

Кинетика и катализ, 2021, T. 62, № 2, стр. 187-197
Гетерогенная реакция диметилсульфида с оксидом йода в области температур 291–365 К
И. К. Ларин a, Т. И. Белякова a, Н. А. Мессинева a, *, А. И. Спасский a, Е. М. Трофимова a, **
a Институт энергетических проблем химической физики им. В.Л. Тальрозе Федерального исследовательского центра химической физики им. Н.Н. Семенова РАН
117829 Москва, Ленинский просп., 38, корп. 2, Россия
* E-mail: nat-messineva@yandex.ru
** E-mail: eltrofimova@yandex.ru
Поступила в редакцию 26.04.2020
После доработки 21.09.2020
Принята к публикации 30.11.2020
Аннотация
Методом резонансной флуоресценции атомов йода была изучена реакция радикала оксида йода (IO) с диметилсульфидом (ДМС) в диапазоне температур 291–365 К. Показано, что данная реакция в условиях наших экспериментов, в основном, является гетерогенной, и константа скорости гетерогенной реакции почти на порядок превышает константу скорости гомогенной реакции. Зависимость константы скорости данной реакции от температуры можно представить следующим выражением k(T) = 1.5 × 10–14 [±3.0 × 10–15 см3 молекула–1 с–1] exp(7150[±800 Дж/моль]/RT). Обсуждены условия, при которых гетерогенная реакция ДМС с радикалом IO может играть заметную роль в окислении ДМС в атмосфере.
Диметилсульфид (CH3)2S (ДМС), является продуктом биогенных процессов разложения морских водорослей и фитопланктона [1]. Его концентрация в тропосфере составляет, по крайней мере, половину от концентраций всех поступающих в атмосферу серосодержащих веществ [2]. После эмиссии ДМС в основном окисляется в тропосфере, на что требуется 1–2 дня [3, 4]. В результате окисления ДМС образуются такие соединения серы, как серная (H2SO4) и метансульфоновая кислоты (CH3SO3H), что приводит к формированию природных аэрозолей и облаков и влияет на климат Земли [5–7]. Основными окислителями ДМС в тропосфере являются радикалы OH [8–11], озон [12], атомы галогенов [13, 14], а также радикалы BrO [15] и оксида йода (IO) [16–18].
Значительным источником ядер конденсации в тропосфере является также фотохимическая трансформация биогенного ДМС, что влияет не только на радиационный баланс вблизи морей и океанов, но и на климат планеты в целом [19, 20].
Данная работа посвящена исследованию гетерогенной реакции ДМС с радикалом IO в области температур 291–365 К. В естественных условиях исходным веществом для образования радикала IO служит йодистый метан (CH3I), являющийся продуктом разложения биомассы морей и океанов. В результате в атмосферу поступает ∼1.5 мегатонны CH3I в год [21]. Фотолиз йодистого метана приводит к образованию атомов йода, при окислении которых образуются радикалы IO. В работе [22] было показано, что в морской атмосфере могут образовываться значительные концентрации оксида йода, достигающие значений, превышающих 108 молекула/см3, в то время как концентрация основного “чистильщика атмосферы” – радикала OH, в дневное время достигает максимальной концентрации 9 × 106 молекула/см3 [23].
Ранее нами было показано [24], что реакция радикала IO с ДМС может протекать на поверхности реактора, что приводит к значительному увеличению константы скорости данной реакции.
Полученные в последнее время данные о высоких концентрациях радикала IO в морской атмосфере возобновили наш интерес к исследованию его взаимодействия с ДМС, и данная работа посвящена изучению гетерогенной реакции ДМС с оксидом йода в интервале температур 291–365 К.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Описание установки
Экспериментальная установка включала в себя проточный реактор, источник атомов йода и систему их регистрации.
Реактор представлял собой стеклянный цилиндр, внутренний диаметр которого равнялся 1.7 см. Он был заключен в водяную рубашку, присоединенную к термостату (рис. 1). Это позволило изменять температуру реактора в диапазоне температур 291–365 К. Поверхность реактора была обработана фторопластом Ф-32Л. Такая обработка позволила снизить величину константы скорости гетерогенной гибели поступающих в реактор атомов йода и образующихся в реакторе радикалов IO до значений, не превышающих 10 с–1.
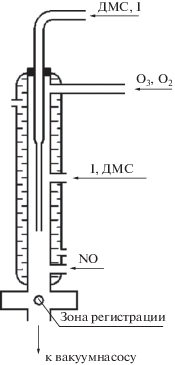
Атомы йода поступали в реактор или через подвижную трубку, герметично соединенную с реактором тефлоновым уплотнением, или через трубку, припаянную к боковой поверхности реактора. К этим трубкам была присоединена бактерицидная кварцевая ртутная лампа низкого давления, более 90% излучения которой приходилось на λ = 253.7 нм, и через них пропускали смесь гелия с газообразным йодистым метилом. Для регистрации атомов йода по их резонансной флуоресценции применялась резонансная лампа, изготовленная из кварца и излучающая резонансную линию атомов йода 178.3 нм. Коротковолновая граница пропускания кварцевых окон лампы составляла 160 нм. Через лампу прокачивали смесь гелия с молекулярным йодом в соотношении ~10 000 : 1. Для возбуждения разряда в лампе использовали СВЧ-резонатор Бройда.
Регистрацию квантов, поглощенных и вновь излученных атомами йода, осуществляли с помощью счетчика Гейгера, снабженного кварцевыми окнами, коротковолновая граница пропускания которых составляла 160 нм. Счетчик заполняли смесью 10 Торр NO и 230 Торр Ar. В стеклянный отросток, припаянный к корпусу счетчика, помещали каплю диэтилферроцена. Длинноволновая граница счетчика определялась потенциалом ионизации диэтилферроцена– 6.3 эВ [25] и составляла ~185 нм. Таким образом, счетчик служил еще и монохроматором, выделяя спектральный участок от ~160 до ~185 нм.
Накопление сигналов осуществляли через частотомер Ч3-63/1. В каждой экспериментальной точке могло накапливаться от 500 до 1.2 × 103 имп. Радикалы IO получали в реакции атомов йода с озоном. Константа скорости этой реакции в диапазоне температур от 291 до 365 К приведена в работах [26, 27]. Усредненные значения данной константы в этом диапазоне изменялись от 1.16 × 10–12 до 2.07 × 10–12 см3 молекула–1 с–1.
Озон, использованный для получения радикалов IO, поступал в реактор через боковое отверстие в верхней части реактора. Его получали в потоке O2, проходившем через озонатор. После очистки в низкотемпературной ловушке с сухим льдом кислород через клапан управления потоком подавали в озонатор. Далее смесь кислорода с озоном поступала в стеклянную ячейку, размещенную в спектрометре Spectromom-204, для измерения концентрации озона. Давление в ней можно было изменять от 10 до 100 Toрр, не изменяя массовый поток. Это давало возможность выбирать интервал оптического поглощения, удобный для измерений. Затем смесь O2 + O3 вводили в реактор. Концентрацию озона вычисляли по изменению оптического поглощения на длине волны 253.7 нм.
Для подачи газов-разбавителей и CH3I в реактор использовали регуляторы расходов, позволявшие поддерживать постоянный расход газа с точностью 2–3% в течение 10–15 ч. Массовые потоки реагентов и газов-носителей измеряли методом истечения из калиброванного объема в единицу времени. Во всех опытах использовали гелий и кислород квалификации “в. ч.” и CH3I – “ч. д. а.” Диметилсульфид получали в реакции йодистого метила CH3I с Na2S [28] и очищали многократной низкотемпературной перегонкой.
МЕТОДИКА ЭКСПЕРИМЕНТА И РЕЗУЛЬТАТЫ
Атомы йода в смеси с гелием, образовавшиеся при фотолизе CH3I, можно было подавать в реактор как через подвижную трубку (когда надо было измерить гетерогенную гибель атомов йода и радикалов IO), так и через неподвижный ввод. В реакции с озоном атомы йода превращались в радикалы IO:
Это приводило к снижению интенсивности сигнала резонансной флуоресценции (РФ) атомов йода до своих фоновых значений. Затем в реактор поступал ДМС, что снова приводило к появлению сигнала РФ атомов йода:
Если в реакторе одновременно происходят и гомогенная и гетерогенная реакции, то для процессов, протекающих в нем, следует записать следующие уравнения:Атомы йода, образующиеся в реакции (IIгет), поступают в газовую фазу. Скорость их генерации описывается следующим выражением:
(1)
$W = k_{{{\text{II}}}}^{{{\text{гом}}}}[{\text{IO}}][{\text{ДМС}}] + {{k}_{{{\text{эфф}}}}}[{\text{IO}}],$(2)
${1 \mathord{\left/ {\vphantom {1 {{{k}_{{{\text{эфф}}}}}}}} \right. \kern-0em} {{{k}_{{{\text{эфф}}}}}}} = {\text{ }}{1 \mathord{\left/ {\vphantom {1 {k_{{{\text{II}}}}^{{{\text{гет}}}}}}} \right. \kern-0em} {k_{{{\text{II}}}}^{{{\text{гет}}}}}}[{\text{ДМС}}] + {1 \mathord{\left/ {\vphantom {1 {{{k}_{{{\text{диф}}}}}}}} \right. \kern-0em} {{{k}_{{{\text{диф}}}}}}},$Стационарную концентрацию атомарного йода определяли из следующего выражения:
(3)
${{k}_{{\text{I}}}}{{[{\text{I}}]}_{{{\text{стац}}}}}[{{{\text{O}}}_{3}}] = k_{{{\text{II}}}}^{{{\text{гом}}}}{{{\text{[IO]}}}_{{{\text{стац}}}}}{\text{[ДМС}}] + {{k}_{{{\text{эфф}}}}}{\text{[IO}}].$Выражение (3) справедливо только тогда, когда в реакторе не протекают химические процессы, приводящие к дополнительному, кроме реакции (II), образованию атомов йода. Таким процессом могла бы быть реакция озона с радикалом IO:
По данным работы [30], константа этой реакции не превышает величину 10–15 см3 молекула–1 с–1. При максимальной концентрации озона в наших экспериментах (1.1 × 1015 молекула/см3) характерное время реакции (III) составляло ~1 с, а время контакта реагентов было на 2 порядка меньше. То есть вклад этой реакции в увеличение концентрации атомов йода не превышал 1%.
Гораздо более быстрым процессом является квадратичная реакция радикалов IO
Константа скорости реакции (IV), по данным работы [31], равна 3.8 × 10–11 см3 молекула–1с–1. При таком высоком значении константы скорости данной реакции требовалось, во-первых, проводить эксперименты при низкой концентрации радикалов IO. Поэтому перед опытами была определена зависимость сигнала РФ атомов йода от их концентрации. Калибровка абсолютной чувствительности кинетической установки к атомам йода подробно описана в работе [24]. Полученная нами зависимость сигнала РФ атомов йода от их концентрации представлена на рис. 2.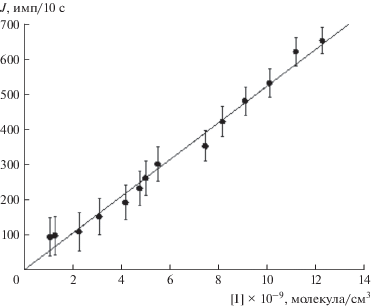
Максимальный сигнал РФ атомов йода в наших опытах составлял 650 имп/10 с, что соответствует концентрации атомов йода 1.2 × 1010 молекулa/см3, а концентрация радикалов IO во всех опытах была ниже этой величины. Наименьшая концентрация ДМС в наших опытах соответствовала величине 1.1 × 1014 молекул/см3, т.е. почти на 4 порядка превышала концентрацию радикалов IO, поэтому, по нашим оценкам, вклад реакции (IV) в сигнал РФ атомов йода не превышал 2.5%.
Второе требование к применимости выражения (3) заключалось в необходимости достаточно малой скорости гетерогенной гибели атомов I и радикалов IO в условиях наших экспериментов. Именно для этой цели внутреннюю поверхность реактора обрабатывали жидким фторопластом Ф-32Л, а затем прогревали реактор в специальной печи при температуре 160°C в течение нескольких часов.
Константу скорости гетерогенной гибели атомов йода измеряли по изменению сигнала РФ в зависимости от расстояния до зоны регистрации. Величина этой константы в наших экспериментах не превышала значения (5 ± 1) с–1.
Для измерения константы скорости гетерогенной гибели радикалов IO по реакции
атомы йода направляли в реактор через подвижную трубку, герметично соединенную с реактором тефлоновым уплотнением. Через реактор проходил поток кислорода, содержащий озон в концентрации 1.1 × 1015 молекула/cм3. При такой концентрации озона атомы йода практически полностью превращались в радикалы IO на расстоянии 1 см от точки поступления атомов йода в реактор. Непосредственно перед зоной регистрации в реактор подавали NO, по реакции с которым снова образовывались атомы йода: Сигнал РФ атомов йода измеряли на разных расстояниях от точки превращения атомов йода в радикалы IO в реакции с озоном до зоны регистрации вновь образованных атомов йода по реакции (VI). График зависимости логарифма сигнала РФ атомов I от времени пребывания в реакторе радикалов IO представлен на рис. 3. По тангенсу угла наклона прямой на этом графике мы оценили константу скорости реакции (V) величиной (6 ± 1) с–1.Рис. 3.
График зависимости логарифма сигнала РФ атомов йода от времени контакта реагентов. Условия реакции: T = 291 К, [O3] = 1.1 × 1015 молекула/см3, [NO] = 9.1 × 1014 молекула/см3.
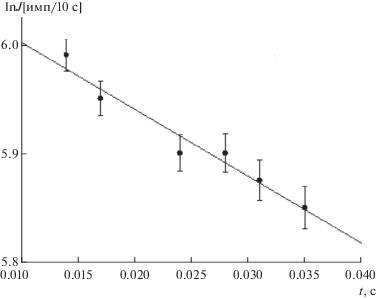
Таким образом, было показано, что в условиях наших экспериментов выражение (3) применимо.
Учитывая, что [I]0 = [I]стац + [IO]стац, из выражений (2) и (3) получаем следующее соотношение:
(4)
$\frac{{{{{[{\text{I }}]}}_{0}}}}{{{{{[{\text{I}}]}}_{{{\text{стац}}}}}}} = \frac{{[{\text{O3}}]}}{{[{\text{ДМС}}]}}\frac{{{{k}_{{\text{I}}}}}}{{k_{{{\text{II}}}}^{{{\text{гом}}}} + k_{{{\text{II}}}}^{{{\text{гет}}}}\frac{{{{k}_{{{\text{диф}}}}}}}{{\left( {k_{{{\text{II}}}}^{{{\text{гет}}}}[{\text{ДМС}}] + {{k}_{{{\text{диф}}}}}} \right)}}}} + 1.$(5)
$\frac{{{{{[{\text{I }}]}}_{0}}}}{{{{{[{\text{I}}]}}_{{{\text{стац}}}}}}} = \frac{{[{\text{O3}}]}}{{[{\text{ДМС}}]}}\frac{{k{\text{I}}}}{{(k_{{{\text{II }}}}^{{{\text{гом}}}} + k_{{{\text{II}}}}^{{{\text{гет}}}})}} + 1.$(6)
$\frac{{J0}}{{{{J}_{{{\text{стац}}}}}}} = \frac{{[{\text{O3}}]}}{{[{\text{ДМС}}]}}\frac{{{{k}_{{\text{I}}}}}}{{\left( {k_{{{\text{II }}}}^{{{\text{гом}}}} + k_{{{\text{II}}}}^{{{\text{гет}}}}} \right)}} + 1.$Рис. 4.
График зависимости J0/Jстац от [O3]/[ДМС]. Условия реакции: [ДМС] = 1.1 × 1014 молекула/см3, T = 291 К, давление в реакторе 1.2 Торр.
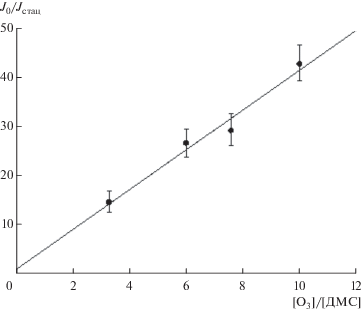
Выгорание ДМС в реакции с озоном не учитывали по следующим причинам. По данным работы [32], константа скорости этой реакции не превышает величину 10–18 см3 молекула–1 с–1. Однако мы дополнительно проверили, не возрастает ли константа скорости этого процесса при протекании его на поверхности реактора. Для этого были измерены сигналы РФ образующихся по реакции (II) атомов йода при различных временах контакта ДМС с озоном при температуре в реакторе 291 К, [ДМС] = 1.2 × 1015 молекула/см3 и [O3] = 0.95 × × 1015 молекула/см3. Как было показано, в условиях нашего эксперимента константа скорости реакции ДМС с озоном не превышала значения 10–17 см3 молекула–1 с–1.
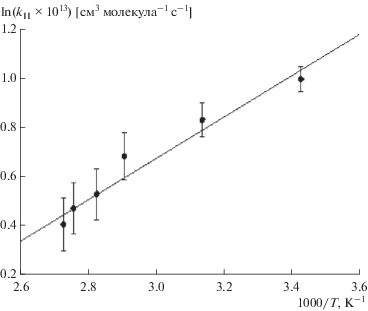
Аналогично были получены значения kII для следующих температур в реакторе: 318, 343, 353, 362 и 365 К (давление в реакторе составляло (1.2 ± ± 0.1) Торр). Данные представлены в табл. 1, а график зависимости ln(kII × 1013) [см3 молекула–1 с–1] от 1000/T приведен на рис. 5. Параметры этого графика (отсечение на оси ординат и тангенс угла наклона прямой) позволили описать зависимость константы скорости данной реакции от температуры следующим выражением:
(7)
$\begin{gathered} \frac{{J0}}{{{{J}_{{{\text{стац}}}}}}}{\text{ }} = \frac{{{\text{[O3] }}}}{{[{\text{ДМС}}]}} \\ \times \,\,\frac{{{{k}_{1}}}}{{(1 - {\alpha }){{k}_{{{\text{II}}}}} + {\alpha }{{k}_{{{\text{II}}}}}\frac{{{{k}_{{{\text{диф}}}}}}}{{({\alpha }{{k}_{{{\text{II}}}}}[{\text{ДМС}}] + {{k}_{{{\text{диф}}}}})}}}} + 1, \\ \end{gathered} $Таблица 1.
Экспериментальные данные, полученные в диапазоне температур в реакторе 291–365 К, давлении 1.2 Торр и [ДМС] = 1.1 × 1014 молекула/см3
| T, К |
[O3] × 10–14, молекула/см3 |
[ДМС] × 10–14, молекула/см3 |
J0, имп/10 с |
Jстац, имп/10 с | [O3]/[ДМС] | J0/Jстац | kII × 1013, см3 молекула–1 с–1 |
|---|---|---|---|---|---|---|---|
| 291 | 3.4 | 1.1 | 641 | 45.8 | 3.1 | 14.0 | 2.7 ± 0.15 |
| 6.6 | 1.1 | 24.1 | 6.0 | 26.6 | |||
| 8.2 | 1.1 | 22.2 | 7.5 | 28.8 | |||
| 10.9 | 1.1 | 14.7 | 9.9 | 43.6 | |||
| 318 | 2.2 | 1.1 | 630 | 44.7 | 2.0 | 14.1 | 2.3 ± 0.2 |
| 4.9 | 1.1 | 23.3 | 4.5 | 27 | |||
| 5.8 | 1.1 | 18 | 5.3 | 34.9 | |||
| 6.5 | 1.1 | 16 | 5.9 | 39.5 | |||
| 343 | 1.6 | 1.1 | 645.4 | 40.3 | 1.5 | 16 | 2.0 ± 0.2 |
| 3.8 | 1.1 | 22.2 | 3.4 | 29.1 | |||
| 4.5 | 1.1 | 17.4 | 4.1 | 37 | |||
| 5.3 | 1.1 | 13.6 | 4.8 | 47.5 | |||
| 353 | 1.8 | 1.1 | 639 | 35.7 | 1.6 | 17.9 | 1.7 ± 0.15 |
| 2.5 | 1.1 | 21.9 | 2.3 | 29.2 | |||
| 3.2 | 1.1 | 18.2 | 2.9 | 35 | |||
| 3.6 | 1.1 | 17.5 | 3.3 | 36.5 | |||
| 362 | 2.0 | 1.1 | 642.5 | 27.3 | 1.8 | 23.5 | 1.6 ± 0.15 |
| 2.4 | 1.1 | 22.9 | 2.2 | 28 | |||
| 2.7 | 1.1 | 20 | 2.5 | 32 | |||
| 3.2 | 1.1 | 18.3 | 2.9 | 37.5 | |||
| 365 | 1.2 | 1.1 | 648.6 | 34.1 | 1.1 | 17.2 | 1.5 ± 0.12 |
| 2.1 | 1.1 | 23.4 | 1.9 | 27.5 | |||
| 2.7 | 1.1 | 19.3 | 2.5 | 33.6 | |||
| 3.1 | 1.1 | 15.3 | 2.8 | 42.5 |
При температурах в реакторе 291 и 365 К нами было проведено сравнение расчетной зависимости отношения J0/Jстац от [O3]/[ДМС] при значениях α от 0 до 1 с экспериментальными значениями, полученными в условиях, когда величина $k_{{{\text{II}}}}^{{{\text{гет}}}}$[ДМС] сравнима с величиной kдиф или превышает ее. В этих условиях часть атомов йода, образованных в гетерогенной реакции (II), не успевают продиффундировать от стенки реактора к центральной оси, где измеряется сигнал РФ. Это приводит к снижению концентрации атомов йода на оси реактора, а значит, и к ослаблению сигнала РФ атомов йода по сравнению с таковым при гомогенном протекании реакции (II).
Коэффициент диффузии радикала IO в кислороде при давлении 760 Торр и температуре 298 К (D0) составляет величину 0.12 см2/с [33]. Он был поправлен на температуру в соответствии с формулой, приведенной в публикации [33]: D0(T) = = $D_{0}^{{298}}$ (T/298)1.5.
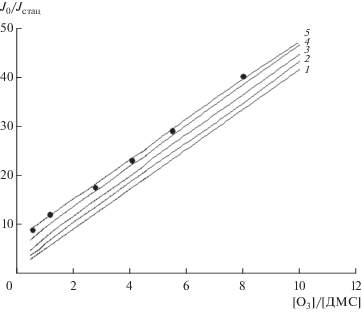
Результаты расчета для температуры 291 К, [O3] = 9.8 × 1014 молекула/см3, давления в реакторе 2.9 Торр, $D_{0}^{{291}}$ = 0.12 и J0 = 655 имп/10 с приведены в табл. 2 и на рис. 6.
Таблица 2.
Экспериментальные данные, полученные при температуре в реакторе 291 К, давлении 2.9 Торр и J0 = 655 имп/10 c
| T, К |
P, Toрр |
[O3] × 10–14, молекула/см3 |
[ДМС] × 10–14, молекула/см3 |
[O3]/[ДМС] | Jстац, имп/10 с |
J0/Jстац | tреакц × 103, с* |
tдиф × 103, с** |
|---|---|---|---|---|---|---|---|---|
| 291 | 2.9 | 9.8 | 15.8 | 0.62 | 72 | 9.1 | 2.3 | 7.6 |
| 291 | 2.9 | 9.8 | 8.9 | 1.1 | 56 | 11.7 | 4.2 | 7.6 |
| 291 | 2.9 | 9.8 | 3.5 | 2.8 | 39.2 | 16.7 | 10.5 | 7.6 |
| 291 | 2.9 | 9.8 | 2.4 | 4.1 | 28 | 23 | 15.4 | 7.6 |
| 291 | 2.9 | 9.8 | 1.8 | 5.4 | 22.5 | 29.1 | 20.5 | 7.6 |
| 291 | 2.9 | 9.8 | 1.2 | 8 | 16.3 | 40 | 30.8 | 7.6 |
Рис. 6.
Расчетная зависимость J0/Jстац от [O3]/[ДМС], полученная при разных значениях α = ${{k_{{{\text{II}}}}^{{{\text{гет}}}}} \mathord{\left/ {\vphantom {{k_{{{\text{II}}}}^{{{\text{гет}}}}} {\left( {k_{{{\text{II}}}}^{{{\text{гет}}}} + k_{{{\text{II}}}}^{{{\text{гом}}}}} \right)}}} \right. \kern-0em} {\left( {k_{{{\text{II}}}}^{{{\text{гет}}}} + k_{{{\text{II}}}}^{{{\text{гом}}}}} \right)}}{\kern 1pt} :$ 1 – 0, 2 – 0.5, 3 – 0.7, 4 – 0.9, 5 – 1. Условия реакции: [О3] = 9.8 × 1014 молекула/см3, давление в реакторе 2.9 Торр. Символом (⚫) показаны экспериментальные значения, полученные при 291 К.
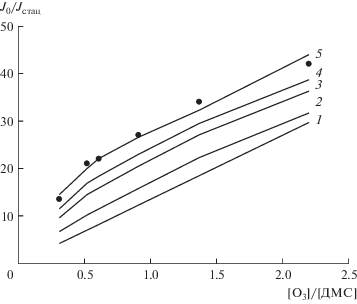
Результаты расчета для температуры 365 К, [O3] = 1.1 × 1015 молекула/см3, давления в реакторе 3.7 Торр, $D_{0}^{{365}}$ = 0.16 и J0 = 648 имп/10 с приведены в табл. 3 и на рис. 7. На обоих рисунках данные экспериментов представлены символами (⚫).
Таблица 3.
Экспериментальные данные, полученные при температуре в реакторе 365 К, давлении 3.7 Торр и J0 = 648 имп/10 c
| T, К |
P, Toрр |
[O3] × 10–14, молекула/см3 |
[ДМС] × 10–14, молекула/см3 |
[O3]/[ДМС] | Jстац, имп/10 с |
J0/Jстац | tреакц × 103, с* |
tдиф × 103, с** |
|---|---|---|---|---|---|---|---|---|
| 365 365 365 365 365 365 |
3.7 3.7 3.7 3.7 3.7 3.7 |
11 11 11 11 11 11 |
25 21 18 12 8 5 |
0.31 0.52 0.61 0.91 1.37 2.2 |
48 30.8 29.4 24 19 15.4 |
13.5 21 22 27 34 42 |
2.7 3.2 3.7 5.6 8.0 13.3 |
5.8 5.8 5.8 5.8 5.8 5.8 |
Рис. 7.
Расчетная зависимость J0/Jстац от [O3]/[ДМС], полученная при разных значениях α = ${{k_{{{\text{II}}}}^{{{\text{гет}}}}} \mathord{\left/ {\vphantom {{k_{{{\text{II}}}}^{{{\text{гет}}}}} {\left( {k_{{{\text{II}}}}^{{{\text{гет}}}} + k_{{{\text{II}}}}^{{{\text{гом}}}}} \right)}}} \right. \kern-0em} {\left( {k_{{{\text{II}}}}^{{{\text{гет}}}} + k_{{{\text{II}}}}^{{{\text{гом}}}}} \right)}}{\kern 1pt} :$ 1 – 0, 2 – 0.2, 3 – 0.5, 4 – 0.9, 5 – 1. Условия реакции: [О3] = 1.1 × 1015 молекула/см3, давление в реакторе 3.7 Торр. Символом (⚫) показаны экспериментальные значения, полученные при 365 К.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Реакцию радикала IO• с диметилсульфидом (ДМС) изучали многие исследователи. Интерес к ней был привлечен еще в 1987 г., когда в работе [34] было получено значение константы скорости этой реакции, равное (3 ± 0.5) × 10−11 см3 молекула–1 с–1 при 298 К и давлении 1 Торр. Авторы [34] сделали вывод, что данная реакция ответственна за малое время жизни ДМС в атмосфере. Этот результат был подтвержден и в работе [35].
Однако вскоре появились исследования, показавшие, что константа скорости реакции IO с ДМС намного меньше, чем в упомянутых выше работах. Так, измеренная в работе [36] константа скорости данной реакции не превышала значения 3.5 × 10−14 см3 молекула–1 с–1. Еще меньшее значение (1.5 × 10−14 см3 молекула–1 с–1) было получено в работе [37]. Значение константы скорости этой же реакции (0.88 × 10−14 см3 молекула–1 с–1), приведенное в [38], тем более не объясняло краткость времени жизни ДМС в атмосфере.
В ХХI в. возобновился интерес к исследованию реакции ДМС с радикалом IO. В работе [39] при температуре 298 К было получено значение константы скорости реакции (II), равное $\left( {2.0_{{ - 0.6}}^{{ + 0.4}}} \right)$ × × 10−14 см3 молекула–1 с–1, а в работе [40] это значение составило величину (1.44 ± 0.15) × 10−14 см3 молекула–1 с–1. В обеих работах была установлена аррениусовская зависимость константы скорости реакции от температуры, т.е. константа скорости реакции росла с повышением температуры, и был сделан вывод [39, 40], что эта реакция не играет существенной роли в химии атмосферы.
В 2004 г. теоретической работе Sayin и McKee [41] в результате проведенного компьютерного исследования реакций (CH3)2S с BrO и IO было достигнуто разумное согласие между расчетными и экспериментальными коэффициентами скорости для реакции ДМС с BrO. Для реакции же ДМС с IO коэффициент скорости был оценен ими как 1.5 × 10–11 см3 молекула−1 с−1 и предположено, что реакция протекает через образование ассоциативного комплекса, за которым следует перенос атома кислорода от радикала IO к (CH3)2S с образованием (CH3)2SО.
В работе [42] экспериментально исследовали зависимость константы скорости реакции (II) от давления в интервале температур 273–312 К и обнаружили увеличение константы скорости реакции при понижении температуры и повышении давления. При давлении ~200 Торр и температуре 298 К было получено следующее значение константы скорости реакции (II):
В нашей работе значение константы скорости реакции ДМС с радикалом IO при температуре 291 К составило величину:
Уменьшение константы скорости реакции (II) с ростом температуры реактора, по-видимому, указывает на тот факт, что при более высокой температуре прочность связи адсорбированной молекулы ДМС с поверхностью реактора уменьшается.
Условия для более заметной роли гетерогенной реакции ДМС с радикалом IO в окислении ДМС могут создаваться в морской атмосфере. Там имеются природные аэрозоли и облака, в том числе содержащие серную (H2SO4) и метансульфоновую кислоты (CH3SO3H). На их поверхностях и могут происходить быстрые гетерогенные процессы. Как упоминалось выше, там же образуются значительные концентрации оксида йода (более 108 молекула/см3) [22], тогда как концентрация радикала OH в дневное время не превышает значения 9 × 106 молекула/см3 [23].
ВЫВОДЫ
1. Измерена константа реакции радикала IO с диметилсульфидом в диапазоне температур 291–365 К: kII(T) = 1.5 × 10–14 [±3.0 × 10–15 см3 молекула–1 с–1] exp(7150[±800 Дж/моль]/RT).
2. Показано, что в экспериментальных условиях, созданных в реакторе, нет дополнительных источников или стоков активных центров, кроме реакций (I) и (II).
3. Путем сравнения расчетных кривых, построенных по формуле (7), и экспериментальных данных доказано, что реакция (II) является гетерогенной.
4. Высказано предположение, что адсорбция одного из реагентов (ДМС) на стенке реактора приводит к уменьшению энергии активации реакции (II) и, как следствие этого, к увеличению скорости реакции по сравнению с гомогенным процессом.
5. Предположено, что уменьшение константы скорости реакции (II) при повышении температуры в реакторе связано с ослаблением прочности связи адсорбированной молекулы с поверхностью реактора, а значит, и уменьшением выигрыша в энергии активации за счет адсорбции.
6. Обсуждена возможная роль гетерогенной реакции (II) в химических процессах, которые возможны на морских аэрозолях, содержащих серную и другие кислоты.
Список литературы
Kloster S., Feichter J., Maier-Reimer E., Six K.D., Stier P., Wetzel P. // Biogeosciences. 2006. V. 3. P. 29.
Andreae M.O. // Mar. Chem. 1990. V. 30. P. 1.
Boucher O., Moulin C., Belviso S., Aumont O., Bopp L., Cosme E., von Kuhlmann R., Lawrence M.G., Pham M., Reddy M.S. // Atmos. Chem. Phys. 2003. V. 3. P. 65.
Breider T.J., Chipperfield M.P., Richards N.A.D., Carslaw K.S., Mann G.W., Spracklen D.V. // Geophys. Res. Lett. 2010. V. 37. L02807.
Charlson R.J., Lovelock J.E., Andreae M.O., Warren S.G. // Nature. 1987. V. 326. P. 655.
Thomas M.A., Suntharalingam P., Pozzoli L., Rast S., Devasthal A., Kloster S., Feichter J., Lenton T.M. // Atmos. Chem. Phys. 2010. V. 10. P. 7425.
Carslaw K.S., Lee L.A., Reddington C.L., Pringle K.J., Rap A., Forster P.M., Mann G.W., Spracklen D.V., Woodhouse T., Regayre L.A., Pierce J.R. // Nature. 2013. V. 503. P. 67.
Chen Q., Sherwen T., Evans M., Alexander B. // Atmos. Chem. Phys. 2018. V. 18. P. 13617.
Abbat P.D., Fenter F.F., Anderson J.G. // J. Phys. Chem. 1992. V. 96. P. 1780.
Baron S.B., Turnipseed A.A., Ravishankara A.R. // J. Phys. Chem. 1996. V. 100. P. 4694.
Williams M.B., Campuzano-Jost P., Bauer D., Hynes A. // J. Phys. Chem. Lett. 2001. V. 344. P. 61.
Du L., Xu Y., Ge M., Jia L., Yao L., Wang W. // Chem. Phys. Lett. 2007. V. 436. P. 36.
O’Dowd C.D., Jimenez J.L., Bahreini R., Flagan R.C., Seinfeld J.H., Hameri K., Pirjola L., Kulmala M., Jennings S.G., Hoffmann T. // Nature. 2002. V. 417. P. 632.
Butkovskaya N.I., Poulet G., LeBras G. // J. Phys. Chem. 1995. V. 99. P. 4536.
Breider T.J., Chipperfield M.P., Richards N.A.D., Carslaw K.S., Mann G.W., Spracklen D.V. // Geophys. Res. Lett. 2010. V. 37. P. L02807.
Knight G.P., Crowley J.N. // Phys. Chem. Chem. Phys. 2001. V. 3. P. 393.
Gravestock T., Blitz M.A., Heard D.E. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 2173.
Dillon T.J., Karunanandan R., Crowley J.N. // Phys. Chem. Chem. Phys. 2006. V. 8. P. 847.
Алоян А.Е., Ермаков А.Н., Арутюнян В.О. // Изв. РАН. Физика атмосферы и океана. 2015. Т. 51. № 3. С. 276.
Алоян А.Е., Ермаков А.Н., Арутюнян В.О. // Изв. РАН. Физика атмосферы и океана. 2016. Т. 52. № 4. С. 402.
Barrie L.A., Bottenheim J.W., Hart W.R. // J. Geophys. Res. 1994. V. 99. P. 25 313.
Huang R.J., Seitz K., Neary T., O’Dowd C.D., Platt U., Hoffmann T. // Geophys. Res. Lett. 2010. V. 37. P. L03803.
Vaughan S., Ingham T., Whalley L.K., Stone D., Evans M.J., Read K.A., Lee J.D., Moller S. J., Carpenter L.J., Lewis A.C., Fleming Z.L., Heard D.E. // Atmos. Chem. Phys. 2012. V. 12. P. 2149.
Ларин И.К., Мессинева Н.А., Спасский А.И., Трофимова Е.М., Туркин Л.Е. // Кинетика и катализ. 2000. Т. 41. С. 485. (Larin I.K., Messineva N.A., Spasskii A.I., Trofimova E.M., Turin L.E. // Kinet. Catal. 2000. V. 41. P. 437.)
Кикоин И.К. Таблицы физических величин. Справ. М.: Атомиздат, 1976. С. 200. (Kikoin I.K. Tablitsy fizicheskikh velichin. Sprav. M.: Atomizdat, 1976. P. 200)
Atkinson R., Baulch D.L., Cox R.A., Crowley J.N., Hampson R.F., Hynes R.G., Jenkin M.E., Rossi M.J., Troe J. // Atmos. Chem. Phys. 2007. V. 7. P. 981.
Бубен С.Н., Ларин И.К., Мессинева Н.А., Трофимова Е.М. // Хим. физика. 1990. Т. 9. С. 116. (Buben S.N., Larin I.K., Messineva N.A., Trofimova E.M. // Chim. Fisika. 1990. V. 9. P. 116.)
McAllen D.T., Gullum T.V., Dean R.A., Fidler F.A. // J. Am. Chem. Soc. 1951. V. 73. P. 3626.
Vinogradov P.S., Borisenko D.M., Hansel A., Taucher J., Ferguson E.E., Lindinger W. // Int. J. Mass Spectrom. 2005. V. 243. P. 127.
Ларин И.К., Невожай Д.В., Спасский А.И., Трофимова Е.М., Туркин Л.Е. // Кинетика и катализ. 1999. Т. 40. С. 487. (Larin I.K., Nevozhai D.V., Spasskii A.I., Trofimova E.M., Turin L.E. // Kinet. Catal. 1999. V. 40. P. 487.)
Atkinson R., Baulch D.L., Cox R.A., Crowley J.N., Hampson R.F., Hynes R.G., Jenkin M.E., Rossi M.J., Troe J. // Atmos. Chem. Phys. 2007. V. 7. P. 984.
Atkinson, R., Baulch D.L., Cox R.A., Crowley J.N., Hampson R.F., Hynes R.G., Jenkin M.E., Ross M.J., Troe J. // Atmos. Chem. Phys. 2004. V. 4. P. 1461.
Fuller E.M., Ensue K., Giddins J.Q. // J. Phys. Chem. 1969. V. 73. P. 3679.
Martin D., Jourdain J.L., Laverdet G., Lebras G. // Int. J. Chem. Kinet. 1987. V. 19. P. 503.
Barnes I., Becker K.H., Carlier P., Mouvier G. // Int. J. Chem. Kinet. 1987. V. 19. P. 489.
Daykin E.P., Wine P.H. // J. Geophys. Res. 1990. V. 95. P. 18547.
Maguin F., Mellouki A., Laverdet G., Poulet G., LeBras G. // Int. J. Chem. Kinet. 1991. V. 23. P. 237.
Barnes I., Bastian V., Becker K.H., Overath R.D. // Int. J. Chem. Kinet. 1991. V. 23. P. 579.
Gravestock T., Blitz M.A., Heard D.E. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 2173.
Dillon T.J., Karunanandan R., Crowley J.N. // Phys. Chem. Chem. Phys. 2006. V. 8. P. 847.
Sayin H., McKee M.L. // J. Phys. Chem. A. 2004. V. 108. P. 7613.
Nakano J., Enamy S., Nakamichi S., Aloisio S., Hashimoto S., Kawasaki M. // J. Phys. Chem. 2003. V. 107. P. 6381.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ