

Кинетика и катализ, 2021, T. 62, № 1, стр. 109-119
Гибридный катализатор селективного синтеза углеводородов топливного ряда методом Фишера−Тропша
Р. Е. Яковенко a, *, И. Н. Зубков a, А. П. Савостьянов a, В. Н. Соромотин a, Т. В. Краснякова b, О. П. Папета a, С. А. Митченко a, b
a ФГБОУ ВО Южно-Российский государственный политехнический университет (НПИ) им. М.И. Платова
346428 Новочеркасск, ул. Просвещения, 132, Россия
b ГУ Институт физико-органической химии и углехимии
283114 Донецк, ул. Р. Люксембург, 70, Украина
* E-mail: jakovenko39@gmail.com
Поступила в редакцию 04.06.2020
После доработки 03.08.2020
Принята к публикации 16.08.2020
Аннотация
Представлены результаты исследований по разработке гибридного катализатора для однореакторного синтеза жидких углеводородов по методу Фишера−Тропша. Катализатор получен смешением и формованием порошков катализатора Со−Al2O3/SiO2, цеолита НZSM-5 и бемита. Промотирование цеолита палладием осуществляли ионным обменом (1.0 мас. %). Катализаторы охарактеризованы методами рентгенофазового анализа, БЭТ, сканирующей и просвечивающей электронной спектроскопии, термопрограммированной десорбции аммиака и водорода, испытаны при температуре 240°С, давлении 2.0 МПа, ОСГ 1000 ч–1. Предлагаемый метод приготовления позволяет получать гибридный катализатор со стабильной структурой и средним размером частиц кобальта 8 нм, обладающий высокой производительностью и селективностью образования жидких углеводородов из СО и Н2.
ВВЕДЕНИЕ
В последнее время актуальным направлением развития топливно-энергетического комплекса является получение высококачественных моторных топлив без использования нефтяного сырья. Альтернативным сырьевым источником может служить попутный нефтяной газ (ПНГ), который в России преимущественно сжигается: в 2017 г. таким образом утилизировано около 2.5 млрд м3, тогда как в США, Канаде, Норвегии он почти полностью перерабатывается. Преобразование ПНГ в моторные топлива и масла, парафины и др. может осуществляться при каталитических превращениях полученного из него синтез-газа (смеси СО и Н2) – синтезе Фишера–Тропша (СФТ) [1–3]. В промышленном масштабе технология производства жидких синтетических углеводородов реализована на заводах компаний Shell и Sasol [4] по трехстадийной схеме: получение синтез-газа – СФТ – гидрооблагораживание (ГО) продуктов.
Для снижения капитальных и эксплуатационных затрат целесообразно объединение двух последних стадий посредством применения гибридных катализаторов, позволяющих получать в одном реакторе углеводороды с заданным фракционным и групповым составом [5–7]. Гибридные бифункциональные катализаторы имеют два типа активных центров: на одних синтезируются углеводороды (преимущественно неразветвленные парафины и α-олефины), на других осуществляются их гидрокрекинг и изомеризация. В качестве компонента катализатора ГО продуктов СФТ используют цеолиты разных типов [8–18]. Основным фактором, определяющим эффективность процесса и селективность образования целевых продуктов на гибридном катализаторе, является миграция первичных продуктов СФТ к кислотным центрам цеолита, на которых происходит их последующее ГО. Для этого необходимо обеспечить тесный контакт между каталитическими центрами синтеза углеводородов и их гидрооблагораживания, что реализуется путем применения мелкодисперсных порошков компонентов гибридного катализатора. Однако в промышленных целях использование физической смеси мелкодисперсных порошков невозможно, поскольку они создают существенное гидродинамическое сопротивление. Во избежание большого перепада давления вдоль слоя катализатора СФТ в трубчатом реакторе с неподвижным слоем оптимальными являются частицы катализатора размером 1–3 мм [19].
Среди большого количества разнообразных гибридных катализаторов единственная коммерчески жизнеспособная каталитическая система, которая обеспечивает производство жидких углеводородов в одном реакторе, содержащая одновременно катализатор СФТ и цеолитный компонент, была предложена как часть технологии Chevron Gas Conversion Catalysis (GCCTM) [15]. Эта технология включает изготовление экструдатов необходимого размера из связанного оксидом алюминия цеолита с последующей их пропиткой раствором кобальта. При таком способе приготовления кобальт селективно наносится на связующее (оксид алюминия) с образованием гибридного катализатора, в котором в пределах одного экструдата компонент СФТ (т.е. Co/Al2O3) отделен от частиц цеолита, но находится в наноразмерной близости к нему. Эта гибридная каталитическая система обеспечивает высокую селективность по углеводородам С5+ и исключает появление твердых восков при умеренных условиях 1–3 МПа, 210–225°C, H2/CO = 1.5–2.0, однако процесс протекает при относительно низкой конверсии синтез-газа (около 40%) для защиты гибридного катализатора от высокого давления водяного пара [15].
В настоящей работе применяется подобная GCCTM методика получения гибридного катализатора, но с использованием уже сформированного предварительно катализатора СФТ с нанесенным на силикагель кобальтом. Ранее нами был разработан кобальтсиликагелевый катализатор СФТ [20], содержащий 1 мас. % промотирующей добавки оксида алюминия, распределение частиц кобальта по размерам в котором было оптимальным для обеспечения высокой активности и селективности образования конденсированных углеводородов С5+ [21]. Помимо значительной производительности полученный катализатор продемонстрировал стабильную работу в реальных условиях СФТ при глубокой конверсии синтез-газа и, соответственно, высоком парциальном давлении воды [22]. Можно было полагать, что его использование в качестве компонента СФТ в гибридном бифункциональном катализаторе окажется плодотворным для применения в однореакторном синтезе жидких углеводородов топливного ряда. Цель представленной работы заключалась в проверке этой гипотезы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методика приготовления катализаторов
Образцы гибридных катализаторов получали формованием смеси порошков (менее 0.1 мм) катализатора СФТ и цеолита со связующим веществом. В качестве катализатора СФТ использовали предварительно приготовленный кобальтовый катализатор, промотированный Al2O3 (1 мас. %), на силикагелевом носителе (1 мас. %) – Co–Al2O3/SiO2 [20]. Для формирования кислотных центров в структуре гибридного катализатора был выбран цеолит марки ZSM-5 (Н-форма, Si/Al = 40) производства ООО “Ишимбайский специализированный химический завод катализаторов”. Н-форму цеолита ZSM-5 получали термическим разложением аммонийной формы при температуре 550°С в течение 4 ч в атмосфере воздуха. В качестве связующего вещества применяли бемит (“Sasol”, TH 80). Для пластификации в полученную смесь порошков прибавляли водно-спиртовой раствор триэтиленгликоля (0.03 моль на 1 моль бемита) и азотную кислоту (0.02 моль на 1 моль бемита). Формование гранул катализаторов осуществляли методом экструзии, затем сушили 24 ч при комнатной температуре и подвергали термообработке в режиме: 4 ч при 80°С, затем последовательно по 1 ч при 100, 120, 140°С и, наконец, 4 ч при 400°C.
Цеолит промотировали палладием методом ионного обмена из раствора PdCl2 при температуре 70°С в течении 3 ч и постоянном перемешивании. Концентрация PdCl2 в растворе составляла 6.5 мас. %. После ионного обмена раствор отфильтровывали, а цеолит промывали дистиллированной водой, нагретой до 60°С. Далее цеолит сушили при комнатной температуре в течение 12 ч, при температуре 110°С – 15 ч и прокаливали при температуре 550°С в течение 4 ч. Количество введенного в цеолит ZSM-5 палладия составило 1.0 мас. %.
С целью изучения влияния кислотной составляющей гибридного катализатора на состав образующихся углеводородов С5+ был приготовлен образец, в котором цеолит был заменен на инертный компонент (кварцевая крошка < 0.1 мм) с сохранением массовых отношений к другим компонентам в готовом катализаторе. Состав и обозначения образцов катализаторов приведены в табл. 1.
Методики исследования свойств катализаторов
Исследования каталитической активности осуществляли в проточном режиме в трубчатом реакторе (внутренний диаметр 16 мм) со стационарным слоем катализатора (10 см3, фракция 1–2 мм), разбавленного 30 см3 кварца (фракция 1–2 мм), с использованием синтез-газа с соотношением Н2/СО = 2 при давлении Р = 2.0 МПа, объемной скорости газа ОСГ = 1000 ч–1, температуре Т = 240°С. Предварительно катализаторы восстанавливали при Т = 400°С, ОСГ = 3000 ч–1 в токе Н2 в течение 1 ч. Катализаторы активировали синтез-газом с соотношением Н2/СО = 2 под давлением 2.0 МПа и ОСГ = 1000 ч–1 путем ступенчатого подъема температуры со скоростью 2.5°С/ч от 180 до 240°С. Балансовые опыты проводили в течение 100 ч, анализируя состав и количество газа на выходе из установки каждые 2 ч. Продолжительные исследования для определения стабильности работы катализатора были выполнены в течение 1000 ч. По окончанию опыта продукты синтеза разделяли и фракционировали.
Состав газообразных и жидкофазных продуктов синтеза анализировали хроматографическим и хромато-масс-спектрометрическим методами [22].
Рентгенофазовый анализ (РФА) осуществляли с использованием оборудования Европейского центра синхротронного излучения (“ESRF”, Гренобль) в интервале углов 2θ от 5° до 55° с длиной волны излучения λ = 0.7121 Å. Определение качественного фазового состава было выполнено с помощью PDF-2 [23] в программном комплексе Crystallographica.
Морфологию поверхности катализатора изучали методом сканирующей электронной микроскопии (СЭМ) на сканирующем электронном микроскопе JSM-6490LV (“JEOL”, Япония, ускоряющее напряжение 30 кВ), который был оснащен энергодисперсионным детектором INCA Penta FET 3 (“Oxford Instruments”, Великобритания).
Исследование катализаторов методом просвечивающей электронной микроскопии (ПЭМ) проводили на микроскопе Tecnai G2 Spirit BioTWIN (“FEI”, США) с ускоряющим напряжением 120 кВ. Образец исходного катализатора предварительно восстанавливали азото-водородной смесью (5% Н2 + 95% N2) при линейном нагреве от комнатной температуры до 500°С в течение 1 ч.
Для восстановленных катализаторов методом ПЭМ было рассчитано распределение кристаллитов кобальта по размерам. Средневзвешенный размер кристаллитов и стандартное отклонение вычисляли с использованием формул [24].
Термопрограммированную десорбцию аммиака (ТПД NH3) и водорода (ТПД H2), а также определение степени восстановления кобальта (R(Со)) импульсным окислением восстановленного образца кислородом и удельной поверхности по методу БЭТ (Sуд) проводили с использованием анализатора ChemiSorb 2750 (“Micromeritics”, США), оснащенного детектором по теплопроводности. Предварительно образцы выдерживали в токе гелия (20 мл/мин) в течение 1 ч при температуре 200°С для удаления влаги и других адсорбированных газов.
Значение Sуд находили с использованием аргоно-гелиевой смеси (10 об. %). ТПД H2 осуществляли в интервале температур 25–500°C в токе гелия (20 мл/мин) после насыщения предварительно восстановленного водородом образца катализатора и удаления физически адсорбированного газа. Кислотность определяли после импульсного насыщения образца аммиаком при температуре 100°С, удаления физически адсорбированного аммиака в течение 1 ч при температуре 100°С в токе гелия (20 мл/мин). Десорбцию аммиака регистрировали в интервале температур 100–700°С при линейном нагреве со скоростью 20°С/мин.
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Характеристика катализатора
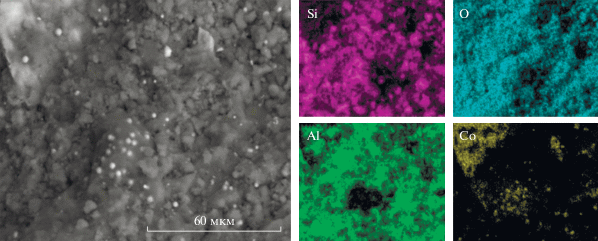
Типичные СЭМ-изображения поверхности гибридного катализатора 2 в окисленной форме (рис. 1) показывают, что распределение кобальта по поверхности образца носит фрагментарный характер, тогда как кремний, алюминий и кислород распределены на ней более равномерно. По данным рентгеновского энергодисперсионного микроспектрального анализа содержание кобальта составляет 7.5 маc. %, что соответствует количеству компонента Co–Al2O3, введенного в состав гибридного катализатора Co–Al2O3/SiO2.
Рентгенофазовый анализ гибридного катализатора 2 в окисленной форме показал (рис. 2), что кобальт входит в состав оксида Co3O4, представленного на дифрактограмме рефлексами в интервале 2θ ≈ 10°–55°. Дифракционные максимумы в области малых углов (2°–12°) соответствуют цеолиту ZSM-5. Три рефлекса при 2θ ≈ 20.3°, 29.5° и 52.0° относятся к фазе Al2O3, образующейся при термическом разложении бемита. Оценка на основании уравнения Шеррера [25] среднего размера кристаллитов Co3O4, осуществленная по уширению наиболее интенсивного дифракционного максимума 2θ = 16.8°, дает значение 10.7 нм, что согласуется с ожидаемым размеру кристаллитов Co0 в восстановленной форме катализатора 8 нм (табл. 2).
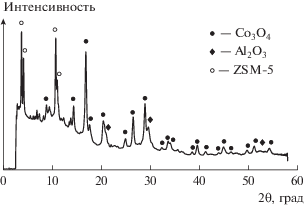
Таблица 2.
Физико-химические характеристики катализаторов
| Образец | Размер частиц, нм | R(Со), % | Кислотность, мкмоль/г | Sуд, м2/г | |||
|---|---|---|---|---|---|---|---|
| d(Co3O4)* | d(Co0)* | d(Co0)** | d(Co0)*** | ||||
| 1 | – | – | 5.2 | 8 ± 2 | 49 | 260 | – |
| 2 | 10.7 | 8.0 | 5.1 | 8 ± 2 | 51 | 480 | 246 |
| 3 | – | – | 5.0 | – | 52 | 440 | – |
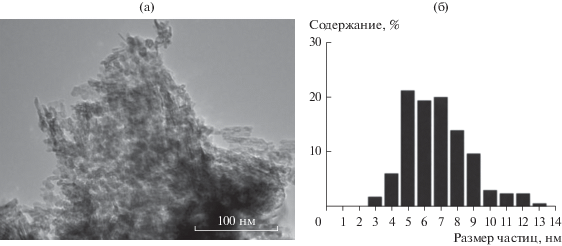
По данным ПЭМ (рис. 3) размер наночастиц кобальта в полученном гибридном катализаторе лежит в диапазоне 3–13 нм со средним значением около 8 нм (табл. 2), что совпадает с данными, приведенными в [26] для исходного катализатора Co–Al2O3/SiO2. Таким образом, в процессе синтеза гибридного катализатора размеры нанесенных на силикагель частиц кобальта не претерпевают существенных изменений, по-прежнему оставаясь оптимальными для обеспечения высокой производительности СФТ по конденсированным углеводородам.
Рис. 3.
ПЭМ-изображение восстановленного гибридного катализатора 2 (а) и гистограмма распределения в нем частиц кобальта по размеру (б).
Для всех катализаторов степень восстановления кобальта R(Со) примерно одинакова (табл. 2). Образец 1 имеет достаточно высокую кислотность, обусловленную, вероятно, присутствием в составе гибридного катализатора оксида алюминия как связующего. Замена инертного компонента (кварцевой крошки) цеолитом ZSM-5 (образец 2) способствует значительному увеличению кислотности. Меньшее значение этого показателя для допированного палладием катализатора 3 может быть обусловлено введением палладия ионным обменом, в ходе которого протоны замещаются на катионы Pd(II).
Каталитические испытания
В табл. 3 приведены значения конверсии СО и селективности реакции СФТ на трех приготовленных нами образцах. Замена инертной кварцевой крошки цеолитом в составе гибридного катализатора приводит к небольшому снижению конверсии СО, которое сильнее заметно при протекании реакции в присутствии катализатора 3, содержащего цеолит с нанесенным на него палладием. При этом селективности образования газообразных и конденсированных углеводородов С5+ остаются практически неизменными в реакциях на обоих гибридных катализаторах 2 и 3. Благодаря большей конверсии СО производительность по углеводородам С5+ на катализаторе 1 была несколько выше, чем на гибридных катализаторах 2 и 3. Таким образом, добавление цеолита или Pd/ZSM-5 в состав гибридного катализатора оказывает незначительное влияние на кинетику СФТ и селективность процесса в отношении как газообразных, так и C5+-углеводородов.
Таблица 3.
Показатели каталитической активности образцов*
| Образец | Конверсия СО, % | TOF × 102, с–1 | Селективность, % | Производительность по углеводородам С5+, кг ${\text{м}}_{{{\text{кат}}}}^{{ - {\text{3}}}}$ ч–1 | |||
|---|---|---|---|---|---|---|---|
| CH4 | C2–C4 | C5+ | CO2 | ||||
| 1 | 79 | 8.0 | 19.6 | 9.0 | 68.1 | 3.2 | 117 |
| 2 | 77 | 7.8 | 18.5 | 11.8 | 67.3 | 2.4 | 108 |
| 3 | 71 | 7.2 | 18.0 | 12.5 | 67.3 | 2.2 | 106 |
Фракционный и групповой составы конденсированных углеводородов, полученных на катализаторах 1–3, существенно различаются (табл. 4, рис. 4). В продуктах, синтезированных на образце 1, превалируют углеводороды линейного строения (табл. 4), в незначительных количествах присутствуют изопарафины и олефины, около трети последних составляют α-олефины. Анализ группового состава продуктов синтеза на образце 2 показал, что вклад протекающих на кислотных центрах цеолита реакций ГО углеводородов довольно высок: наблюдается значительное по сравнению с образцом 1 увеличение содержания изопарафинов и олефинов (преимущественно разветвленных, α-олефины детектируются лишь в следовых количествах), а селективность по углеводородам С19+ снижается втрое. В продуктах синтеза на образце 3 зафиксировано почти трехкратное возрастание содержания изопарафинов и пятикратное уменьшение содержания олефинов по сравнению с углеводородами, полученными на катализаторе 2. Однако выход восков С19+ тоже заметно вырастает.
Таблица 4.
Групповой и фракционный состав углеводородов, полученных на катализаторах
| Образец | Группа углеводородов | Фракционный состав, мас. % | Сумма | изо/н | о/п | |||
|---|---|---|---|---|---|---|---|---|
| C5–C10 | C11–С18 | С19+ | ||||||
| 1 | н-Парафины | 31.8 | 28.1 | 18.1 | 78.0 | 88.4 | 0.13 | 0.13 |
| Изопарафины | 3.5 | 4.7 | 2.2 | 10.4 | ||||
| Олефины | 7.9 | 2.8 | 0.1 | 10.8 | 11.6 | |||
| Разв. Олефины | 0.8 | – | – | 0.8 | ||||
| Сумма | 44.0 | 35.6 | 20.4 | 100 | ||||
| 2 | н-Парафины | 12.5 | 18.4 | 5.2 | 36.1 | 58.1 | 0.76 | 0.72 |
| Изопарафины | 9.5 | 10.8 | 1.7 | 22.0 | ||||
| Олефины | 18.3 | 2.3 | – | 20.6 | 41.9 | |||
| Разв. олефины | 14.0 | 7.3 | – | 21.3 | ||||
| Сумма | 54.3 | 38.8 | 6.9 | 100 | ||||
| 3 | н-Парафины | 20.7 | 15.5 | 7.1 | 43.3 | 91.9 | 1.09 | 0.09 |
| Изопарафины | 28.8 | 16.0 | 3.8 | 48.6 | ||||
| Олефины | 3.8 | 0.8 | – | 4.6 | 8.1 | |||
| Разв. олефины | 2.9 | 0.6 | – | 3.5 | ||||
| Сумма | 56.2 | 32.9 | 10.9 | 100 | ||||
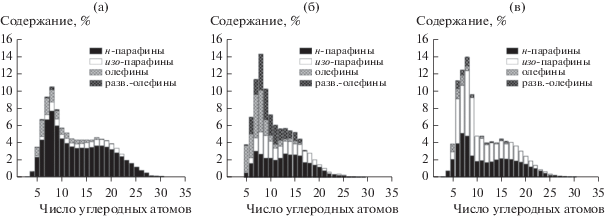
Заметим, что при одинаковых условиях проведения процесса достигаются близкие значения конверсии СО на катализаторе Со(15 мас. %)/ ZSM-5 [27] и на гибридном катализаторе 2. Тот факт, что катализатор 2 содержит вдвое меньше кобальта, свидетельствует о его более высокой активности в СФТ. Примечательно, что он оказался также более активным по сравнению с катализатором Co/ZSM-5 [27] и в ГО углеводородов. Действительно, селективность по отношению к C19+ в реакции на катализаторе 2 (6.9%) почти в два раза ниже таковой для C23+ (11.6%) в работе [27], тогда как селективность образования углеводородов бензинового ряда C5–C10 на образце 2 (54.3%) была в 2.3 раза выше значения, достигнутого в [27] для углеводородов C5–C11 (23.9%).
Отметим, что длина углеродной цепи синтезированных углеводородов на трех изученных катализаторах не превышает тридцати С-атомов. Распределение н-парафинов С5+ для всех образцов является бимодальным с максимумами в области бензиновой и дизельной фракций. В случае катализатора 1 максимумы приходятся на C7–C9 и C15–C17, а для образцов 2 и 3 они немного смещены в сторону более коротких цепей – С7 и С13–С16 соответственно (рис. 4а). Для цеолитсодержащих каталитических систем 2 и 3 бимодальность распределения углеводородов С5+ имеет более сглаженный характер, чем для катализатора 1.
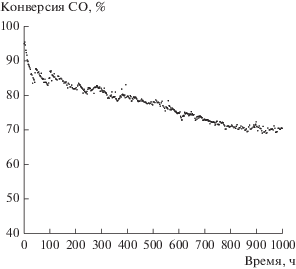
Важной характеристикой потенциального промышленного катализатора является стабильность его работы. Мы испытали активность гибридного катализатора 2 в течение 1000 ч в потоке (рис. 5); условия процесса приведены в подписи к рисунку. Конверсия СО постепенно снижается во времени, приближаясь к асимптоте при XCO ≈ 51%. Эти результаты качественно согласуются с данными о скорости дезактивации компонента Фишера–Тропша гибридной каталитической системы Chevron [15]. Однако, несмотря на более жесткие условия процесса по сравнению с теми, что использовали в работе [15], в нашем случае общее падение конверсии СО оказалось намного меньше. Действительно, после 1000 ч в потоке потеря активности катализатора 2 составила всего 17–20%, тогда как в [15] снижение этого показателя за то же время реакции было почти втрое больше. Таким образом, составляющая СФТ в гибридном катализаторе 2 работает достаточно стабильно при высокой конверсии СО и, соответственно, при высоком парциальном давлении водяного пара: скорость его дезактивации заметно ниже, чем у гибридной каталитической системы Chevron.
Рис. 5.
Изменение конверсии СО во времени в продолжительном испытании катализатора 2. Условия реакции: T = 250°C, P = 1.0 MПa, H2/CO = 2, ОСГ = = 1000 ч–1.
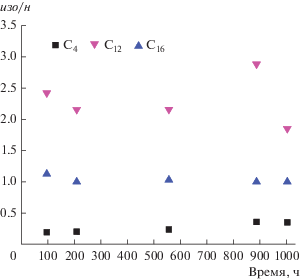
Одной из главных проблем, связанных с промышленным применением цеолитсодержащих бифункциональных катализаторов, является стабильность и срок службы кислотного компонента по сравнению с компонентом СФТ [28]. Основные пути дезактивации цеолитной части гибридных катализаторов – закупоривание пор цеолита восковыми или углеродными отложениями и блокирование кислотных центров коксом [15, 28]. Разветвление первичных углеводородов СФТ может происходить только в результате реакции, катализируемой кислотной составляющей гибридного катализатора, что делает отношение изо/н удобным показателем эффективности цеолита [15]. Мы наблюдали, что степень разветвления, а также выход углеводородов C19+ остаются почти неизменными в течение 1000 ч работы катализатора 2 в потоке. Из рис. 6 видно, что отношения изо/н для углеводородов С4, С12 и С16, полученных на гибридном катализаторе 2, практически постоянны в течение 1000-часового эксперимента. На основании полученных результатов можно заключить, что цеолитный компонент не вносит заметного вклада в общую дезактивацию гибридного катализатора 2.
Рис. 6.
Изменение во времени соотношения изо/н для углеводородов C4 (проанализировано с помощью газовой хроматографии в режиме онлайн), C12 и C16 (анализ продуктов, накопленных в течение 100 ч работы в потоке), полученных на гибридном катализаторе 2. Условия процесса: T = 250°C, P = 1.0 MПa, H2/CO = 2, ОСГ = 1000 ч–1.
Таким образом, гибридный катализатор 2 демонстрирует очевидные преимущества по сравнению с катализатором Шевроном. Во-первых, он стабильно работает при бóльших значениях конверсии CO, что особенно важно для применения в мобильных модульных установках переработки попутного нефтяного газа на удаленных нефтяных месторождениях. Во-вторых, несмотря на работу при высоких конверсиях CO и, соответственно, высоком парциальном давлении паров воды, компонент СФТ гибридного катализатора 2 дезактивируется намного медленнее, чем в гибридной каталитической системе Шеврона [15].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Известно, что синтез углеводородов по методу Фишера–Тропша на монофункциональных катализаторах (к примеру, Со/SiO2, Co/Al2O3 и др.) протекает по полимеризационно-конденсационному механизму, селективность образования продуктов подчиняется распределению Андерсона–Шульца–Флори [29] с унимодальным молекулярно-массовым распределением. Такое унимодальное распределение синтезированных углеводородов с максимумом при C16–C18 было получено в [14] на катализаторе Co/SiO2 в условиях процесса, близких к нашим. Продукты были представлены углеводородами до С50 включительно преимущественно линейного строения с незначительным количеством изопарафинов – продуктов вторичной изомеризации. Селективность процесса авторы объясняли отсутствием сильных кислотных центров на силикагелевом носителе, которые могли бы инициировать образование карбениевых ионов с последующей изомеризацией, β-элиминированием и образованием укороченных изомеров и линейных углеводородов [30, 31].
В наших экспериментах на катализаторе 1 образуются углеводороды с бимодальным распределением (рис. 4а) и существенно меньшей длиной цепи, до 30 С-атомов. Доля изопарафинов при этом вдвое выше (изо/н = 0.13), чем в системе, исследованной в [14] (изо/н = 0.06). Эти факты свидетельствуют о заметном вкладе вторичных реакций гидрокрекинга углеводородов на кислотных центрах оксида алюминия, входящего в состав катализатора 1 [32]. Максимум на графике распределения углеводородов в области C15–C17 на рис. 4а практически совпадает с тем, что наблюдался для Co/SiO2 при C16–C18 в работе [14]. Поскольку у катализатора Co/SiO2 отсутствуют сильные кислотные центры, ответственные за реакции гидрокрекинга и изомеризации углеводородов, этот максимум можно считать присущим распределению углеводородов, образующихся в СФТ в указанных условиях проведения процесса.
Замена инертного кварца на цеолит H-ZSM-5 приводит к значительному увеличению выхода разветвленных парафинов и олефинов (при этом α-олефины обнаруживаются лишь в следовых количествах) и существенному снижению выхода углеводородов С19+ (табл. 3, рис. 4б). Продукты характеризуются более узким распределением вследствие подавления реакций образования углеводородов с длиной цепи более С16. Такая ситуация обусловлена ростом количества кислотных центров при введении цеолита в состав катализатора (табл. 2).
Нанесенные гибридные катализаторы, как правило, проявляют более низкую активность в реакциях синтеза углеводородов. Так, причиной меньшей активности в СФТ на Co/ZSM-5 [27] по сравнению с катализатором 2 может быть сильное взаимодействие между активным металлом и цеолитом, приводящее к ухудшению восстанавливаемости кобальта [7]. С другой стороны, пропитка цеолита раствором кобальта способствует снижению активности также и компоненты ГО углеводородов за счет блокирования кислотных центров цеолита наносимым металлом, что уменьшает общую кислотность катализатора. Очевидно, эти факторы и обусловливают более высокую производительность катализатора 2 по сравнению с описанным в [27] катализатором Co/ZSM-5 как в СФТ, так и в ГО образующихся углеводородов.
В настоящее время установлены основные закономерности превращения углеводородов на кислотных центрах цеолита [31, 33]. Ключевыми интермедиатами превращений парафинов (изомеризация, крекинг, олигомеризация и др.) на кислотных катализаторах являются ионы карбения. Они легко образуются в присутствии следовых количеств олефинов за счет обратимого протонирования последних на бренстедовских кислотных центрах цеолита. Карбениевые ионы претерпевают скелетную перестройку, а последующее взаимодействие перегруппированных ионов со спилловерными формами водорода приводит к появлению изопарафинов и регенерации кислотного центра цеолита. Ионы карбения с числом атомов углерода 7 и более подвергаются β-расщеплению с получением алкенов и новых R+ с меньшим числом атомов углерода (реакция крекинга), а алкены легко реагируют с R+, давая ионы с более длинными углеродными цепями (реакция олигомеризации) [31].
Поскольку олефины, включая α-олефины, являются одними из продуктов синтеза на катализаторе 1 (рис. 4а), можно полагать, что промежуточные ионы карбения при использовании катализатора 2 образуются преимущественно путем протонирования олефинов, адсорбированных на бренстедовских кислотных центрах цеолита. Данное предположение подтверждается тем, что α-олефины в продуктах синтеза на образце 2 обнаружены только в следовых количествах. Спилловер водорода в гибридном катализаторе, вероятно, может способствовать ГО первичных углеводородов, генерируемых в СФТ [27]. Молекулы H2, диссоциативно адсорбированные на активных центрах Co0, в основном расходуются на синтез углеводородов, но некоторая их часть может перетекать с поверхности металла на носитель, а затем на ближайшие частицы цеолита. Различают две формы спилловерного водорода – гидридоподобный ${\text{H}}_{{{\text{so}}}}^{ - }$ и протоноподобный ${\text{H}}_{{{\text{so}}}}^{{\text{ + }}}$ [33, 34]. Первый быстро гидрирует промежуточный ион карбения в парафин, а второй регенерирует бренстедовский кислотный центр [31, 33].
Однако водород, будучи диссоциативно адсорбированным на компоненте СФТ гибридного катализатора, преимущественно идет на синтез углеводородов, и его перетекание на цеолит недостаточно эффективно из-за пространственного разделения компонент СФТ (Co–Al2O3/SiO2) и ГО углеводородов (Н-ZSM-5). Адсорбированные на поверхности цеолита промежуточные ионы карбения при дефиците спилловерного водорода подвергаются β-расщеплению с образованием олефинов [31]. По-видимому, именно последними реакциями объясняется существенное увеличение доли олефинов на гибридном катализаторе 2.
Повышенная способность металлического палладия к диссоциативной сорбции водорода по сравнению с Со0 в сочетании с более тесным контактом Pd и цеолита благоприятствует возрастанию концентрации спилловерного водорода на поверхности ZSM-5. Это приводит к увеличению количества бренстедовских кислотных центров, и, как следствие, к росту поверхностной концентрации ионов карбения, являющихся ключевыми интермедиатами в процессе ГО углеводородов. В итоге гибридный катализатор 3, содержащий в своем составе Pd-ZSM-5, должен проявлять бóльшую активность в реакциях ГО углеводородов по сравнению с катализатором 2. Действительно, сравнение продуктов синтеза, полученных на образцах 2 и 3 (табл. 3, рис. 4), подтверждает такое предположение. Продукты, синтезированные в присутствии катализатора 3, имеют более короткие цепи, до С26 (рис. 4в), повышенное содержание изопарафинов (отношение изо/н увеличивается в полтора раза) и включают существенно меньшее количество непредельных углеводородов (отношение олефины/парафины (о/п) уменьшается в 8 раз). Наблюдаемые изменения в составе продуктов свидетельствуют о большей активности катализатора 3 в реакциях гидрокрекинга, гидроизомеризации и гидрирования первичных углеводородов по сравнению с образцом 2.
Вместе с тем, в продуктах синтеза на катализаторе 3 возрастает содержание углеводородов С19+ при несколько меньшей конверсии СО. Это объясняется тем, что водород в синтез-газе расходуется не только на получение углеводородов в СФТ, но и на их гидрокрекинг, что приводит к обеднению синтез-газа водородом. Известно [35], что снижение парциального давления водорода уменьшает скорость гидрирования СО, но увеличивает вероятность роста цепи, повышая в результате селективность образования длинноцепочечных углеводородов (табл. 4, рис. 4в). К такому же эффекту могут приводить и процессы ГО углеводородов, что связано с протеканием реакций их олигомеризации вследствие взаимодействия олефинов и ионов карбения [31].
ЗАКЛЮЧЕНИЕ
Разработан новый гибридный бифункциональный кобальтсодержащий катализатор синтеза Фишера–Тропша и гидрооблагораживания первичных продуктов с получением углеводородов топливной фракции, демонстрирующий бóльшую каталитическую активность и стабильность работы по сравнению с литературными данными. Методика его приготовления предполагает использование готового катализатора Co–Al2O3/SiO2, что устраняет потерю активного металла в виде трудновосстанавливаемых соединений Co и сохраняет оптимальные для высокой производительности в отношении конденсированных углеводородов размеры наночастиц кобальта 8 нм. С другой стороны, отсутствие стадии пропитки гибридного катализатора раствором кобальта предотвращает блокирование кислотных центров цеолита, сохраняя тем самым его активность в гидрооблагораживании первичных углеводородов. Размещение в наноразмерной близости кобальтового компонента СФТ и цеолитной составляющей обеспечивает повышенную активность гибридного катализатора как в СФТ, так и в гидрооблагораживании (гидрокрекинге/гидроизомеризации) углеводородов. Оксид алюминия в разработанной гибридной каталитической системе служит не только связующим веществом, но и катализатором гидрокрекинга/гидроизомеризации первичных углеводородов. Гибридный катализатор, содержащий нанесенный на цеолит палладий, проявляет или бóльшую? активность в реакциях гидрооблагораживания углеводородов, следствием чего является более компактное распределение углеводородов, резкое уменьшение содержания олефинов и увеличение выхода изомерных соединений в продуктах синтеза, что улучшает эксплуатационные характеристики топлива.
Список литературы
Tso W.W., Niziolek A.M., Onel O., Demirhan C.D., Floudas C.A.,Pistikopoulos E.N. // Comput. Chem. Eng. 2018. V. 113. P. 222.
Fu T., Chang J., Shao J., Li Z. // J. Energy Chem. 2017. V. 26. P. 139.
Савостьянов А.П., Нарочный Г.Б., Яковенко Р.Е., Астахов А.В., Земляков Н.Д., Меркин А.А., Комаров А.А. // Катализ в промышленности. 2014. № 3. С. 43. (Savost’yanov A.P., Narochnyi G.B., Yakovenko R.E., Astakhov A.V., Zemlyakov N.D., Merkin A.A., Komarov A.A. // Catalysis in Industry. 2014. V. 6. № 3. Р. 212.)
Яковенко Р.Е., Нарочный Г.Б., Зубков И.Н., Непомнящих Е.В., Савостьянов А.П. // Кинетика и катализ. 2019. Т. 60. № 2. С. 235. (Yakovenko R.E., Narochnyi G.B., Zubkov I.N., Nepomnyashchikh E.V., Savost’yanov A.P. // Kinet. Catal. 2019. V. 60. № 2. Р. 212.)
Синева Л.В., Асалиева Е.Ю., Мордкович В.З. // Успехи химии. 2015. Т. 84. № 11. С. 1176. (Sineva L.V., Asalieva E.Y., Mordkovich V.Z. // Russ. Chem. Rev. 2015. V. 84. № 11. P. 1176.)
Flores C., Batalha N., Ordomsky V.V., Zholobenko V.L., Baaziz W., Marcilio N.R., Khodakov A.Y. // ChemCatChem. 2018. V. 10. P. 2291.
Adeleke A.A., Liu X., Lu X., Moyo M., Hildebrandt D. // Rev. Chem. Eng. 2020. V. 36. I. 4. P. 437.
Yao M., Yao N., Liu B., Li S., Xu L., Li X. // Catal. Sci. Technol. 2015. V. 5. P. 2821.
Valero-Romero M.J., Sartipi S., Sun X., Rodríguez-Mirasol J., Cordero T., Kapteijn F.,Gascon J. // Catal. Sci. Technol. 2016. V. 6. P. 2633.
Kang J., Wang X., Peng X., Yang Y., Cheng K., Zhang Q.,Wang Y. // Indus. Eng. Chem. Res. 2016. V. 55. P. 13 008.
Nakanishi M., Uddin M.A., Kato Y., Nishina Y., Hapipi A.M. // Catal. Today. 2017. V. 291. P. 124.
Wang Y., Jiang Y., Huang J., Wang H., Li Z.,Wu J. // RSC Adv. 2016. V. 6. P. 107 498.
Kruse N., Machoke A.G., Schwieger W.,Güttel R. // ChemCatChem. 2015. V. 7. P. 1018.
Subramanian V., Zholobenko V.L., Cheng K., Lancelot C., Heyte S., Thuriot J., Paul S., Ordomsky V.V., Khodakov A.Y. // ChemCatChem. 2016. V. 8. P. 380.
Kibby C., Jothimurugesan K., Das T., Lacheen H.S., Rea T., Saxton R.J. // Catal. Today. 2013. V. 215. P. 131.
Martínez A., Valencia S., Murciano R., Cerqueira H.S., Costa A.F.,S.-Aguiar E.F. // Appl. Catal. A: General. 2008. V. 346. P. 117.
Martinez A., Rollan J., Arribas M., Cerqueira H., Costa A., Saguiar E. // J. Catal. 2007. V. 249. P. 162.
Tsubaki N., Yoneyama Y., Michiki K., Fujimoto K. // Catal. Commun. 2003. V. 4. P. 108.
Rytter E., Tsakoumis N.E.,Holmen A. // Catal. Today. 2016. V. 261. P. 3.
Нарочный Г.Б., Яковенко Р.Е., Савостьянов А.П., Бакун В.Г. // Катализ в промышленности. 2016. № 1. С. 37. (Narochnyi G.B., Yakovenko R.E., Savost’yanov A.P., Bakun V.G. // Catalysis in Industry. 2014. V. 6. № 2. Р. 139.)
Diehl F.,Khodakov A.Y. // Oil & Gas Science and Technology – Revue de l’IFP. 2008. V. 64. P. 11.
Савостьянов А.П., Нарочный Г.Б., Яковенко Р.Е., Митченко С.А., Зубков И.Н. // Нефтехимия. 2018. Т. 58. № 1. С 80. (Savost’yanov A.P., Narochnyi G.B., Yakovenko R.E., Mitchenko S.A., Zubkov I.N. // Petrol. Chem. 2018. V. 58. № 1. Р. 76.)
PDF-2. The powder diffraction file TM. International Center for Diffraction Data (ICDD). PDF-2 Release 2012. web site: www.icdd.com (2014).
Prieto G., Martínez A., Concepción P.,Moreno-Tost R. // J. Catal. 2009. V. 266. P. 129.
Young R.A. The Rietveld Method. Oxford University Press, 1995. P. 298.
Яковенко Р.Е., Зубков И.Н., Нарочный Г.Б., Папета О.П., Денисов О.Д., Савостьянов А.П. // Кинетика и катализ. 2020. Т. 61. № 2. С. 278. (Yakovenko R.E., Zubkov I.N., Narochniy G.B., Papeta O.P., Denisov O.D., Savost’yanov A.P. // Kinet. Catal. 2020. V. 61. № 2. P. 310.)
Wu L., Li Z., Han D., Wu J.,Zhang D. // Fuel Process. Technol. 2015. V. 134. P. 449.
Sartipi S., Makkee M., Kapteijn F., Gascon J. // Catal. Sci. Technol. 2014. V. 4. P. 893.
Anderson R.B., Friedel R.A.,Storch H.H. // J. Chem. Phys. 1951. V. 19. P. 313.
Komvokis V.G., Karakoulia S., Iliopoulou E.F., Papapetrou M.C., Vasalos I.A., Lappas A.A., Triantafyllidis K.S. // Catal. Today. 2012. V. 196. P. 42.
Ono Y. // Catal. Today. 2003. V. 81. P. 3.
Gafurov M.R., Mukhambetov I.N., Yavkin B.V., Mamin G.V., Lamberov A.A., Orlinskii S.B. // J. Phys. Chem C. 2015. V. 119. P. 27 410.
Fujimoto K. // Stud. Surf. Sci. Catal. 1999. V. 127. P. 37.
Ueda R., Kusakari T., Tomishige K., Fujimoto K. // J. Catal. 2000. V. 194. P. 14.
Tristantini D., Lögdberg S., Gevert B., Borg Ø., Holmen A. // Fuel Process. Technol. 2007. V. 88. P. 643.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ