

Кинетика и катализ, 2021, T. 62, № 2, стр. 254-262
Исследование стадии активации алкена в условиях “безлигандного” катализа реакции Мицороки–Хека с нереакционноспособными арилхлоридами
А. А. Курохтина a, Е. В. Ларина a, Е. В. Видяева a, Н. А. Лагода a, А. Ф. Шмидт a, *
a ФГБОУ ВО Иркутский государственный университет, химический факультет
664003 Иркутск, ул. К. Маркса, 1, Россия
* E-mail: aschmidt@chem.isu.ru
Поступила в редакцию 08.07.2020
После доработки 24.08.2020
Принята к публикации 22.10.2020
Аннотация
В работе представлены результаты кинетического исследования характера (быстрый или медленный) стадии активации алкена в каталитическом цикле реакции Мицороки–Хека с нереакционноспособными арилхлоридами в присутствии так называемой “безлигандной” каталитической системы на основе палладия. С помощью анализа интегральных кинетических зависимостей расходования двух конкурирующих алкенов в их реакции с арилхлоридом показано, что алкены участвуют в быстрой стадии каталитического цикла.
ВВЕДЕНИЕ
Катализируемая соединениями палладия реакция Мицороки–Хека представляет собой инструмент тонкого органического синтеза, широко применяемый в современной малотоннажной химической промышленности [1, 2]. При этом область исследований, связанная с установлением фундаментальных особенностей функционирования каталитических систем этого процесса, активно развивается в течение последних 20–25 лет. С одной стороны, эти многолетние исследования, безусловно, являются успешными. На сегодняшний день можно уверенно говорить об установлении ключевых особенностей функционирования как основного каталитического цикла, так и сопряженных с ним процессов формирования и дезактивации каталитически активных соединений в реакциях с относительно реакционноспособными арилиодидами и арилбромидами [3–5]. С другой стороны, в случае наименее реакционноспособных, но при этом наиболее доступных в ряду арилгалогенидов субстратов – арилхлоридов – утверждать существование определенной единой точки зрения на механизм реакции Мицороки–Хека к настоящему моменту не представляется возможным в силу отсутствия систематических исследований реакции с арилхлоридами. Многими исследователями “по умолчанию” принимается идентичность процессов внутри и за пределами каталитического цикла для всех типов арилгалогенидов, хотя в литературе существуют свидетельства, как минимум, различного состава активных частиц палладия, активирующих арилиодиды, арилбромиды и арилхлориды [6]. Существующий дефицит данных, в том числе об особенностях процессов формирования и дезактивации каталитически активных соединений, которые, как известно, могут оказывать определяющую роль в функционировании каталитических систем [7], создает дополнительные трудности в исследовании механизма реакции Мицороки–Хека. Однако уже сейчас можно констатировать, что отсутствие серьезных прорывов в решении проблемы активации арилхлоридов указывает на то, что реакции с ними имеют некие принципиальные отличия от реакций с арилиодидами и арилбромидами. Это не позволяет добиться успеха в конверсии арилхлоридов путем применения методов, разработанных ранее и неоднократно апробированных на более реакционноспособных субстратах (см., например, [8, 9]). Сформировавшаяся несколько лет назад точка зрения о том, что существующие трудности с активацией арилхлоридов в реакции Мицороки–Хека обусловлены резким падением скорости стадии их окислительного присоединения, лимитирующей протекание всей каталитической реакции из-за роста энергии связи в ряду C–I < C–Br $ \ll $ C–Cl [10, 11], до сих пор принимается верной “по умолчанию” без получения каких-либо экспериментальных подтверждений подавляющим большинством исследователей за небольшим исключением [12, 13]. Следует отметить, что аналогичные объяснения пониженной реакционной способности арилбромидов в сравнении с арилиодидами долгое время были доминирующими при интерпретации экспериментальных данных сравнительных экспериментов в реакциях с арилиодидами и арилбромидами. Однако было однозначно показано, что и в случае арилиодидов, и при использовании арилбромидов стадия окислительного присоединения арилгалогенида не является скоростьопределяющей в каталитическом цикле реакции Мицороки–Хека [3, 14], а также родственных процессов сочетания арилгалогенидов, включая реакцию так называемого прямого арилирования гетероароматических соединений [15]. С предположением о быстром характере стадии окислительного присоединения согласуются хоть и немногочисленные, но заслуживающие внимания данные об относительно небольшой величине ее активационных барьеров в случае арилхлоридов в условиях реакции Мицороки–Хека, оцененных квантово-химическими методами, в совокупности с экспериментальными данными, указывающими, по мнению авторов, на то, что скорость реакции определяется отличной от окислительного присоединения элементарной стадией каталитического цикла [12]. Поэтому, на наш взгляд, выяснение скоростьопределяющих стадий каталитического цикла реакции Мицороки–Хека с малореакционноспособными арилхлоридами в специальных экспериментах без использования аналогий с реакциями арилбромидов и арилиодидов является абсолютно необходимым. Следует отметить, что активация всех типов арилгалогенидов (иодидов, бромидов и хлоридов) в каталитическом цикле реакции Мицороки–Хека путем их окислительного присоединения к частицам нольвалентного палладия (схема 1 , стадия А) не вызывает сомнения и является довольно хорошо изученной экспериментально в модельных условиях [6, 16–20], реальных каталитических условиях для арилиодидов и арилбромидов [21, 22], а также с применением методов квантово-химического моделирования [12, 23]. В то же время экспериментальные исследования природы стадии активации алкена проводились только с использованием модельных условий (т.е. при низких соотношениях субстрат/катализатор [24], либо при частичном исключении компонентов реакционной системы [16]).
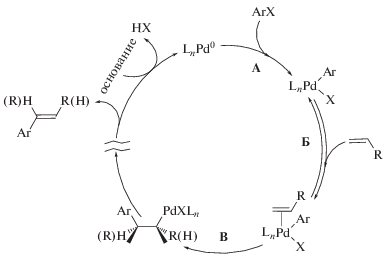
Схема 1 . Общепринятая схема механизма реакции Мицороки–Хека.
Учитывая значительно меньший объем накопленного экспериментального материала по исследованию стадий активации алкенов в сравнении со стадией активации арилгалогенидов, предсказать характер влияния смены природы арилирующего реагента на эту стадию не представляется возможным. Даже в самом простом случае принципиальной неизменности механизма этой элементарной стадии смена галогенидного противоиона в интермедиате ArPdX (схема 1 , стадия Б), очевидно, способна оказать значительное влияние на величину скорости его реакции с алкеном. Безусловно, надежные выводы о быстром или скоростьопределяющем характере той или иной элементарной стадии могут быть сделаны только на основе исследования реакции в условиях реального каталитического процесса (присутствие всех компонентов каталитической системы, высокое соотношение субстрат/катализатор). В связи с этим целью настоящей работы являлось экспериментальное установление характера (быстрый или медленный) стадии активации алкена в условиях “безлигандного” катализа реакции Мицороки–Хека (т.е. при отсутствии в реакционной системе добавок фосфиновых, аминовых, карбеновых или иных сильных органических лигандов) при использовании арилхлоридов в качестве арилирующих реагентов. Для этого нами был применен простой кинетический подход, предполагающий анализ интегральных кинетических кривых, описывающих расходование двух алкенов в условиях их конкуренции [25]. Ранее с помощью этого подхода было показано, что в реакции Мицороки–Хека с арилбромидами, а также ангидридами ароматических кислот в условиях применения “безлигандных” и фосфинсодержащих каталитических систем алкены участвуют в быстрой стадии каталитического цикла [25, 26]. Для проверки сохранения этой закономерности механизма реакции в условиях использования арилхлоридов данный метод применен впервые в ходе настоящего исследования.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пробы реакционной смеси анализировали на газожидкостном хроматографе Кристалл 5000 (“Хроматэк”, Россия, ДИП, колонка HP-5 15 м) и хромато-масс-спектрометре GCMS-QP2010 Ultra (“Shimadzu”, Япония) с ионизацией электронным ударом (энергия ионизации − 70 эВ, колонка GsBP-5MS размером 0.25 мкм × 0.25 мм × 30 м, газ-носитель – гелий) с программированным нагревом от 100 до 250°С. Полученные масс-спектры сравнивали с библиотечными (библиотеки сравнения Wiley, NIST, NIST05). Количественный состав проб вычисляли методом внутреннего стандарта (внутренний стандарт – нафталин) с применением калибровки по аутентичным образцам.
Каталитические эксперименты
Все эксперименты проводили без использования инертной атмосферы на воздухе. В круглодонный стеклянный реактор, снабженный резиновой мембраной и магнитной мешалкой, при комнатной температуре помещали 4-хлорацетофенон (7.5 ммоль), конкурирующие стирол и н‑бутилакрилат (по 1.25 или 0.625 ммоль каждого), NaOAc в качестве основания (3.25 ммоль), нафталин (0.5 ммоль) в качестве внутреннего стандарта и смесь ДМФА−NBu4Br (2.4 мл/2.5 г) в качестве растворителя. Далее реактор помещали в предварительно нагретую до 140 или 160°С масляную баню при перемешивании (477.5 об/мин). Реакцию начинали, вводя в нагретый реактор с помощью шприца раствор PdCl2 в ДМФА (0.01 ммоль, 50 мкл раствора концентрации 0.2 М). Пробы реакционной смеси для хроматографического анализа периодически отбирали из реактора с помощью шприца с металлической иглой. Каждый эксперимент проводили 3 раза для проверки воспроизводимости. Скорости расходования субстратов определяли по разности их концентраций в последовательно отбираемых пробах реакционной смеси. Материальный баланс по реагентам оценивали в каждой из проб с помощью ГЖХ. В качестве побочного продукта наблюдалось образование ацетофенона в результате восстановления 4-хлорацетофенона (до 0.5% от исходного количества арилхлорида).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В настоящий момент считается, согласно общепринятой схеме механизма каталитического цикла реакции Мицороки−Хека (схема 1 ), что активация алкенов осуществляется в стадии их координации с интермедиатом типа ArPdX (стадия Б), формирующимся в результате окислительного присоединения арилгалогенида к Pd(0) (стадия А), с последующим их внедрением по связи Pd–Ar и образованием σ-комплекса (стадия В) [3]. Соответственно, использование пары конкурирующих алкенов в реакции Мицороки–Хека позволяет исследовать свойства стадий координации и внедрения, определяющих селективность по конкурирующим алкенам. Следует, тем не менее, отметить, что результаты применения используемого нами подхода (краткое обоснование которого будет дано ниже) позволяют сделать вывод о быстром или медленном характере стадий активации конкурирующих субстратов вообще без каких-либо предварительных предположений об их природе. Единственным необходимым условием для корректности этого вывода является одинаковая природа стадии активации двух конкурирующих субстратов и идентичность механизмов превращения конкурирующих субстратов в конкурирующих каталитических циклах, что в случае использования пары близких по свойствам алкенов в реакции Мицороки−Хека не вызывает сомнений.
Подробное описание предлагаемого нами кинетического подхода для установления быстрого или медленного характера отдельных стадий каталитических циклов дано в [25]. Рассмотрим схему конкурентной реакции Мицороки−Хека в условиях применения пары алкенов, конкурирующих за общий катализатор ArPdX (схема 2 ).
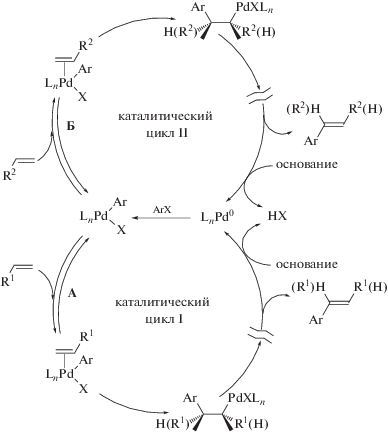
Схема 2 . Схема конкурентной реакции Мицороки–Хека с участием двух конкурирующих алкенов.
Возможны два принципиально отличающихся друг от друга случая. В первом случае катализатор находится в основном не в общих интермедиатах Pd(0) и ArPdX, а в интермедиатах, образующихся после разветвления конкурирующих каталитических циклов (общие интермедиаты не доминируют). Во втором случае катализатор присутствует наоборот в основном в виде общих интермедиатов. Если один из конкурирующих алкенов является более реакционноспособным по сравнению со вторым, то соотношение их концентраций будет изменяться с течением реакции. При этом при реализации первого варианта (общие интермедиаты не доминируют) будет происходить постепенный переход катализатора из интермедиатов каталитического цикла, в котором осуществляется превращение более активного алкена, в каталитический цикл превращения менее активного алкена, заканчивающийся при полном расходовании более активного субстрата. Во втором же варианте (доминирование общих интермедиатов) даже полное расходование более активного алкена не скажется на распределении катализатора в каталитическом цикле превращения менее активного алкена (общие интермедиаты продолжат доминировать). В [25] нами было продемонстрировано, что в случае значительной разницы в реакционных способностях конкурирующих субстратов при реализации первого варианта (общие интермедиаты не доминируют) в момент практически полного расходования более активного субстрата происходит резкий переход катализатора из одного каталитического цикла в другой, что приводит к значительному росту скорости расходования менее активного субстрата, проявляющемуся на кинетической кривой его расходования. Если же соотношение скоростей стадий каталитических циклов таково, что катализатор в основном находится в форме общих интермедиатов, то после расходования более активного субстрата эти интермедиаты продолжат доминировать в распределении катализатора, и в этом случае никаких видимых изменений скорости расходования менее активного субстрата наблюдаться не будет. Таким образом, при наблюдении эффекта ускорения расходования менее активного субстрата в момент полного расходования более активного можно сделать обоснованный вывод о том, что лишь небольшая часть катализатора находится в виде общих интермедиатов, в том числе, и в виде интермедиата ArPdX, реагирующего с конкурирующими субстратами. Такая ситуация возможна только тогда, когда лимитирующая скорость каталитического цикла стадия находится после разветвления конкурирующих каталитических циклов, то есть после превращений общих интермедиатов, включая и стадию взаимодействия ArPdX с алкеном. Следовательно, стадии с участием алкенов А и Б (схема 2 ) не могут быть медленными. Отсутствие эффекта ускорения в свою очередь означает доминирование общих для конкурирующих каталитических циклов интермедиатов, указывая на их участие в относительно медленных стадиях конкурирующих каталитических циклов.
Как следует из описанных выше особенностей предлагаемого кинетического подхода, необходимым условием его успешной реализации является применение пары конкурирующих субстратов, имеющих значительное отличие в реакционных способностях. В качестве таких субстратов нами были использованы стирол и н-бутилакрилат в реакции их арилирования 4-хлорацетофеноном (I).
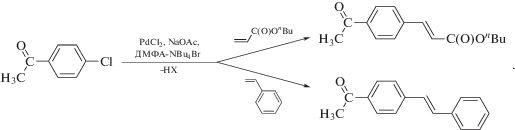
Ранее было показано, что избыток арилгалогенида положительно сказывается на конверсии и выходе продуктов в случае использования неактивированных арилбромидов [27]. Кроме того, эффективными были добавки NBu4Br [28, 29]. Положительное влияние добавок галогенидных солей тетраалкиламмония на протекание реакции Мицороки–Хека (т.н. условия Jeffery) обычно связывается с увеличением доли каталитически активных растворенных соединений палладия, которое обусловлено ростом скорости процессов растворения палладия, стабилизацией растворенных соединений палладия и образующихся в ходе реакции агломератов восстановленного палладия [5, 28]. Действительно, избыток арилхлорида в комбинации с добавками NBu4Br позволял получать количественные выходы продуктов за очень короткие времена (7−15 мин). При этом для всех экспериментов кинетические кривые расходования алкенов представляли собой типичные зависимости, наблюдаемые в каталитических реакциях с формированием активного катализатора из загружаемого в реактор прекурсора. Протекание процесса формирования катализатора in situ проявлялось на кривых в виде периода автоускорения реакции (от 2 до 3 мин), связанного с восстановлением исходного хлорида палладия(II) до соединений Pd(0). Кроме того, на кинетику реакции в некоторых случаях оказывала влияние и дезактивация катализатора, проявляясь в неполной конверсии менее активного стирола, которое, однако, было значительно менее выраженным в используемой нами каталитической системе в сравнении с экспериментами, проводимыми без применения добавок NBu4Br.
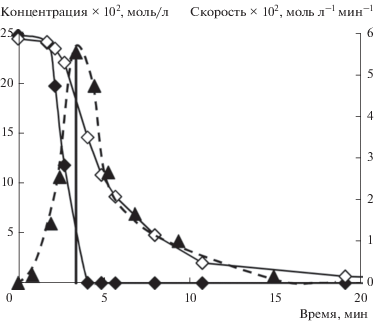
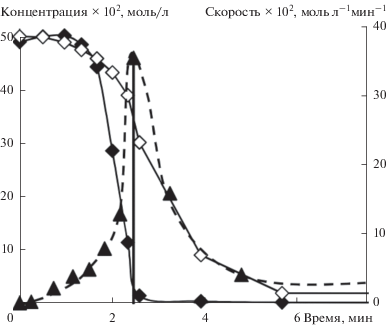
В реакции с 4-хлорацетофеноном на кривой расходования менее реакционноспособного стирола наблюдалось ускорение, совпадающее по времени с моментом полного расходования более активного н-бутилакрилата (рис. 1). Для лучшего представления получаемого результата на рис. 1 также показана зависимость “мгновенной” скорости расходования стирола от времени (последнюю оценивали по убыли концентрации стирола за временной промежуток между отборами двух проб). На этой зависимости виден четкий максимум, совпадающий по времени с точностью, определяемой промежутком времени между двумя пробами, с моментом расходования н-бутилакрилата (рис. 1). Таким образом, ускорение расходования менее активного стирола в момент полного расходования более активного н-бутилакрилата однозначно указывает, что конкуренция алкенов осуществляется не в медленной стадии, а общий интермедиат, с которым реагируют алкены (ArPdX в соответствии с общепринятым механизмом), не доминирует среди интермедиатов каталитических циклов. Это означает, что активация алкенов не может являться скоростьопределяющей в каталитическом цикле реакции Мицороки–Хека с 4-хлорацетофеноном. Необходимо отметить, что в присутствии большого количества бромид-ионов в реакционной системе, вероятно, реализуется равновесие между комплексами типа ArPdX с бромидными и хлоридными противоионами [30], что, однако, никак не влияет на вывод о быстром характере стадий каталитических циклов с их участием.
Рис. 1.
Зависимости концентраций конкурирующих н-бутилакрилата (◆) и стирола (◇), а также “мгновенной” скорости расходования стирола (▲) от времени в конкурентной реакции (I) при 140°С и начальных концентрациях конкурирующих алкенов 0.25 М (стрелкой показан момент достижения максимальной скорости расходования стирола).
Для подтверждения наблюдаемых закономерностей аналогичные зависимости были получены для конкурентной реакции стирола и н-бутилакрилата, проводимой при более высоких начальных концентрациях конкурирующих алкенов (рис. 2) (0.5 М каждого вместо используемых в эксперименте на рис. 1 0.25 М), а также при повышенной температуре (160°С, рис. 3). В обоих случаях вновь наблюдалось ускорение расходования стирола, совпадающее по времени с полным расходованием н-бутилакрилата. Необходимо отметить, что скорость расходования обоих конкурирующих алкенов из-за начального периода автоускорения проходила через максимум. Однако во всех экспериментах была зафиксирована значительная разница (более полутора минут) между временами достижения максимальных скоростей расходования конкурирующих алкенов, что в совокупности с достаточным количеством отобранных в промежутке между максимумами скоростей проб и воспроизводимостью кинетических кривых позволяет сделать уверенный вывод о разном времени достижения максимальной скорости конкурирующими алкенами. Таким образом, полученные данные однозначно свидетельствуют о том, что, как и в реакции Мицороки−Хека с более реакционноспособными арилиодидами, арилбромидами и ангидридами ароматических кислот [5, 25, 26], в реакции с арилхлоридами алкены участвуют в быстрой стадии каталитического цикла.
Рис. 2.
Зависимости концентраций конкурирующих н-бутилакрилата (◆) и стирола (◇), а также “мгновенной” скорости расходования стирола (▲) от времени в конкурентной реакции (I) при 140°С и начальных концентрациях конкурирующих алкенов 0.5 М (стрелкой показан момент достижения максимальной скорости расходования стирола).

Рис. 3.
Зависимости концентраций конкурирующих н-бутилакрилата (◆) и стирола (◇), а также “мгновенной” скорости расходования стирола (▲) от времени в конкурентной реакции (I) при 160°С и начальных концентрациях конкурирующих алкенов 0.5 М (стрелкой показан момент достижения максимальной скорости расходования стирола).
ЗАКЛЮЧЕНИЕ
Представленные в работе данные о быстром характере стадии активации алкена в совокупности с результатами квантово-химических расчетов работы [12] о низких активационных барьерах стадии активации арилхлорида указывают на то, что скорость всего каталитического процесса определяется стадиями, не включающими прямое взаимодействие катализатора с молекулами субстратов. При этом авторами [12] предполагается, что скорость каталитической реакции лимитируется некими стадиями с участием молекул основания. Необходимо отметить, что последний вывод сделан на основании анализа закономерностей изменения селективности при варьировании природы основания в конкурентных экспериментах. Однако исследования селективности позволяют делать обоснованные заключения исключительно о закономерностях селективностьопределяющих, а не скоростьопределяющих стадий, которые совсем не обязательно должны совпадать [5]. Поэтому для уверенного исключения стадии окислительного присоединения арилхлоридов как возможной скоростьопределяющей стадии и обоснованного вывода о ее природе в реакции Мицороки−Хека с арилхлоридами необходимо получение дополнительных данных в условиях реального каталитического процесса. Однако на основании представленного в настоящей работе анализа интегральных кинетических зависимостей расходования пары конкурирующих алкенов, наблюдаемых в условиях реального каталитического процесса, можно уверенно констатировать быстрый характер стадии активации алкенов в “безлигандных” условиях реакции Мицороки–Хека с арилхлоридами. При этом данный результат, означающий, что общий интермедиат, участвующий в активации алкенов, не доминирует в распределении интермедиатов каталитического цикла, справедлив вне зависимости от гипотезы о природе (составе) общего интермедиата. Таким образом, быстрое протекание активации алкенов сохраняется в реакции Мицороки−Хека в условиях использования широкого ряда арилирующих агентов (арилиодидов, арилбромидов [5, 25] и арилхлоридов, а также ангидридов ароматических кислот [26]). Учитывая специфику реакции Мицороки−Хека и низкую вероятность высокой концентрации π-комплексов алкенов, основная доля катализатора в ходе ее протекания, скорее всего, находится в виде продукта стадии внедрения (алкильного σ-комплекса, схема 1 ). Такое предположение согласуется с результатами, полученными ранее для реакции Мицороки−Хека с арилиодидами и арилбромидами на основании данных измерений кинетического изотопного эффекта [31].
Список литературы
Torborg C., Beller M. // Adv. Synth. Catal. 2009. V. 351. P. 3027.
Dumrath A., Lübbe C., Beller M. in Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments. Wiley-VCH Verlag GmbH & Co. KGaA. 2013. P. 445.
The Mizoroki–Heck Reaction / Ed. Oestreich M. Munster: John Wiley & Sons Ltd. 2009. 587 p.
Eremin D.B., Ananikov V.P. // Coord. Chem. Rev. 2017. V. 346. P. 2.
Schmidt A.F., Al Halaiqa A., Smirnov V.V. // Synlett. 2006. № 18. P. 2861.
Barrios-Landeros F., Carrow B.P., Hartwig J.F. // J. Am. Chem. Soc. 2009. V. 131. P. 8141.
Темкин О.Н. // Кинетика и катализ. 2019. Т. 60. С. 683. (Temkin O.N. // Kinet. Catal. 2019. V. 60. P. 689.)
Röhlich C., Köhler K. // Adv. Synth. Catal. 2010. V. 352. P. 2263.
Hayouni S., Ferlin N., Bouquillon S. // Mol. Catal. 2017. V. 437. P. 121.
Kleist W., Pröckl S.S., Köhler K. // Catal. Lett. 2008. V. 125. P. 197.
Veerakumar P., Thanasekaran P., Lu K.-L., Lin K.-C., Rajagopal S. // ACS Sustain. Chem. Eng. 2017. V. 5. P. 8475.
Fernandez E., Rivero-Crespo M.A., Dominguez I., Rubio-Marques P., Oliver-Meseguer J., Liu L., Cabrero-Antonino M., Gavara R., Hernandez-Garrido J.C., Boronat M., Leyva-Perez A., Corma A. // J. Am. Chem. Soc. 2019. V. 141. P. 1928.
Knowles J.P., Whiting A. // Org. Biomol. Chem. 2007. V. 5. P. 31.
Шмидт А.Ф., Аль-Халайка А., Смирнов В.В. // Кинетика и катализ. 2007. Т. 48. С. 766. (Schmidt A.F., Al-Halaiqa A., Smirnov V.V. // Kinet. Catal. 2007. V. 48. P. 716.)
Ларина Е.В., Ярош Е.В., Лагода Н.А., Курохтина А.А., Шмидт А.Ф. // Кинетика и катализ. 2019. Т. 60. С. 358. (Larina E.V., Yarosh E.V., Lagoda N.A., Kurokhtina A.A., Schmidt A.F. // Kinet. Catal. 2019. V. 60. P. 337.)
Amatore C., Jutand A. // Acc. Chem. Res. 2000. V. 33. P. 314.
Jutand A., Negri S., de Vries J.G. // Eur. J. Inorg. Chem. 2002. V. 479. P. 1711.
Maes B.U.W., Verbeeck S., Verhelst T., Ekomié A., von Wolff N., Lefèvre G., Mitchell E. A., Jutand A. // Chem. Eur. J. 2015. V. 21. P. 7858.
Kurbangalieva A., Carmichael D., Hii K.K., Jutand A., Brown J.M. // Chem. Eur. J. 2014. V. 20. P. 1116.
Kolter M., Boeck K., Karaghiosoff K., Koszinowski K. // Angew. Chem. Int. Ed. 2017. V. 56. P. 13 244.
Beller M., Riermeier T.H. // Eur. J. Inorg. Chem. 1998. V. 1998. P. 29.
Шмидт А.Ф., Смирнов. В.В. // Кинетика и катализ. 2002. Т. 43. С. 215. (Shmidt A.F., Smirnov V.V. // Kinet. Catal. 2002. V. 43. P. 195.)
Polynski M.V., Ananikov V.P. // ACS Catal. 2019. V. 9. P. 3991.
Carrow B.P., Hartwig J.F. // J. Am. Chem. Soc. 2010. V. 132. P. 79.
Шмидт А. Ф., Курохтина А. А., Смирнов В.В., Ларина Е.В., Чечиль Е.В. // Кинетика и Катализ. 2012. Т. 53. С. 223. (Schmidt A.F., Kurokhtina A.A., Smirnov V.V., Larina E.V., Chechil E.V. // Kinet. Catal. 2012. V. 53. P. 214.)
Kurokhtina A.A., Larina E.V., Yarosh E.V., Schmidt A.F. // J. Mol. Catal. A: Chem. 2016. V. 425. P. 43.
Schmidt A.F., Smirnov V.V. // J. Mol. Catal. A: Chem. 2003. V. 203. P. 75.
Köhler K., Kleist W., Pröckl S.S. // Inorg. Chem. 2007. V. 46. P. 1876.
Reetz M.T., Westermann E. // Angew. Chem. Int. Ed. 2000. V. 39. P. 165.
Smirnov V.V., Pröckl S.S., Schmidt A.F., Köhler K. // ARKIVOC. 2011. V. viii. P. 225.
Шмидт А.Ф., Аль-Халайка А., Смирнов. В.В. // Кинетика и Катализ. 2007. Т. 48. С. 766. (Schmidt A.F., Al-Halaiqa A., Smirnov V.V. // Kinet. Catal. 2007. V. 48. P. 716.)
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ