

Кинетика и катализ, 2021, T. 62, № 1, стр. 38-43
Каталитическое жидкофазное окисление циклооктена молекулярным кислородом в 1,2-эпоксициклооктан
Д. Р. Шангареев a, Т. Н. Антонова a, *, И. Г. Абрамов a, Т. С. Сивова a, А. С. Данилова a
a Ярославский государственный технический университет
150999 Ярославль, Московский просп., 88, Россия
* E-mail: antonovatn@ystu.ru
Поступила в редакцию 14.11.2019
После доработки 10.06.2020
Принята к публикации 17.06.2020
Аннотация
Исследованы закономерности жидкофазного окисления циклооктена молекулярным кислородом в присутствии ряда катализаторов в сравнении с его инициированным окислением. Показано, что использование каталитической системы, включающей соединения молибдена, увеличивает содержание эпоксида в продуктах окисления в 1.5 раза при той же конверсии циклоолефина, что и при инициированном окислении.
В последние годы большое внимание исследователей и разработчиков методов получения циклоолефинов привлекает процесс селективного гидрирования циклодиенов, получаемых в ряде синтезов [1–7]. Гидрированные аналоги циклодиенов – соответствующие циклоолефины – представляют интерес для последующего получения на их основе производных различной функциональности. К ним относятся, прежде всего, эпоксиды – важнейшие продукты промышленного органического синтеза [8].
1,2-Эпоксициклооктан, получаемый на основе циклооктена, может быть использован для создания эпоксидных смол, которые имеют хорошие диэлектрические показатели, стабильны в широком интервале температур, а также отличаются высокой тепло- и атмосферостойкостью. Они находят применение в производстве лакокрасочных материалов, пленочных покрытий для радиоэлектроники и вычислительной техники. Кроме того, высокая реакционная способность 1,2-эпоксициклооктана позволяет использовать его для получения циклооктанона, циклооктанола, 1,2-циклооктандиола и далее пробковой (1,8-октандиовой, субериновой, гександикарбоновой) кислоты как исходного продукта для получения пластификаторов, придающих изделиям из пластмасс морозостойкость [9].
Как известно из литературных источников [10, 11], для получения 1,2-эпоксициклооктана на основе циклооктена можно использовать окислители различной природы – органические гидропероксиды, надкислоты, пероксид водорода, а также молекулярный кислород.
При жидкофазном окислении циклооктена молекулярным кислородом (в отличие от циклогексена и других циклоолефинов) 1,2-эпоксициклооктан образуется в качестве основного продукта реакции и его выход достигает 65–70% [11–14]. По-видимому, это объясняется тем, что циклооктен является алициклическим углеводородом со средним размером цикла. В тесном цикле средней величины в результате трансаннулярных, а также питцеровских взаимодействий [9] двойная связь циклооктена напряжена, регибридизована, а соседняя с ней α-С–Н-связь ослаблена, что и определяет относительно высокий выход эпоксида по сравнению с циклогексеном. Тем не менее наряду с эпоксидом в продуктах окисления циклооктена образуются и нежелательные побочные продукты: пероксидные и карбонильные соединения, включая корковый альдегид [15], а также карбоновые кислоты.
С целью оценки возможности повышения выхода 1,2-эпоксициклооктана в настоящей работе изучены кинетические закономерности процесса окисления циклооктена молекулярным кислородом в присутствии ряда катализаторов в сопоставлении с его инициированным окислением.
Применение катализаторов в химической технологии, как известно, способствует не только ускорению процесса, но и направляет его течение в сторону образования целевых продуктов. Использование катализаторов с ярко выраженной регулирующей функцией упрощает схему выделения целевых продуктов из реакционной смеси и обеспечивает высокую степень их чистоты.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Циклооктен с содержанием основного вещества не менее 98% получали селективным гидрированием 1,5-цис,цис-циклооктадиена. Условия гидрирования и чистота полученного циклооктена приведены в работе [1]. Перед использованием в реакции окисления циклооктен очищали, испаряя в токе азота (Тпл = 261.2 К, Ткип = 411.2 К, $\rho _{4}^{{20}}$ = = 0.8472, $n_{{\text{д}}}^{{20}}$ = 1.4698).
В качестве инициаторов окисления использовали индивидуальные соединения. Гидропероксид трет-бутила (ГПТБ, ТУ 6-01-463-80) дополнительно очищали от примесей трет-бутилового спирта перегонкой на лабораторной ректификационной колонке со стеклянной насадкой (массовая доля основного вещества составила 98–100%, Ткип = 319 К/2.27 кПа, $\rho _{4}^{{20}}$ = 0.8930, $n_{{\text{д}}}^{{20}}$ = 1.4013). N-гидроксифталимид (N-ГФИ) и пероксид бензоила (ПБ) произведены фирмой “Alfa Aesar” с чистотой 97–98%. Гидропероксид кумола (ГПК, МРТУ-38-25-86) очищали от примесей через натриевую соль (содержание основного вещества 98–100%, Ткип = 333 К/0.026 кПа, $\rho _{4}^{{20}}$ = 1.061).
В качестве катализаторов применяли стеарат кобальта (ТУ 2494-02-53904859-01), резинат марганца (ТУ 6-09-4659-78), гептамолибдат аммония (ГОСТ 3765-78) и пропандиолат молибденила, предоставленный ПАО “Нижнекамскнефтехим”.
Окисление циклооктена молекулярным кислородом в 1,2-эпоксициклооктан осуществляли в отсутствие растворителей при атмосферном давлении на волюмометрической установке замкнутого типа в реакторе типа “утка”, а также в проточной системе в термостатированном реакторе (моделирующем реактор идеального смешения), снабженном мешалкой, термометром, диффузором для подачи кислорода и обратным холодильником с каплеотбойником.
Опыты оценивали по количеству поглощенного кислорода и содержанию в реакционной смеси продуктов реакции, которые анализировали химическими и инструментальными методами (потенциометрией, газожидкостной хроматографией (ГЖХ), ИК-спектроскопией).
Метод ГЖХ использовали для количественного определения исходного циклооктена, целевого эпоксида и циклооктенола на приборе Хроматэк-Кристалл 5000.2 (СКБ “Хроматэк”, Россия), снабженном капиллярной колонкой CR-5 (30 м × × 0.32 мм), заполненной в качестве жидкой фазы смесью 5% фенил- и 95% диметилэпоксилоксана (толщина пленки 0.5 мкм). Температура испарителя 493 К, температура колонки 343–433 К), скорость подъема температуры 10 град/мин, скорость газа-носителя (азота) 60 см3/мин. Содержание исследованных веществ рассчитывали по методу внутреннего стандарта, которым служил 1-гептанол. Относительная погрешность ГЖХ-анализа не превышала 5.0%.
Наличие в продуктах окисления карбонильных соединений определяли методом потенциометрического титрования по разработанной нами методике [16], позволяющей анализировать их в присутствии эпоксидов.
Целевой эпоксид циклооктена после выделения идентифицировали методом 1Н ЯМР-спектрометрии на приборе Bruker DRX-400 (Германия) в ДМСО-d6 при 30°С. В качестве эталона для отсчета химических сдвигов использовали сигналы остаточных протонов растворителя в протонных спектрах (ΔδH = 2.50 м. д.), а в качестве маркера – сигнал тетраметилсилана.
1,2-Эпоксициклооктан. 1Н ЯМР-спектр (400 MHz, DMSO-d6), δ, м. д.: 0.67–0.80 (m, 1H), 1.20–1.40 (m, 3H), 1.80–1.95 (m, 1H), 2.65–2.75 (m, 1H), 2.80 (br. s., 2H), 4.84 (d, J = 10.26 Hz, 1H), 4.95 (s, 1H), 4.99 (s, 1H), 5.36–5.51 (m, 1H), 5.90–5.98 (m, 1H), 6.17 (dd, J = 5.13, 2.69 Hz, 1H).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Процесс жидкофазного инициированного окисления циклооктена молекулярным кислородом в целевой эпоксид, как показано ранее [14] и подтверждается нами, протекает по радикально-цепному механизму. Он затормаживается при использовании ингибиторов различной природы, а именно, ионола (2,6-ди(трет-бутил)-4-метилфенола) и стабильного нитроксильного радикала (2,2,6,6-тетраметилпиперидин-1-оксила) (рис. 1).
Рис. 1.
Кинетические кривые расходования циклооктена в процессе инициированного окисления в зависимости от природы и концентрации инициатора, моль/л: 1 – ПБ (0.03), 2 – ГПТБ (0.06), 3 – N-ГФИ (0.06), 4 – ПБ (0.02), 5 – ГПТБ (0.03), 6 – без инициатора. Ингибированное окисление: 7 – 2,2,6,6-тетраметилпиперидин-1-оксил, 8 – 2,6-ди(трет-бутил)-4-метилфенол. Синг = 0.01 моль/л, СПБ = 0.03 моль/л, 398 К.
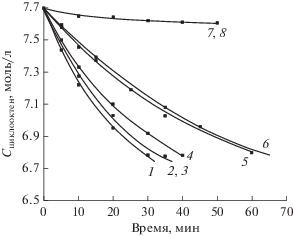
Начальная скорость окисления (W0), полученная графическим дифференцированием кинетических кривых расходования циклооктена (рис. 1), определяется природой инициатора, заметно возрастая по сравнению с неинициированным окислением (табл. 1).
Таблица 1.
Составы продуктов инициированного окисления циклооктена и при проведении процесса в присутствии молибденсодержащей каталитической системы (степень превращения циклооктена 12%, 398 К)
| Инициатор (Синиц, моль/л) |
Начальная скорость окисления (W0 × 102, моль л–1 мин–1) |
Состав продуктов, мас. д. % | Время, мин | Выход*, мол. % | ||||
|---|---|---|---|---|---|---|---|---|
| эпоксид | гидропероксид | кетон | кислота | |||||
| Инициированное окисление | ||||||||
| – | – | 1.68 | 7.10 | 1.56 | 1.32 | 1.05 | 55 | 52.70 |
| ПБ | 0.01 | 2.00 | 6.50 | 1.84 | 1.12 | 0.82 | 60 | 48.20 |
| 0.015 | 2.90 | 50 | ||||||
| 0.02 | 4.24 | 40 | ||||||
| 0.03 | 5.10 | 30 | ||||||
| ГПТБ | 0.03 | 1.68 | 7.20 | 1.68 | 1.30 | 1.12 | 60 | 52.10 |
| 0.06 | 4.68 | 8.30 | 2.01 | 0.97 | 0.65 | 35 | 61.50 | |
| N-ГФИ | 0.06 | 4.68 | 7.10 | 2.19 | 1.25 | 1.10 | 35 | 52.70 |
| Каталитическое окисление | ||||||||
| каталитическая система | 10.80 | 0.31 | 0.90** | 0.43 | 25 | 80.10 | ||
| инициатор | катализатор | |||||||
| N-ГФИ (0.03 моль/л) | гептамолибдат аммония (0.001моль/л) |
|||||||
| ГПТБ (0.03 моль/л) | 12.45 | 0.25 | 1.15** | 0.48 | 30 | 92.40 | ||
Как видно из данных, представленных в табл. 1, инициированное окисление циклооктена протекает не селективно и выход целевого эпоксида в расчете на превращенный циклооктен составляет 50–62 мол. % при степени его превращения 12%. В качестве побочных продуктов образуются гидропероксид циклооктена, циклооктенон и кислоты.
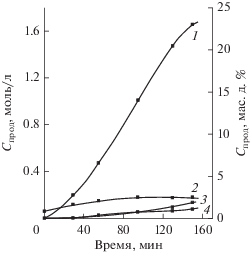
Образование кетонов в радикально-цепном процессе окисления углеводородов обычно связывают с атакой образующегося гидропероксида алкильными радикалами R•, что особенно характерно для вторичных гидропероксидов [17]. Дальнейшее окисление ненасыщенного кетона при повышенных температурах приводит к образованию кислот, преимущественно пимелиновой. Эту кислоту, выделенную из продуктов окисления циклооктена, идентифицировали по температуре ее плавления и на основании данных потенциометрического титрования. Кинетические кривые накопления целевого эпоксида циклооктена и побочных продуктов в процессе окисления циклооктена в проточном реакторе в присутствии гидропероксида кумола как инициатора при температуре 393 К показаны на рис. 2.
Рис. 2.
Кинетические кривые накопления продуктов инициированного окисления циклооктена, моль/л: 1 – эпоксициклооктан, 2 – гидропероксид циклооктена, 3 – пимелиновая кислота, 4 – циклооктенон. Инициатор – ГПК (0.06 моль/л), 393 К.
В настоящей работе изучено каталитическое окисление циклооктена, где в качестве катализаторов применяли соединения кобальта и марганца – типичные катализаторы реакции жидкофазного окисления. Использование стеарата кобальта при указанной концентрации увеличивает скорость окисления в той же мере, что и использование инициаторов процесса (рис. 3), но приводит к значительному снижению содержания эпоксида в продуктах окисления с одновременным повышением содержания кислот (табл. 2).
Рис. 3.
Влияние природы катализатора на процесс окисления циклооктена молекулярным кислородом, моль/л: 1 – каталитическая система ГПТБ (0.06)–гептамолибдат аммония (0.001), 2 – стеарат кобальта (0.01), 3 – ГПТБ (0.06), 4 – гептамолибдат аммония (0.001), 5 – пропандиолат молибденила (0.001), 6 – пропандиолат молибденила (0.001)–ингибитор 2,2,6,6-тетраметилпиперидин-1-оксил (0.01), 398 К.
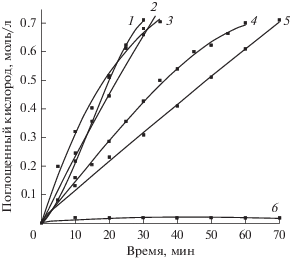
Таблица 2.
Количественные показатели процесса окисления циклооктена в зависимости от природы катализатора (степень превращения циклооктена 12%, 398 К)
| Каталитическая система | С, моль/л |
Wэф × 102, моль л–1 мин–1 |
Содержание эпоксида, мас. д. % | Содержание кислот, мас. д. % | Время реакции, мин |
|---|---|---|---|---|---|
| ГПТБ | 0.06 | 2.60 | 8.30 | 0.65 | 35 |
| Стеарат Со | 0.01 | 2.30 | 4.58 | 3.45 | 30 |
| Резинат Mn | 0.01 | 1.94 | 5.20 | 2.87 | 35 |
| Пропандиолат Mo | 0.001 | 0.90 | 10.20 | 0.39 | 70 |
| Гептамолибдат аммония | 0.001 | 1.45 | 10.30 | 0.30 | 60 |
| ГПТБ–гептамолибдат аммония | 0.03 | 2.40 | 12.45 | 0.48 | 30 |
| 0.001 |
При использовании соединений молибдена как органических, так и неорганических (пропандиолат молибденила, гептамолибдат аммония) в качестве катализаторов окисления сохраняется радикально-цепной механизм окисления (рис. 3, кривая 6) и повышается содержание эпоксида циклооктена в продуктах окисления практически в 1.2 раза до 10.2 мас. д. % по сравнению с инициированным окислением (8.3 мас. д. %) при той же степени превращения циклооктена (табл. 2), но без повышения скорости процесса окисления (рис. 3, кривые 4 и 5).
Установлено, что применение в процессе окисления циклооктена каталитической системы, включающей наряду с соединением молибдена дополнительно инициатор окисления (в частности, гидропероксид трет-бутила), способствует росту скорости процесса (рис. 3, кривая 1) и дальнейшему повышению содержания эпоксида в продуктах окисления до 12.5 мас. д. %, т.е. в 1.5 раза по сравнению с инициированным окислением. Выход эпоксида на превращенный циклооктен при его окислении в присутствии каталитической системы гептамолибдат аммония–ГПТБ повышается до 92 мол. % (табл. 1).
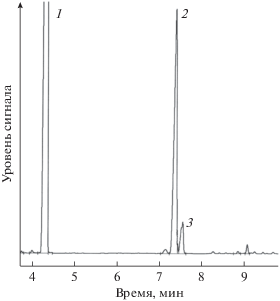
Согласно полученным экспериментальным данным, содержание циклооктенола в продуктах окисления по отношению к образующемуся целевому эпоксиду не превышает 5–7% (рис. 4).
Рис. 4.
ГЖ-хроматограмма продуктов жидкофазного окисления циклооктена, полученных в присутствии каталитической системы ГПТБ (0.03 моль/л) – пропандиолат молибденила (0.001моль/л): 1 – циклооктен, 2 – 1,2-эпоксициклооктан, 3 – циклооктенол.
В процессах жидкофазного окисления углеводородов молекулярным кислородом селективность образования целевого продукта (выход на превращенный углеводород) во многом определяется степенью превращения исходного соединения. Данные, представленные в табл. 3, показывают, что при увеличении степени превращения циклооктена от 5.4 до 24.5% содержание эпоксида в продуктах его каталитического окисления молекулярным кислородом растет, а селективность образования эпоксида непрерывно снижается с 88.7 до 84.4 мол. %. В этой связи для сохранения высокой селективности образования эпоксида на уровне 85–86 мол. % становится целесообразным ограничить конверсию циклооктена в пределах 18–20%.
Таблица 3.
Влияние степени превращения циклооктена на селективность образования эпоксида в процессе жидкофазного окисления в присутствии молибденсодержащей каталитической системы ГПТБ (0.03 моль/л)–пропандиолат молибденила (0.001 моль/л), 398 К
| Время реакции, мин |
Содержание эпоксида, мас. д. % |
Выход эпоксида на загруженный циклооктен, мол. % |
Селективность образования эпоксида, мол. % | Степень превращения циклооктена, % |
|---|---|---|---|---|
| 20 | 5.5 | 4.8 | 88.7 | 5.4 |
| 45 | 13.8 | 12.0 | 86.0 | 14.0 |
| 55 | 17.8 | 15.5 | 85.1 | 18.2 |
| 65 | 23.2 | 20.7 | 84.4 | 24.5 |
Таким образом, изучены кинетические закономерности жидкофазного окисления циклооктена молекулярным кислородом в 1,2-эпоксициклооктан и предложена каталитическая система, позволяющая обеспечить высокий выход целевого продукта на превращенный циклооктен.
Список литературы
Верещагина Н.В., Антонова Т.Н., Копушкина Г.Ю., Абрамов И.Г. // Кинетика и катализ. 2017. Т. 58. № 3. С. 266.
Бермешев М.В., Антонова Т.Н., Шангареев Д.Р., Данилова А.С., Пожарская Н.А. // Нефтехимия. 2018. Т. 58. № 5. С. 580.
Верещагина Н.В., Антонова Т.Н., Ильин А.А., Чиркова Ж.В. // Нефтехимия. 2016. Т. 56. № 1. С. 46.
Behr A., Manz V., Lux A., Ernst A. // Catal. Lett. 2013. V. 143. № 3. P. 241.
Пaт. 1577281PФ, 1997.
Уалиханова А., Темирбулатова А.Е., Майлюбаев Б.Т. // Нефтехимия. 1990. Т. 30. № 4. С. 458.
Zou J.-J., Zhang X., Kong J., Wang L. // Fuel. 2008. V. 87. P. 3655.
Сулимов А.В., Овчарова А.В., Флид М.Р., Леонтьева С.В. // Национальная ассоциация ученых. 2014. № 4. С. 97.
Антонова Т.Н. Алициклические соединения со средним размером цикла. Окислительные превращения. Реакционная способность. Саарбрюккен: Lambert Academic Publishing GmbH & Co, 2011. С. 128.
Bäckvall J.E. Modern Oxidation Methods. Wiley-VCH Verlag & Co. KGaA, 2010. 465 p.
Кучер Р.В., Тимохин В.И., Шевчук И.П. Жидкофазное окисление непредельных соединений в окиси олефинов. Киев: Наук. думка, 1986. 160 с.
Филиппова Т.В., Блюмберг Э.А. // Успехи химии. 1982. Т. 51. № 6. С. 1017.
Алимарданов Х.М., Гарибов Н.И. // Журн. общ. химии. 2013. Т. 83. № 11. С. 1822.
Рубайло В.Л., Маслов С.А. Жидкофазное окисление непредельных соединений. М.: Химия, 1989. С. 224.
Могилевич М.М., Плисс Е.М. Окисление и окислительная полимеризация непредельных соединений. М.: Химия, 1990. 240 с.
Козлова О.С., Яськина В.Г., Машина С.А., Антонова Т.Н. // Вестн. ЯГТУ. 1999. № 2. С. 59.
Эмануэль Н.М., Денисов Е.Т., Майзус З.К. Цепные реакции окисления углеводородов в жидкой фазе. М.: Наука, 1965. С. 375.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ