — смесь гелия (80 об. %) и кислорода (20 об. %) – со скоростью 30 мл/мин. В ходе эксперимента в реактор, помещенный в печь, импульсно подавали CO; длительность импульса – 0.75 с, объем подаваемого за один импульс СО равен 0.1 мл. Мольное соотношение O2 : CO в потоке было непостоянным и составляло не менее 3.5, что связано с методикой импульсного окисления. Изменение концентрации CO на выходе из реактора определяли с помощью катарометра. При тестировании, температуру в реакторе изменяли от 100 до 600°С со скоростью 5°С/мин. Мерой каталитической активности была выбрана температура достижения 50% конверсии CO – T50. Абсолютная ошибка определения активности составляла 5°С. Тестирование катализаторов в проточно-циркуляционно режиме проводили на установке “ПЦКУ-1” (ООО “СОЛО”, Россия), состоящей из системы формирования потоков газов, реактора, холодильника-сепаратора, системы автоматического управления и сбора данных. В ходе экспериментов навеску исследуемого образца помещали в реактор объемом 20 мл, который соединен с циркуляционным насосом в проточно-циркуляционный контур. В контур подавали поток исходной реакционной смеси – 1 об. % CO и 1 об. % O2 в N2. Скорость потока варьировали в диапазоне 100–1000 мл/мин. Концентрацию CO определяли с помощью многокомпонентного газоанализатора ТЕСТ-1 (“БОНЭР”, Россия) с оптическими датчиками CO и CO2. Скорость превращения CO (W) рассчитывали по формуле:
Фазовый состав катализаторов определяли методом порошковой рентгеновской дифракции. Дифрактограммы получали на дифрактометре D8 Advance (“Bruker”, Германия) с использованием монохроматического излучения CuKα (λ = 1.5418 Å). Размер доменов – области когерентного рассеяния (ОКР) – оценивали по формуле Шеррера.
Удельную площадь поверхности катализаторов (Sуд) определяли по методу Брунауэра–Эммета–Теллера с помощью изотерм адсорбции азота, измеренных при температуре жидкого азота. Исследования проводили с помощью автоматизированной системы ASAP 2400 (“Micromeritics Instrument Corp.”, США).
Запись спектров рентгеновского поглощения К-краев Fe и Cu проводили на станции “Структурное материаловедение” Курчатовского специализированного источника синхротронного излучения (НИЦ “Курчатовский институт”, Москва). Энергия электронного пучка в накопительном кольце составляла 2.5 ГэВ, средний ток пучка – 100 мА. Генерацию синхротронного излучения (СИ) осуществляли на поворотном магните. Для монохроматизации СИ использовали кремниевый монокристалл, сделанный в виде моноблока с вырезанным каналом; двукратное отражение СИ от плоскостей Si(111) обеспечивало энергетическое разрешение ΔЕ/Е = 2 × 10–4. Для получения спектров исследуемый образец облучали пучком рентгеновского излучения; интенсивность падающего и прошедшего излучения детектировали с помощью ионизационных камер, заполненных смесью азота и аргона. Оцифровку сигналов проводили с помощью пикоамперметров Keithley 6487 (“Keithley Instruments”, США). Для калибровки монохроматора перед измерением каждой серии спектров рентгеновского поглощения К-краев Fe и Cu записывали спектры рентгеновского поглощения реперных образцов – железной или медной фольг соответственно. Положение края поглощения E0 определяли по максимуму первой производной спектра по энергии вблизи К-края поглощения – 7112.0 эВ для железа и 8979.0 эВ для меди [14].
Для проведения in situ исследований порошок изучаемых катализаторов смешивали с гексагональным нитридом бора в соотношении 1 : 15, полученную смесь спрессовывали в таблетку толщиной 1 мм. Для регистрации спектров рентгеновского поглощения в режиме in situ таблетку помещали в высокотемпературную камеру, через которую продували смесь газов заданного состава при атмосферном давлении. Конструкция камеры позволяла нагревать катализатор и газовую смесь до температуры 600°C [15]. In situ спектры рентгеновского поглощения записывали в потоке 5% CO в N2 и в потоке смеси CO и O2 с молярным соотношением CO : O2 = 2 : 1 и 1 : 1 при ступенчатом нагреве от комнатной температуры до 600°C. Суммарный поток газов составлял 200 см3/мин.
Экспериментальные спектры обрабатывали с применением стандартных процедур выделения фона и нормирования на величину скачка K-края поглощения с использованием пакета программ DEMETER [16]. Фурье-преобразование EXAFS-спектров χ(k) проводили в интервале волновых чисел 2.0–11.0 Å–1 с весовой функцией k2. Для корректной интерпретации EXAFS-данных осуществляли моделирование спектров. Для этого строили предварительную модель на основании кристаллографических данных [17]. Затем рассчитывали теоретическую кривую EXAFS-осцилляций, представляющую собой сумму однократных и многократных путей рассеяния с учетом нескольких координационных сфер. Минимизация функционала невязки теоретического и экспериментального спектров с применением программы FEFF6 [18] позволяла уточнять параметры используемой модели и определять координационные числа, радиусы координационных сфер, факторы Дебая–Валлера и химический сдвиг.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Каталитические свойства нанокомпозитов
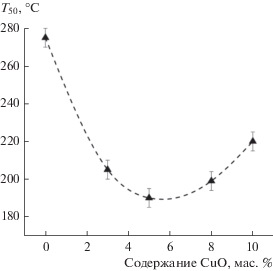
Нанокомпозитные Fe–Al-оксидные катализаторы были детально исследованы в предыдущих работах [11, 12]. По данным РФА и просвечивающей электронной микроскопии Fe–Al-катализаторы состоят из наночастиц Fe2O3 со структурой гематита и аморфного Al2O3. Катионы Al3+ частично растворяются в решетке Fe2O3, образуя твердый раствор (Fe,Al)2O3. Установлено, что активность катализаторов в реакции окисления CO зависит от их химического состава. Наибольшую активность проявляет катализатор, содержащий 82 мас. % Fe2O3 и 18 мас. % Al2O3 [11]. Для этого катализатора (Fe82Al18) температура зажигания T50, измеренная в проточном реакторе, составляет 275°С. Ранее было показано, что введение меди способствует увеличению активности катализатора [11]. Для выяснения природы этого эффекта были синтезированы катализаторы с тем же соотношением Fe2O3 : Al2O3 = 82 : 18, содержащие 3, 5, 8 и 10 мас. % CuO (табл. 1). Результаты каталитических испытаний в проточном реакторе показали, что добавление меди приводит к снижению температуры зажигания (рис. 1). Минимальная температура зажигания (190°С) наблюдается при окислении СО на катализаторе, в состав которого входит 5 мас. % CuO (Cu5Fe78Al17).
Таблица 1.
Состав, температура зажигания (T50), удельная площадь поверхности (Sуд) и размер областей когерентного рассеяния (ОКР) нанокомпозитных катализаторов
| Катализаторы | Состав, мас. % | T50, °C | Sуд, м2/г | ОКР, нм | ||
|---|---|---|---|---|---|---|
| CuO | Fe2O3 | Al2O3 | ||||
| Fe82Al18 | 0 | 82 | 18 | 275 | 60 | 46 |
| Cu3Fe79Al18 | 3 | 79 | 18 | 205 | 65 | 42 |
| Cu5Fe78Al17 | 5 | 78 | 17 | 190 | 60 | 48 |
| Cu8Fe75Al17 | 8 | 75 | 17 | 199 | 57 | 45 |
| Cu10Fe74Al16 | 10 | 74 | 16 | 220 | 55 | 48 |
Рис. 1.
Зависимость температуры зажигания (Т50) в реакции окисления CO на Cu–Fe–Al-нанокомпозитах от содержания CuO.
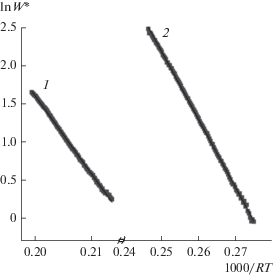
Детальные кинетические исследования наиболее активных Fe–Al- и Cu–Fe–Al-нанокомпозитов в проточно-циркуляционном реакторе показали, что скорость окисления СО при 250°С на катализаторе Cu5Fe78Al17 в 260 раз превышает таковую для катализатора Fe82Al18 (табл. 2). Скорость окисления СО практически не зависит от концентрации кислорода в газовой фазе, т.е. порядок по кислороду при протекании реакции на обоих катализаторах близок к нулю; при этом порядок по СО равен 1.10–1.11. Зависимости скорости окисления CO при 50% конверсии от температуры в координатах Аррениуса для исследуемых катализаторов представлены на рис. 2. Наблюдается линейная зависимость lnW* от 1/T, что позволяет оценить величину кажущейся энергии активации (Еа). Для катализатора Cu5Fe78Al17 энергии активации составляет 88 кДж/моль. Для катализатора Fe82Al18 наблюдается существенно бóльшая Еа – 102 кДж/моль. Это указывает на то, что введение меди приводит к изменению природы активных центров.
Таблица 2.
Результаты кинетических исследований катализаторов Fe82Al18 и Cu5Fe78Al17 в проточно-циркуляционном реакторе: порядки реакции, кажущаяся энергия активации (Ea) и скорость окисления CO при 250°С, нормированная на удельную поверхность катализаторов $(W_{{{\text{250}}}}^{{\text{*}}})$
| Катализатор | Порядок реакции по CO | Порядок реакции по O2 | Ea, кДж/моль | 107 × $W_{{{\text{250}}}}^{{\text{*}}}{\text{,}}$ моль м–2 с–1 |
|---|---|---|---|---|
| Fe82Al18 | 1.11 | 0 | 102 ± 8 | 0.35 ± 0.03 |
| Cu5Fe78Al17 | 1.10 | 0 | 88 ± 6 | 91 ± 9 |
Структура и химический состав Cu–Fe–Al-нанокомпозитов
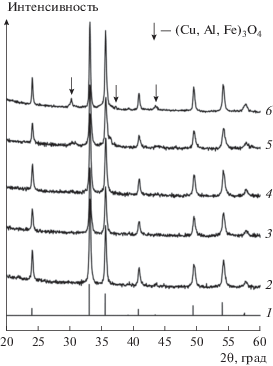
Как и в случае Fe–Al-катализаторов, на дифрактограммах Cu–Fe–Al-катализаторов с содержанием оксида меди 3–8 мас. % наблюдаются пики, соответствующие Fe2O3 со структурой гематита, пространственная группа (пр. гр.) $R\bar {3}c$ (PDF 330664) (рис. 3). Дополнительных рефлексов, соответствующих кристаллическим медьсодержащим фазам, не было обнаружено. Увеличение содержания оксида меди до 10 мас. % приводит к появлению на дифрактограмме дополнительных низкоинтенсивных пиков, соответствующих смешанному оксиду (Cu,Al,Fe)3O4 со структурой шпинели, пр. гр. Fd3m, параметр решетки которого составляет 8.333 Å, что находится между значениями, характерными для CuFe2O4 (8.400 Å, ICSD № 39132) и AlFe2O4 (8.204 Å, ICSD № 86571).
Рис. 3.
Дифрактограммы нанокомпозитных Cu–Fe–Al-катализаторов с различным содержанием меди: 1 – α-Fe2O3 (PDF 330664), 2 – Fe82Al18, 3 – Cu3Fe79Al18, 4 – Cu5Fe78Al17, 5 – Cu8Fe75Al17, 6 – Cu10Fe74Al16.
Для получения более детальной информации катализаторы были исследованы методами XANES и EXAFS. Оба метода являются структурно-чувствительными. XANES позволяет определить химическое состояние атомов, EXAFS – структуру локального окружения атомов. В отличие от дифракционных методов, EXAFS дает возможность изучать строение наноматериалов и аморфных тел. На рис. 4а приведены XANES-спектры K-края меди Cu–Fe–Al-катализаторов с содержанием 3–10 мас. % CuO. Спектры XANES CuK-края всех исследуемых образцов представлены одним интенсивным пиком на краю поглощения (переход 1s → 4p). Сравнивая положение CuK-края и форму спектров металлической меди, Cu2O, CuO, CuAl2O4, CuFe2O4 и катализаторов (рис. 4б), можно сделать вывод, что медь в катализаторах присутствует преимущественно в состоянии Cu2+. Действительно, форма спектров XANES K-края Cu существенно зависит от химического состояния меди. В спектрах К-края поглощения металлической меди собственно край поглощения Е0 находится на 8979 эВ. Окисление меди приводит к его сдвигу в сторону бóльших энергий, что указывает на наличие положительного заряда на атомах меди. Величина сдвига зависит от зарядового состояния меди. Край поглощения в спектре Cu2O, где медь находится в состоянии Cu1+, смещается в сторону более высоких энергий фотонов (~8980 эВ); в спектре также присутствует интенсивное плечо в районе 8981 эВ [19]. Появление плеча на краю поглощения обусловлено переходом 1s → 4p, возникающим в результате переноса валентных электронов на внутреннюю вакансию, так называемые “shakedown” электронные переходы [20, 21]. Атомы Cu2+ имеют частично заполненную 3d-электронную оболочку. Рентгеновские спектры поглощения таких соединений характеризуются наличием слабоинтенсивного пика в предкраевой области спектра и плеча на краю поглощения. Предпик соответствует дипольно-запрещенными квадрупольными переходами 1s → 3d, возникающими за счет гибридизации p- и d-орбиталей меди [22]. Край поглощения CuO наблюдается на 8983 эВ, край поглощения CuFe2O4 – на 8989 эВ.
Рис. 4.
XANES-спектры K-края меди: а – спектры нанокомпозитных Cu–Fe–Al-катализаторов; б – спектры массивных оксидов меди и медной фольги; на врезке приведена предкраевая область XANES спектров CuFe2O4 (1), CuAl2O4 (2) и CuO (3).
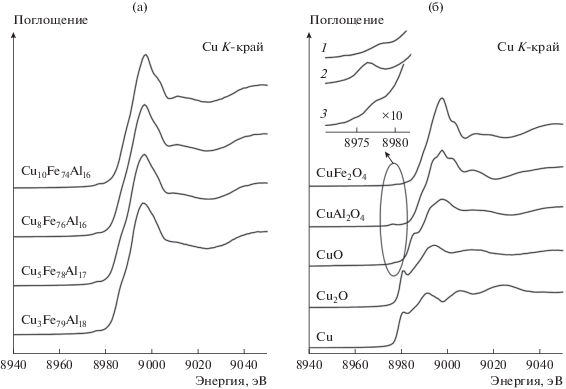
Дипольно-запрещенный переход можно рассматривать в качестве идентификатора состояния Cu2+, поскольку в соединениях, где медь находится в состоянии Cu1+, переход 1s → 3d полностью запрещен вследствие заполненной 3d-электронной оболочки, и в спектрах поглощения соединений Cu(I) предпик не регистрируется. Интенсивность предкраевого пика возрастает по мере того, как атом меди отклоняется от центросимметричного положения из-за увеличения перемешивания 3d- и 4p-орбиталей и, следовательно, зависит от заполнения 3d-орбиталей, координационной геометрии и локальной симметрии. Такой предпик присутствует в спектрах CuO, CuAl2O4 и CuFe2O4 (рис. 4б). Интенсивность предпика больше для атомов меди в тетраэдрической координации (в соединениях типа CuAl2O4) и значительно меньше для атомов меди в центросимметричном плоско-квадратном или октаэдрическом окружении (в соединениях типа CuO и CuFe2O4) [23].
В низкоэнергетической области края поглощения спектров CuO и CuAl2O4 присутствует заметное плечо. Эта особенность обусловлена дипольно-разрешенным переходом 1s → 4p, возникающим в результате переноса валентных электронов в несвязанное состояние непрерывного спектра и “shakedown” переходов. Эти переходы зависят от центросимметричности окружения поглощающего атома, а также чувствительны к природе лиганда. В спектре CuO наблюдается довольно интенсивное плечо вследствие плоско-квадратной координации атомов меди, в то время как в спектре CuAl2O4, плечо малоинтенсивное, что может объясняться октаэрической симметрией окружения атомов меди. Спектр CuFe2O4 характеризуется резко возрастающим краем поглощения с интенсивным пиком на 8998 эВ [24, 25]. В отличие от CuAl2O4, в спектре CuFe2O4 практически отсутствует плечо на низкоэнергетическом склоне края поглощения и расщепления основного пика поглощения, что свидетельствует о сильной степени инверсии структуры шпинели [23, 24].
Таким образом, сравнение спектров нанокомпозитов (рис. 4а) со спектрами реперных соединений (рис. 4б) указывает на то, что медь в катализаторах присутствует преимущественно в окисленном состоянии Cu2+ – край поглощения Е0 находится в диапазоне 8985–8987 эВ, что близко к Е0 для CuFe2O4. Спектры нанокомпозитов по своей структуре похожи на спектры CuFe2O4. Наличие небольшого плеча на краю поглощения для нанокомпозитов с содержанием 3–8 мас. % оксида меди может говорить о некотором количестве катионов меди с локальным окружением близким к объемному CuO.
Для получения количественной информации XANES спектры катализаторов аппроксимировали линейной комбинацией спектров реперных соединений, используя пакет программ DEMETER [16]. Этот метод получил название “linear combination fitting — LCF” и активно применяется для анализа XANES-спектров [11]. Теоретическим обоснованием метода LCF является аддитивность природы поглощения рентгеновского излучения различными компонентами, из которых состоит исследуемый материал. Следовательно, спектр поглощения изучаемого вещества представляет собой сумму спектров поглощения от компонент, образующих исследуемое вещество. Весовые коэффициенты в линейной комбинации спектров эталонных соединений подбираются методом наименьших квадратов. Из анализа LCF установлено, что возникновение плеча на краю поглощения в спектрах Cu–Fe–Al-катализаторов связано с присутствием некоторого количества меди в составе CuO, медь в металлическом состоянии и Cu2O отсутствуют. В качестве примера на рис. 5 приведен результат аппроксимации XANES-спектра катализатора Cu3Fe79Al18 линейной комбинацией спектров CuO (43 мас. %) и CuFe2O4 (57 мас. %). На рисунке представлен экспериментальный спектр катализатора Cu3Fe79Al18 (1), а также теоретически построенный модельный спектр (2), полученный путем варьирования весовых коэффициентов каждого отдельного спектра массивного оксида меди CuFe2O4 (3) и CuO (4) для достижения наилучшего соответствия теоретического спектра экспериментальному. При добавлении спектров металлической меди либо Cu2O в линейную комбинацию для построения модельного спектра их весовые коэффициенты обращались в ноль. Поэтому данные вещества были исключены из рассмотрения в качестве компонент Cu–Fe–Al-катализаторов.
Рис. 5.
Аппроксимация спектра XANES K-края меди катализатора Cu3Fe79Al18 линейной комбинацией спектров CuO и CuFe2O4: 1 – экспериментально полученный спектр катализатора Cu3Fe79Al18, 2 – модельный спектр (пунктирная кривая), 3 – спектр CuFe2O4, 4 – спектр CuO.
EXAFS-анализ Cu–Fe–Al-катализаторов дает более детальную информацию о локальном окружении атомов меди, включая межатомные расстояния и координационные числа каждой координационной сферы. На рис. 6а представлены модули Фурье-трансформант (МФТ) спектров EXAFS металлической меди, CuO, Cu2O, CuAl2O4 и CuFe2O4); МФТ спектров EXAFS нанокомпозитных катализаторов, а также результаты оптимизации их структурных моделей показаны на рис. 6б. Найденные значения параметров локального окружения атомов Cu (межатомные расстояния и координационные числа) приведены в табл. 3. Следует отметить, что вид Фурье-образа EXAFS-сигнала напоминает парную функцию радиального распределения атомов, что иногда приводит к его неправильной трактовке. Фурье-образ отличается от функции радиального распределения тем, что содержит информацию как о парных, так и многоатомных функциях распределения; положения пиков не соответствуют реальным межатомным расстояниям из-за сдвига фаз, а форма пиков сильно искажена из-за нетривиальной зависимости амплитуды обратного рассеяния от волнового числа k. Фурье-образ может дать лишь приближенное представление о геометрической структуре, и его полная интерпретация возможна только на основе моделирования.
Рис. 6.
Фурье-трансформанты EXAFS-спектров соединений меди (а) и нанокомпозитных Cu–Fe–Al-катализаторов (б). Сплошной линией показаны Фурье-трансформанты экспериментальных спектров, пунктирной — Фурье-трансформанты модельных спектров.
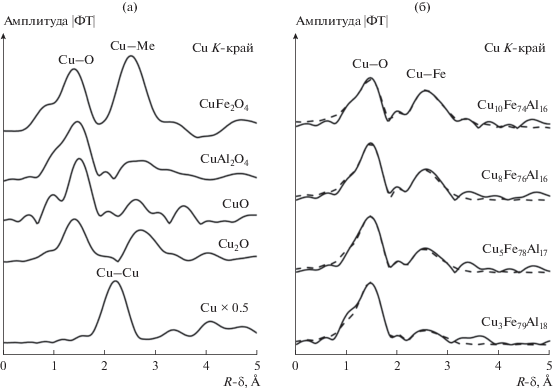
Таблица 3.
Межатомные расстояния (R), координационные числа (N) для локального окружения меди в Cu–Fe–Al-катализаторах, полученные из моделирования EXAFS-спектров на К-крае Cu
| Катализаторы | Cu3Fe79Al18 | Cu5Fe78Al17 | Cu8Fe75Al17 | Cu10Fe74Al16 | ||||
|---|---|---|---|---|---|---|---|---|
| R, Å | N | R, Å | N | R, Å | N | R, Å | N | |
| Cu–O | 1.842 | 2.5 | 1.879 | 2.7 | 1.895 | 3.1 | 1.954 | 4.3 |
| Cu–O | 2.010 | 4.1 | 2.038 | 2.8 | 2.066 | 2.9 | 2.149 | 4.0 |
| Cu–O | 2.315 | 3.4 | 2.344 | 2.9 | 2.354 | 3.0 | 2.412 | 3.6 |
| Cu–O | 2.586 | 3.0 | 2.638 | 2.8 | 2.650 | 3.0 | 2.728 | 4.5 |
| Cu–Cu | 2.901 | 1.7 | 2.917 | 2.3 | 2.919 | 2.8 | 2.948 | 3.4 |
| Cu–Fe | 3.490 | 0.7 | 3.496 | 1.7 | 3.469 | 1.3 | 3.472 | 1.6 |
Для получения структурной информации из EXAFS-спектров используется метод сопоставления теоретической и экспериментальной EXAFS-функций. Функции амплитуд и фаз рассчитываются теоретически для предполагаемого строения атомов ближайшего окружения поглощающего атома. По этим данным строится теоретический EXAFS-спектр и минимизируется разность теоретического и экспериментального EXAFS-спектров путем варьирования параметров: координационных чисел, межатомных расстояний, факторов Дебая–Валлера и порога ионизации E0. “Подгонка” модельного спектра к экспериментальному в большой мере позволяет определить степень соответствия предложенных локальных структур реальным.
Результаты моделирования, приведенные на рис. 6б, указывают на то, что первый пик, лежащий примерно на 1.5 Å на кривой Фурье-трансформанты спектров катализаторов, относится к первой координационной сфере и характеризует межатомные расстояния Cu–O; второй пик в районе 2.5 Å соответствует межатомным расстояниям Cu–Cu и Cu–Fe. Хорошо известно, что уменьшение размеров наночастиц ведет к изменению соотношения атомов, находящихся в объеме частицы и на ее поверхности. Это приводит к изменению значения координационного числа в сравнении со значением, характерным для массивных стандартных соединений. Из рис. 6б видно, что интенсивность пика на 2.5 Å постепенно увеличивается с ростом содержания в катализаторах меди. По результатам моделирования координационное число рассеивающих атомов Cu (на расстоянии ~2.9 Å) и Fe (на расстоянии ~3.5 Å) и межатомные расстояния Cu–O и Cu–Cu также монотонно возрастают с повышением содержания меди (табл. 3). Такое изменение координационных чисел и межатомных расстояний свидетельствует об увеличении размеров кластеров CuFeOx и CuO. Полученные результаты подтверждают сделанное выше предположение о том, что атомы меди в катализаторах находятся в составе кластеров CuO и CuFeOx, которые не могли быть обнаружены методом РФА вследствие их малых размеров.
In situ XANES/EXAFS исследование
Для выявления природы активных центров проведено три серии in situ экспериментов, в ходе которых проводили запись спектров поглощения К-краев Cu и Fe при ступенчатом нагреве катализатора Cu5Fe78Al17 от комнатной температуры до 600°C в потоке чистого CO и смеси газов CO и O2 с соотношением CO : O2 = 2 : 1 и 1 : 1. Исследования осуществляли с использованием катализатора Cu5Fe78Al17, проявляющего наибольшую активность в реакции окисления CO (рис. 1). На рис. 7 представлена серия спектров XANES К-краев Cu и Fe, полученных in situ при обработке катализатора в потоке чистого СО. В табл. 4 приведены результаты моделирования спектров поглощения методом LCF. Форма спектров XANES на K-крае Cu и Fe постепенно изменяется с повышением температуры. Спектр XANES К-края Fe исходного катализатора аналогичен спектру α-Fe2O3, где атомы железа находятся в состоянии Fe3+. Спектр имеет широкий предкраевой пик в районе 7114 эВ и интенсивный пик на краю поглощения на 7133 эВ, что характерно для катионов Fe3+ в октаэдрическом кислородном окружении [26, 27]. После нагрева катализатора в потоке СО до 400°С в спектре появляется небольшое плечо на крае поглощения, интенсивность которого растет с повышением температуры. Из анализа LCF было установлено, что при температуре около 400°С происходит превращение Fe2O3 → Fe3O4, т. е. происходит частичное восстановление катионов железа Fe3+ до Fe2+. Одновременно с этим процессом происходит дальнейшее восстановление Fe3O4 до FeO и металлического железа, однако Fe3O4 восстанавливается не полностью, даже при 600°С около 25% железа находится в шпинельной структуре Fe3O4. Аналогичные результаты были получены ранее методом РФА при in situ исследовании восстановления катализатора Cu5Fe78Al17 в потоке СО [28]. Восстановление Fe3O4 → Fe было зафиксировано лишь при 500°С, небольшое количество FeO было обнаружено при 650–700°C. Такое расхождение результатов XANES и РФА может быть связано с формированием рентгеноаморфных фаз, к которым метод РФА нечувствителен. Таким образом, восстановление Fe3O4 → FeO → → Fe в СО происходит при 400–500°С.
Рис. 7.
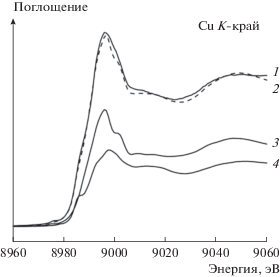
XANES-спектры К-края Fe (а) и Cu (б) катализатора Cu5Fe78Al17, полученные in situ в ходе нагрева в потоке CO.
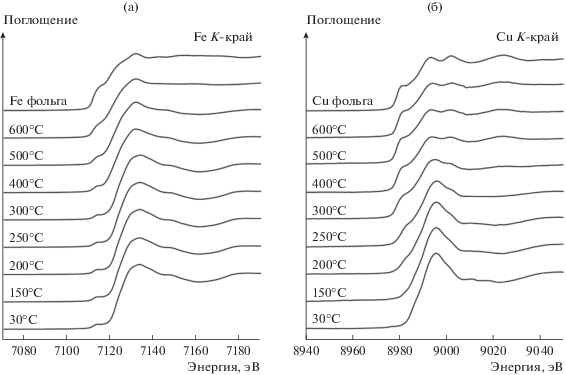
Таблица 4.
Фазовый состав, рассчитанный по данным LCF катализатора Cu5Fe78Al17, восстановленного при нагревании в потоке CO
| Температура, °С | Медьсодержащие фазы, % (Cu K-край) | Железосодержащие фазы, % (Fe K-край) | ||||||
|---|---|---|---|---|---|---|---|---|
| Cu | Cu2O | CuO | CuFe2O4 | Fe | FeO | Fe3O4 | Fe2O3 | |
| 30 | 0 | 0 | 21 | 79 | 0 | 0 | 0 | 100 |
| 150 | 0 | 0 | 35 | 65 | 0 | 0 | 0 | 100 |
| 200 | 0 | 0 | 40 | 60 | 0 | 0 | 2 | 98 |
| 250 | 35 | 0 | 35 | 30 | 0 | 0 | 3 | 97 |
| 300 | 60 | 0 | 29 | 11 | 0 | 0 | 21 | 79 |
| 400 | 80 | 15 | 5 | 0 | 0 | 27 | 73 | 0 |
| 500 | 80 | 20 | 0 | 0 | 28 | 22 | 50 | 0 |
| 600 | 80 | 20 | 0 | 0 | 65 | 10 | 25 | 0 |
Спектр XANES K-края Cu исходного катализатора Cu5Fe78Al17 представляет собой суперпозицию спектров CuO и CuFe2O4. При нагреве катализатора до 250°C в потоке CO наблюдается увеличение количества CuO и появление при 250°C металлической меди. Восстановление меди с Cu2+ до Cu1+ и Cu0 начинается при температуре около 200–250°C; при 400–600°C медь находится преимущественно в металлическом состоянии (табл. 4).
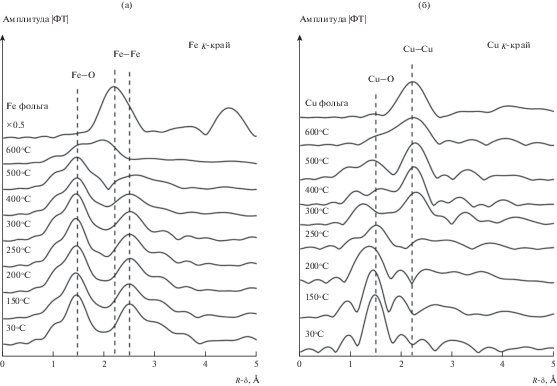
На рис. 8 представлены МФТ спектров EXAFS для K-краев поглощения Fe и Cu катализатора Cu5Fe78Al17. На кривой МФТ K-краев поглощения Fe исходного катализатора присутствует два пика в районе 1.5 и 2.5 Å, которые относятся к расстояниям Fe–O и Fe–Fe в Fe2O3 соответственно. На рис. 8а хорошо видно, что интенсивность второго пика, относящегося к расстояниям Fe–Fe в Fe2O3, монотонно снижается при повышении температуры от комнатной до 500°C. Это свидетельствует о том, что уменьшается заполнение катионных позиций железа вследствие теплового и структурного разупорядочения. При 600°C появляется пик на 2.25 Å, соответствующий межатомному расстоянию Fe–Fe в объемно-центрированной кубической решетке металлического железа. Это означает, что восстановление оксида железа до металла происходит при температуре выше 500°C. Для K-края поглощения меди на кривых исходного катализатора имеется интенсивный пик на 1.5 Å, что характерно для расстояния Cu–O (рис. 8б). При повышении температуры наблюдается резкое уменьшение интенсивности этого пика. После нагрева до 250°C появляется дополнительный пик примерно на 2.2 Å, относящийся к расстоянию Cu–Cu в металлической меди; особенно четко этот пик становится заметен при температуре 300°C. Таким образом, результаты EXAFS подтверждают сделанный ранее вывод о том, что высокодисперсный оксид меди восстанавливается в потоке CO до металла при температуре около 300°C.
Рис. 8.
Модули Фурье-трансформант спектров EXAFS железа (а) и меди (б) катализатора Cu5Fe78Al17, полученные in situ в ходе нагрева в потоке CO.
In situ спектры XANES К-краев Cu и Fe, полученные при обработке катализатора Cu5Fe78Al17 в потоке смеси газов CO и O2 с соотношением CO : O2 = 2 : 1, представлены на рис. 9. Сдвига K-края поглощения Fe не наблюдается вплоть до 600°C, в то время как XANES-спектры K-края Cu выше 400°C изменяются аналогично спектрам, полученным при исследовании катализатора в потоке чистого CO, но со сдвигом на 200°C в высокотемпературную область. Следовательно, присутствие кислорода в газовой смеси затрудняет восстановление. Следует отметить, что по XANES-спектрам сложно определить, находится медь в состоянии Cu0 или Cu1+. Более детальную информацию можно получить методом EXAFS.
Рис. 9.
Спектры XANES К-края Fe (а) и Cu (б) катализатора Cu5Fe78Al17, полученные in situ в ходе нагрева в потоке реакционной смеси (CO : O2 = 2 : 1).
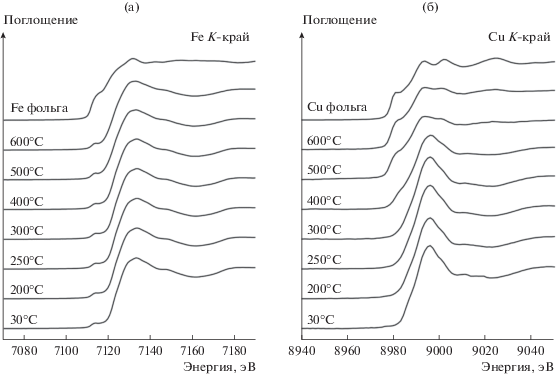
На рис. 10 приведены соответствующие кривые МФТ спектров EXAFS К-края железа и меди. На кривых МФТ спектров EXAFS меди при 500 и 600°C отчетливо видны пики на 1.5 и 2.25 Å, соответствующие расстояниям Cu–O в оксиде меди и Cu–Cu в металлической меди. Следовательно, при окислении СО при 500–600°C происходит восстановление меди, и в катализаторе медь находится как в окисленном, так и в металлическом состоянии. На кривых МФТ спектров К-края железа наблюдаются два интенсивных пика на 1.5 и 2.5 Å, характерные для межатомных расстояний Fe–O и Fe–Fe в Fe2O3 соответственно. С повышением температуры происходит постепенное уменьшение интенсивности пика на 2.5 Å. Это свидетельствует о том, что при нагреве до 500–600°C в потоке смеси CO и кислорода, как и в случае восстановления в потоке CO, в катализаторе Cu5Fe78Al17 образуются катионные вакансии. Однако даже при 500–600°C никаких дополнительных пиков в районе 2.25 Å, соответствующих межатомному расстоянию Fe–Fe в металлическом железе, на кривых МФТ не наблюдается. То есть в присутствии кислорода в газовой смеси железо не восстанавливается до металлического состояния.
Рис. 10.
Модули Фурье-трансформант спектров EXAFS железа (а) и меди (б) катализатора Cu5Fe78Al17, полученные in situ в ходе нагрева в потоке реакционной смеси (CO : O2 = 2 : 1).
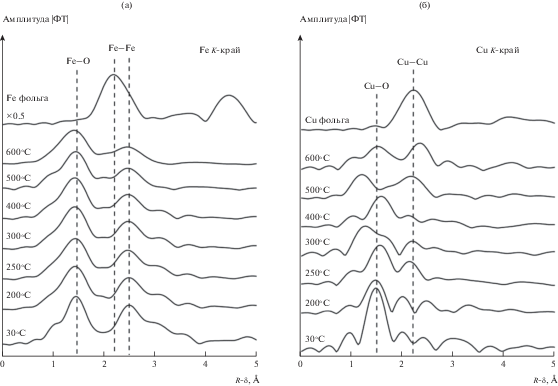
Таким образом, результаты анализа XANES и EXAFS показывают, что при наличии кислорода в реакционной смеси температура восстановления Cu2+ → Cu1+ смещается в область высоких температур: восстановление начинается при ~400°C, затем при 500°C начинается формирование металлической меди. Следует подчеркнуть, что медь находится в двух состояниях – Cu1+ и Cu0. В то же время степень окисления железа остается без изменения даже при 600°C (рис. 9).
В заключении было проведено in situ исследование катализатора методом XANES при окислении СО в избытке кислорода (соотношение CO : O2 = 1 : 1). Установлено, что форма спектров XANES К-краев Cu и Fe остается неизменной в процессе нагрева до 600°C, т.е. состояния железа и меди не меняются в реакционных условиях (спектры не приведены). Следовательно, можно предположить, что в ходе окисления СО на нанокомпозитных катализаторах Cu–Fe–Al происходит частичное восстановление Fe3+ → Fe2+ и Cu2+ → Cu1+ молекулами СО, однако в присутствие кислорода протекает быстрое обратное окисление до Fe3+ и Cu2+. Скорость окисления выше скорости восстановления, что характерно для окислительно-восстановительного механизма Марса-ван Кревелена. Это хорошо согласуется с результатами кинетических исследований, указывающих на нулевой порядок реакции по кислороду и первый порядок реакции по СО при окислении СО на Fe–Al- и Cu–Fe–Al-нанокомпозитах (табл. 2). Окисление СО на медьсодержащих нанокомпозитах происходит преимущественно на оксиде меди – СО адсорбируется на поверхности кластеров CuO, затем реагирует с решеточным кислородом с образованием СО2 и кислородной вакансии Cu1+–≤; последующая адсорбция кислорода приводит к заполнению вакансий и окислению Cu1+ до Cu2+. Существует другая возможность окисления СО на Cu–Fe–Al-катализаторах, которая заключается в адсорбции СО на катионах Fe3+ с последующей реакцией с решеточным кислородом Fe3+–[O2-]. Однако скорость альтернативного маршрута окисления СО в 260 раз меньше скорости окисления СО на оксиде меди (табл. 2). Это позволяет предположить, что именно кластеры CuO и CuFeOx являются активными центрами в реакции окисления СО на Cu–Fe–Al-нанокомпозитах.
ЗАКЛЮЧЕНИЕ
Синтезированы две серии Fe–Al- и Cu–Fe–Al-оксидных нанокомпозитов, перспективных для разработки катализаторов сжигания твердых топлив в кипящем слое. Проведены их испытания в реакции окисления СО. Показано, что введение меди существенно повышает активность катализаторов в окисления СО. Наиболее активным катализатором является нанокомпозит, содержащий 5 мас. % CuO, 78 мас. % Fe2O3 и 17 мас. % Al2O3. По данным РФА катализаторы состоят из наночастиц гематита, α-Fe2O3, и аморфного оксида алюминия. Медь находится в высокодисперсном состоянии, образуя кластеры CuO и CuFeOx. Увеличение содержания меди до 10 мас. % приводит к формированию шпинели CuxAlyFe3-x-yO4, что сопровождается падением активности катализатора. Результаты in situ исследований методами спектроскопии рентгеновского поглощения показали, что окисление СО на Cu–Fe–Al происходит преимущественно на кластерах оксида меди по окислительно-восстановительному механизму – СО адсорбируется на поверхности кластеров оксида меди, затем реагирует с решеточным кислородом с образованием СО2 и кислородной вакансии Cu1+–≤; последующая адсорбция кислорода приводит к заполнению вакансий и окислению Cu1+ до Cu2+. Скорость окисления СО на оксиде железа примерно в 260 раз меньше скорости окисления СО на оксиде меди. Оксид железа и оксид алюминия являются структурными промоторами, обеспечивающими высокую удельную поверхность катализаторов и стабильность высокодисперсных частиц оксида меди.
Список литературы
Prasad R., Kennedy L.A., Ruckenstein E. // Catal. Rev. 1984. V. 26. P. 1.
Ismagilov Z.R., Kerzhentsev M.A. // Catal. Today. 1999. V. 47. P. 339.
Simonov A.D., Yazykov N.A., Vedyakin P.I., Lavrov G.A., Parmon V.N. // Catal. Today. 2000. V. 60. P. 139.
Симонов А.Д., Федоров И.А., Дубинин Ю.В., Языков Н.А., Яковлев В.А., Пармон В.Н. // Катализ в промышленности. 2012. № 3. С. 50. (Simonov A.D., Fedorov N.A., Dubinin Yu.V., Yazykov N.A., Yakovlev V.A., Parmon V.N. // Catal. Ind. 2013. V. 5. P. 42.)
Пармон В.Н., Симонов А.Д., Садыков В.А., Тихов С.Ф. // Физика горения и взрыва. 2015. Т. 51. № 2. С. 5. (Parmon V.N., Simonov A.D., Sadykov V.A., Tikhov S.F. // Combust. Explo. Shock Waves. 2015. V. 51. P. 143.)
Fedorov A.V., Yazykov N.A., Bulavchenko O.A., Saraev A.A., Kaichev V.V., Yakovlev V.A. // Nanomaterials. 2020. V. 10. P. 1002.
Fedorov A.V., Saraev A.A., Kremneva A.M., Selivanova A.V., Vorokhta M., Šmíd B., Bulavchenko O.A., Yakovlev V.A., Kaichev V.V. // ChemCatChem. 2020. V. 12. In press. https://doi.org/10.1002/cctc.202000852
Kamal M.S., Razzak S.A., Hossain M.M. // Atmos. Environ. 2016. V. 140. P. 117.
Ismagilov Z.R., Shkrabina R.A., Koryabkina N.A. // Catal. Today. 1999. V. 47. P. 51.
Arendarskii D.A., Ismagilov Z.R., Barannik G.B. // Catal. Lett. 1992. V. 15. P. 347.
Fedorov A.V., Tsapina A.M., Bulavchenko O.A., Saraev A.A., Odegova G.V., Ermakov D.Yu., Zubavichus Y.V., Yakovlev V.A., Kaichev V.V. // Catal. Lett. 2018. V. 148. P. 3715.
Bulavchenko O.A., Pochtar’ A.A., Gerasimov E.Yu., Fedorov A.V., Chesalov Yu.A., Saraev A.A., Yakovlev V.A., Kaichev V.V. // Appl. Catal. A. 2020. V. 590. P. 117 364.
Пахаруков И.Ю., Бобров Н.Н., Пармон В.Н. // Катализ в промышленности. 2008. Т. 6. С. 11. (Pakharukov I.Yu., Bobrov N.N., Parmon V.N. // Catal. Ind. 2009. V. 1. P. 38.)
Bearden J.A., Burr A.F. // Rev. Modern Phys. 1967. V. 39. P. 125.
Велигжанин А.А., Зубавичус Я.В., Чернышов А.А., Тригуб А.Л., Хлебников А.С., Низовский А.И., Худорожков А.К., Бекк И.Э., Бухтияров В.И. // Журн. Струк. Химии. 2010. Т. 51. Приложение. С. S26. (Veligzhanin A.A., Zubavichus Ya.V., Chernyshov A.A., Trigub A.L., Khlebnikov A.S., Nizovskii A.I., Khudorozhkov A.K., Beck I.E., Bukhtiyarov V.I. // J. Struc. Chem. 2010. V. 51. Supplement. P. S20.)
Ravel B., Newville M. // J. Synchrotron Rad. 2005. V. 12. P. 537.
ISCD (Inorganic Crystal Structure Database) https://icsd.products.fiz-karlsruhe.de/
Rehr J.J., Mustre de Leon J., Zabinsky S.I., Albers R.C. // J. Am. Chem. Soc. 1991. V. 113. P. 5135.
Kim J.Y., Rodriguez J.A., Hanson J.C., Frenkel A., Lee P.L. // J. Phys. Chem. B. 2003. V. 107. P. 9195.
Kosug N., Yokoyama T., Asakuna K., Kuroda H. // Chem. Phys. 1984. V. 91. P. 249.
Bair R.A., Goddard W.A. // Phys. Rev. B. 1980. V. 22. P. 2767.
Hahn J.E., Scott R.A., Hodgson K.O., Doniach S., Desjardins S.E., Solomon E.I. // Chem. Phys. Lett. 1982. V. 88. P. 595.
Shimizu K.-I., Maeshima H., Yoshida H., Satsuma A., Hattori T. // Phys. Chem. Chem. Phys. 2001. V. 3. P. 862.
Krishnan V., Selvan R.K., Augustin C.O., Gedanken A., Bertagnolli H. // J. Phys. Chem. C. 2007. V. 111. P. 16 724.
Caddeo F., Loche D., Casula M.F., Corrias A. // Sci. Rep. 2018. V. 8. P. 797.
Piquer C., Roca A.G., Laguna-Marco M.A., Boada R., Guglieri C., Chaboy J. //. J. Phys. Chem. C. 2014. V. 118. P. 1332.
Okube M., Oshiumi T., Nagase T., Miyawaki R., Yoshiasa A., Sasaki S., Sugiyama K. // J. Synchrotron Rad. 2018. V. 25. P. 1694.
Bulavchenko O.A., Vinokurov Z.S., Saraev A.A., Tsapina A.M., Trigub A.L., Gerasimov E.Y., Gladky A.Y., Fedorov A.V., Yakovlev V.A., Kaichev V.V. // Inorg. Chem. 2019. V. 58. P. 4842
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ