

Кинетика и катализ, 2021, T. 62, № 1, стр. 29-35
Моделирование кинетики фотоактивированного изотопного обмена O2 $ \rightleftarrows $ ZnO в проточном реакторе
В. В. Титов a, А. А. Лисаченко a, *
a Санкт-Петербургский государственный университет, физический факультет
198504 Санкт-Петербург, ул. Ульяновская, 1, Россия
* E-mail: a.lisachenko@spbu.ru
Поступила в редакцию 05.02.2020
После доработки 20.05.2020
Принята к публикации 21.08.2020
Аннотация
Предложена теоретическая модель разложения взаимодействия газа O2 с оксидом цинка по трем каналам: 1) фотоадсорбция, 2) обмен в газовой фазе (гомообмен) и 3) обмен оксид цинка–газ (гетерообмен), при проведении фотоактивированного изотопного обмена (ФИО) в проточном реакторе. По сравнению с использованными ранее предложенная методика учитывает фотоадсорбцию как увеличение скорости обновления смеси, что устраняет возможные искажения временных зависимостей вычисляемых скоростей. Приведены вывод формул и обработка экспериментальных данных на примере ФИО в системе O2–ZnO при освещении в областях межзонного и экситонного поглощения и поглощения дефектами структуры ZnO.
Фундаментальные исследования изотопного обмена кислорода в системе O2(газ)–оксид металла, выполненные школой акад. Г.К. Борескова, дали ключ к пониманию механизма каталитических redox-реакций на оксидных катализаторах [1]. Моделирование процессов в значительной степени опирается на математический аппарат и вычислительные методы, развитые в работе [2]. Анализ кинетики фотоактивированного изотопного обмена (ФИО) кислорода является одним из эффективных методов исследования механизмов фотокаталитических redox-реакций. С помощью ФИО были получены сведения о механизмах фотоактивации наиболее активных фотокатализаторов TiO2 и ZnO в условиях, не осложненных присутствием исходных, промежуточных и конечных продуктов реакции [3–5]. Изучаемая система O2–оксид металла является простейшей бинарной.
Для исследований обычно используют кислород газовой фазы, обогащенный изотопом 18O, и оксид природного изотопного состава (99.75% 16O). Газовая фаза состоит из молекул 16O16O, 16O18O и 18O18O, имеющих массы 32, 34 и 36 а. е. м. соответственно.
Изотопный состав газа удобно определять тремя параметрами [6]: суммарное давление P и две безразмерные изотопные переменные α и Y (α – доля атомов 18O в газовой фазе, Y – отклонение отношения давлений 32 : 34 : 36 от статистически равновесного (1 – α)2 : 2α(1 – α) : α2.
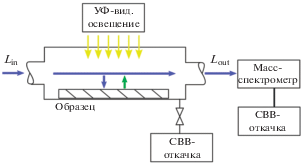
Если проводить изотопный обмен в замкнутом объеме, гомомолекулярный обмен приводит смесь в состояние равновесия (Y = 0), после чего невозможно определять скорость гомомолекулярного обмена. Для снятия этого ограничения можно создать поток газа O2 через реактор (рис. 1). При этом поступающая неравновесная смесь O2 постоянно поддерживает неравновесность смеси в реакторе.
Рис. 1.
Схема проточного режима. Поток газа O2Lin поступает в предварительно откаченный до сверхвысокого вакуума (СВВ) реактор идеального смешения. В нем газ взаимодействует с образцом ZnO. Объем реактора непрерывно вакуумируется через ионный источник масс-спектрометра. При этом измеряются количество выходящего газа Lout и его изотопный состав.
На рис. 2 приведен пример полученных на масс-спектрометре в такой системе показателей при T = 300 К и световых потоках 10–100 мВт (подробно условия измерений см. в [4, 7]). В отсутствие освещения оксид ZnO не влияет на протекающий поток О2, и масс-спектрометр регистрирует изотопный состав поступающего потока (L0). Облучение ZnO светом с λ = 365, 385, 405 и 546 нм вызывает ФИО и фотоадсорбцию кислорода с разнообразными амплитудами, соотношениями и временными зависимостями.
Рис. 2.
а, б – Примеры изотопного обмена кислорода на ZnO при различных потоках газа. Освещение отмечено П-образными контурами (сверху указаны длины волн). Отчетливо видны одновременно значительная фотоадсорбция (падение суммарного давления), гомообмен (рост давления 34О2 за счет падения давлений 32О2 и 36О2) и гетерообмен (различие скоростей падения 32О2 и 36О2). Условия эксперимента описаны в статьях [4, 7].
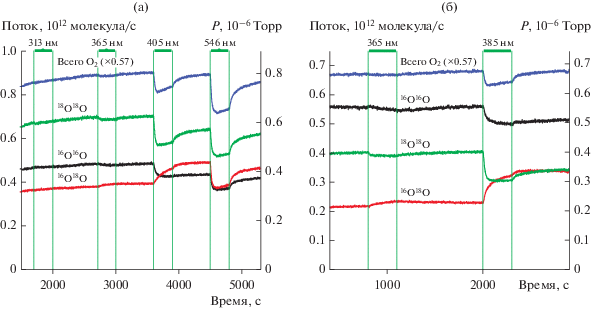
Задача данной работы состояла в нахождении из измеренных зависимостей вытекающего потока и его состава от времени временных зависимостей скоростей фотоадсорбции, фотодесорбции и изотопного обмена двух типов.
Чтобы понять, как именно эти процессы влияют на состав газа в реакторе, построим модель, дадим определения процессов и укажем, как они меняют состав в реакторе идеального смешения.
ОПРЕДЕЛЕНИЯ ПРОЦЕССОВ
1. Фотодесорбция
В газовую фазу выделяется RPDdt молекул O2. Изотопный состав заранее неизвестен, так как возможна десорбция как решеточного кислорода (природного изотопного состава), так и адсорбированного, частично или полностью обмененного с газовой фазой.
На наших образцах оксида цинка рост давления фиксировали только при очень низких давлениях (~ 10−7 Торр/10−5 Па).
2. Фотоадсорбция
Этот процесс забирает RPAdt молекул изотопного состава газовой фазы. К фотоадсорбции отнесем любое поглощение кислорода образцом оксида: как обратимое увеличение покрытия O2 (O2 можно выделить обратно облучением или нагревом), так и необратимое поглощение (например, фотореакция с превращением O2 в другие соединения). В примере на рис. 2 фотоадсорбция необратима: при нагреве обратно выделялась лишь незначительная доля поглощенного O2 (на рисунке не показано).
Фотоадсорбцию и фотодесорбцию регистрировали по падению или росту давления в газовой фазе соответственно. При их одновременном протекании скомпенсированные их доли отнесем к изотопному обмену. Тогда если давление растет, то это только фотодесорбция, а если падает, то это только фотоадсорбция.
3. Гомообмен
Такой процесс забирает Rqdt молекул и возвращает Rqdt молекул уравновешенного изотопного состава (α = αg, но Y = 0). Размерность Rq – молекула/с.
4. Гетерообмен
Гетерообмен забирает 0.5Rxdt молекул из газа и отдает 0.5Rxdt молекул 16O2 (т.е. α = 0*, Y = 0). Множитель 1/2 добавлен для того, чтобы скорость имела размерность ат/с, а не молекула/с как в случае Rq. (*Для природного кислорода α ≈ 0.002. Но на фоне других неточностей (нелинейность, шум, разрешение масс-спектрометра, воспроизводимость состояния образца и т.д.) ошибка, вносимая округлением 0.002 до нуля, незначительна.)
На давление и состав газа в реакторе в проточном режиме влияют еще два процесса – натекание и откачка.
6. Выходящий поток
Выносит Ndt/τ молекул, где N – количество молекул O2 в газовой фазе в реакторе, τ – постоянная времени откачки/обновления смеси (c). Именно этот поток измеряется масс-спектрометром.
В публикации [6] изотопный обмен дикислорода газовой фазы с твердым оксидом металла описан тремя скоростями – K1, K2 и K3. K1 связана с обменом атомами между молекулами газовой фазы без участия кислорода образца оксида. K2 и K3 описывают обмены между оксидом и газовой фазой, вовлекающие в одном акте один или два атома оксида соответственно. К сожалению, из величин изменения парциальных давлений трех масс невозможно получить эти три скорости. Поэтому, учитывая специфику наших экспериментов, для обработки кинетик мы объединили K2 и K3 в одну скорость “гетерообмена” – Rx, а K1 переобозначили как скорость “гомообмена” – Rq.
Формулы для получения скоростей процессов из экспериментальных кинетических кривых в проточном режиме ранее были выведены нами для TiO2 [3] в предположении, что: (1) скорость гетерообмена много меньше скорости гомообмена (Rq$ \gg $ Rx) и (2) суммарное давление O2 не меняется (RPA$ \ll $ Lin). При таких предположениях Rx влияет исключительно на α, а Rq – на Y, что сильно упрощает обработку экспериментальных данных. В отличие от системы O2–TiO2 в системе O2–ZnO скорости гетеро- и гомообмена могут быть сопоставимы, причем наблюдается интенсивная фотоадсорбция: давление кислорода над образцом ZnO существенно падает при освещении в УФ и видимой областях спектра (рис. 2). Поэтому нужны новые формулы, где эти предположения были бы сняты.
ВЫВОД ФОРМУЛ
Запишем уравнение изменения состава газа в реакторе в результате процессов 1–5. Уравнение имеет вид dN = (молекулы, прибывшие за время dt) − (молекулы, убывшие за время dt) по одному уравнению на каждый вариант изотопного состава O2, т.е. всего 3 уравнения. Их удобно записать как одно уравнение в векторной форме.
(1)
$\begin{gathered} \frac{{{\text{d}}\vec {N}}}{{{\text{d}}t}} = {{{\vec {F}}}_{{{\text{in}}}}} - {{{\vec {F}}}_{{{\text{out}}}}} - {{R}_{{{\text{PA}}}}}\frac{{\vec {N}}}{{\left| {\vec {N}} \right|}} + {{{\vec {R}}}_{{{\text{PD}}}}} + \\ + \,\,\frac{1}{2}{{R}_{{\text{x}}}}\left( { - \frac{{\vec {N}}}{{\left| {\vec {N}} \right|}} + \left( \begin{gathered} 1 \hfill \\ 0 \hfill \\ 0 \hfill \\ \end{gathered} \right)} \right) + {{R}_{{\text{q}}}}\left( { - \frac{{\vec {N}}}{{\left| {\vec {N}} \right|}} + \left( \begin{gathered} {{{\bar {\alpha }}}^{2}} \hfill \\ 2\alpha \bar {\alpha } \hfill \\ {{\alpha }^{2}} \hfill \\ \end{gathered} \right)} \right). \\ \end{gathered} $– компоненты векторов – соответствующие величины для масс 32, 34, 36;
– модули векторов – суммы всех компонентов: $N \equiv \left| {\vec {N}} \right| \equiv {{N}_{{32}}} + {{N}_{{34}}} + {{N}_{{36}}}$;
– $\alpha = \frac{{{{N}_{{36}}} + 0.5 \cdot {{N}_{{34}}}}}{{\left| {\vec {N}} \right|}}$ – безразмерная, зависящая от времени изотопная переменная (“обогащение”), которая показывает долю изотопа 18O среди всех молекул газовой фазы;
– $\bar {\alpha } \equiv \left( {1 - \alpha } \right)$ – (обогащение по 16O) безразмерная, зависящая от времени величина;
– $\vec {N},\;{{R}_{{\text{x}}}},\;{{R}_{{\text{q}}}},\;{{\vec {F}}_{{{\text{out}}}}},\;{{\vec {F}}_{{{\text{in}}}}},\;{{R}_{{{\text{PA}}}}}$ – зависят от времени;
– $\vec {N}$ – количество молекул O2 в реакторе c массами 32, 34 и 36;
– ${{\vec {F}}_{{{\text{out}}}}}$ – выходящий поток (молекула/с) 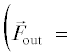 $\left. { = \frac{{\vec {N}}}{\tau }} \right),$ измеряется на масс-спектрометре;
$\left. { = \frac{{\vec {N}}}{\tau }} \right),$ измеряется на масс-спектрометре;
– ${{\vec {F}}_{{{\text{in}}}}}$ – входящий поток (молекула/с), задается в эксперименте.
Первые два члена правой части уравнения (1) отражают процессы 5 и 6 (проточный режим). Третий член представляет процесс 1 (фотоадсорбцию), четвертый – процесс 2 (фотодесорбцию), пятый – процесс 3 (гетерообмен) и шестой – процесс 4 (гомообмен).
Известно, что:
– ${{\vec {F}}_{{{\text{out}}}}}$ измеряется масс-спектрометром. $\vec {N}$ и $\frac{{{\text{d}}\vec {N}}}{{{\text{d}}t}}$ связаны с ним непосредственно как ${{\vec {F}}_{{{\text{out}}}}} = \frac{{\vec {N}}}{\tau }$ (τ измеряли независимо по падению давления после резкого перекрывания входящего потока);
– ${{\vec {F}}_{{{\text{in}}}}}$ – величина постоянная, ее значение определяли по начальному участку ${{\vec {F}}_{{{\text{out}}}}}{\text{.}}$
Надо найти: RPA, ${{\vec {R}}_{{{\text{PD}}}}}{\text{,}}$ Rx и Rq.
Уравнение (1) – дифференциальное нелинейное. Однако величина под дифференциалом известна, а, следовательно, и ее производная, и уравнение становится алгебраическим.
В проведенных опытах фотодесорбцию не наблюдали, поэтому ${{\vec {R}}_{{{\text{PD}}}}}$ считаем равным нулю.
RPA легко получить, просуммировав все компоненты:
Подставив найденное и преобразовав уравнение, имеем:
Левая часть интересна сама по себе. Обозначим ее как A:
A содержит данные о скоростях перераспределения (молекула/с) между изотопами с массами 32, 34 и 36 а. е. м., не осложненные проточным режимом и фотоадсорбцией. Сумма компонентов A равна нулю. Возможно рассчитать такую же величину для кинетики в замкнутом объеме, дальнейшие преобразования универсальны.
Rx легко получить, вычтя уравнение для изотопа 36 а. е. м. из уравнения для изотопа 32 а. е. м.
Rq получается из уравнения для изотопа 34 а. е. м.:
РЕЗУЛЬТАТЫ РАСЧЕТОВ
Итак, предложен следующий алгоритм:
(3)
$\vec {A} = \frac{{{\text{d}}\vec {N}}}{{{\text{d}}t}} - {{\vec {F}}_{{{\text{in}}}}} + {{\vec {F}}_{{{\text{out}}}}} + {{R}_{{{\text{PA}}}}}\frac{{\vec {N}}}{{\left| {\vec {N}} \right|}},$(5)
${{R}_{{\text{q}}}} = \frac{{{{A}_{{34}}} + \alpha \bar {\alpha }{{R}_{{\text{x}}}}}}{Y} - \frac{1}{2}{{R}_{{\text{x}}}}.$Все это справедливо, если предположить:
– быструю диффузию газа по объему реактора. Принципиально, чтобы изотопные составы газа над поверхностью оксида и на входе в масс-спектрометр совпадали (реактор идеального смешения). Это одна из двух главных причин, почему мы работаем при низких (<10−3 Toрр) давлениях, когда длина свободного пробега молекул порядка размеров реактора максимизирует скорость диффузии;
– остаточный вакуум по O2 в масс-спектрометре и в системе откачки много меньше давления в реакторе, что позволяет не учитывать обратные потоки;
– отсутствие фотодесорбции – RPA > RPD;
– образец оксида исходно содержит только кислород изотопа 16O, что заложено в член (1,0,0) в формулах.
У нас на практике Rq обычно существенно больше Rx. Если Rq$ \ll $ L и Y не стремится к нулю, то приближенно ${{R}_{{\text{q}}}} = \frac{{{{A}_{{34}}}}}{Y}{\text{.}}$ Это эквивалентно ранее использованной упрощенной формуле в случае TiO2 [3, 5].
“Константы скорости” обменов двух типов можно получить, разделив Rq и Rx на N: ${{K}_{{{\text{q}},{\text{x}}}}} \equiv $ $ \equiv {{{{R}_{{{\text{q}},{\text{x}}}}}} \mathord{\left/ {\vphantom {{{{R}_{{{\text{q}},{\text{x}}}}}} N}} \right. \kern-0em} N}$ [c–1]. На графиках построены Rx и Kq. Такой выбор связан с тем, что разумно предположить (опираясь на механизм обмена для TiO2), что Rx не зависит от давления, а Rq прямо пропорциональна давлению, тогда Rx и Kq не зависят от рабочего давления и полученные скорости можно сравнить напрямую в экспериментах при разных давлениях.
На рис. 3 показан результат обработки кинетик по формулам (2)–(5) (“новые” формулы), в сравнении со “старыми” формулами из [3, 5], в которых фотоадсорбцией пренебрегали. Хорошо видна разница в кинетиках Kq в момент выключения освещения при 405 нм. По старым формулам кажется, что реакция на выключение света запаздывает, а после выключения скорость гомообмена не падает. По новым формулам, напротив, видно, что нарастание еще не закончилось, реакция на гашение света мгновенна и сразу начинается спад. При последующем освещении при 546 нм, вызывающем сильную фотоадсорбцию, скорость гомообмена Kq сильно падает во время освещения, если считать по старым формулам; форма падения практически повторяет скорость фотоадсорбции. По новым формулам Kq тоже понижается, но существенно меньше, и форма понижения не повторяет кинетику фотоадсорбции. Природа этого явления пока неясна.
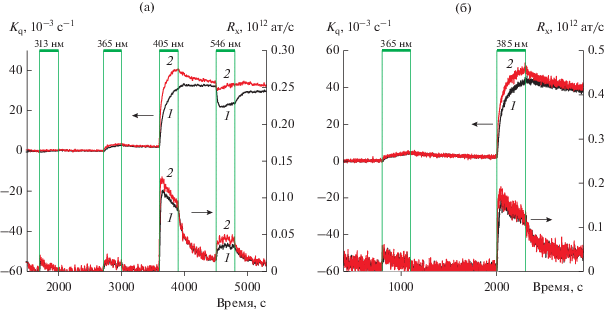
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Реально, скорости гетеро- и гомообмена могут быть связаны различными способами со скоростями реальных процессов, происходящих на поверхности.
Рассмотрим примеры взаимодействия электронов и дырок на фотоактивированной поверхности ZnO с O2:
(III)
$\begin{gathered} *{{{\text{O}}}_{{2({\text{gas}})}}} + *{{{\text{O}}}_{{2({\text{gas}})}}} + {\text{e}} \rightleftarrows *{{{\text{O}}}_{{2({\text{gas}})}}} + \\ + \,\,*{\text{O}}_{{2({\text{ads}})}}^{ - } \rightleftarrows *{\text{O}}_{{4({\text{ads}})}}^{ - }, \\ \end{gathered} $(IV)
$\begin{gathered} *{{{\text{O}}}_{{2({\text{gas}})}}} + {\text{O}}_{{({\text{surf}})}}^{ - } \to \left[ {*{\text{O}}*{\text{OO}}} \right]_{{({\text{surf}})}}^{ - } \to \\ ~ \to {{\left[ {*{\text{OO}}} \right]}_{{({\text{gas}})}}} + *{\text{O}}_{{({\text{surf}})}}^{ - } \\ \end{gathered} $(*атомы O, перешедшие из газовой фазы).
Процесс (I) – фотодесорбция. Если это единственный процесс, то нарушается предположение об отсутствии десорбции и вычислять Rx и Rq бессмысленно. Вычисление RPA даст отрицательную величину, равную скорости этого процесса.
Процесс (II) – фотоадсорбция. Если это единственный процесс, то в результате обработки RPA даст скорость этого процесса, а Rx и Rq должны быть нулевыми.
Если процессы (I) и (II) идут одновременно, то: RPA даст разность их скоростей, Rx будет равна удвоенной скорости процесса (I), а Rq должна остаться равной нулю.
Процесс (III) – это в чистом виде гомообмен. Он вносит вклад только в скорость Rq.
Процесс (IV) несколько сложнее. Рассмотрим два варианта его.
(IV)-1: обмен на одном центре, активируемый одним фотоном, однократен, а центров много. Тогда в начальный момент Rx равна скорости обмена, а Rq примет значение порядка Rx. По мере того как поверхность насыщается изотопом 18О, Rx будет падать (при постоянной скорости процесса (IV)), а Rq будет стремиться к скорости процесса (IV). Когда поверхность полностью насытится изотопом 18О, останется только скорость Rq.
(IV)-2: многократный обмен на одном центре от одного фотона (аналогично обмену на TiO2 [3]). В этом случае первый цикл центра внесет вклад в скорость Rx, а последующие циклы центра будут давать вклад только в скорость Rq. В результате получается, что:
– Rx = скорость активации (количество центров в секунду, переходящих из состояния O2− в O− за счет фотоактивации) в начальный момент; повторная фотоактивация того же центра вклада в Rx не даст;
– Rq ≈ частота циклов процесса (IV);
– Rq/Rx ≈ количество циклов на одном центре от его активации до дезактивации.
ЗАКЛЮЧЕНИЕ
Предложенный механизм разложения позволяет следить за изменением изотопного состава покрытия образца ZnO по Rx и наблюдать за скоростью каталитической пересборки молекул кислорода Rq. Эти скорости удобны для построения зависимостей (от давления, от состояния образца, от длины волны и интенсивности фотовозбуждения, температуры и т.д.). Сопоставляя Rx и Rq, можно судить о механизме обмена.
В предыдущей нашей работе [7] конечные формулы (2)–(5) были использованы для сравнения эффективности активации фотокатализатора ZnO в областях межзонного и экситонного поглощения. Приведенный в настоящей статье вывод и анализ системы уравнений, описывающих ФИО в системе O2–оксид цинка, позволяет количественно анализировать разнообразие каналов взаимодействия газовой фазы с поверхностью оксида и критически оценивать получаемые результаты.
Список литературы
Андрушкевич Т.В., Бухтияров В.И. // Кинетика и катализ. 2019. Т. 60. № 2. С. 152.
Слинько М.М., Макеев А.Г. // Кинетика и катализ. 2020. Т. 61. № 4. С. 447.
Mikhaylov R.V., Lisachenko A.A., Titov V.V. // J. Phys. Chem. C. 2012. V. 116. № 44. P. 23 332.
Titov V.V., Lisachenko A.A., Akopyan I.K., Labzovskaya M.E., Novikov B.V. // Phys. Solid State. 2019. V. 61. № 11. P. 2134.
Titov V.V., Mikhaylov R.V., Lisachenko A.A. // J. Phys. Chem. C. 2014. V. 118. № 38. P. 21 986.
Boreskov G.K., Muzykantov V.S. // Ann. N.Y. Acad. Sci. 1973. V. 213. № 1. P. 137.
Titov V.V., Lisachenko A.A., Labzovskaya M.E., Akopyan I.K., Novikov B.V. // J. Phys. Chem. C. 2019. V. 123. № 45. P. 27 399.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ