

Кинетика и катализ, 2021, T. 62, № 2, стр. 245-253
Причины антибатной зависимости частоты оборотов гидрирования ненасыщенных соединений от концентрации палладиевого катализатора
Н. И. Скрипов a, Л. Б. Белых a, *, Т. П. Стеренчук a, А. С. Левченко a, Ф. К. Шмидт a
a ФГБОУ ВО Иркутский государственный университет
664003 Иркутск, ул. Карла Маркса, 1, Россия
* E-mail: belykh@chem.isu.ru
Поступила в редакцию 02.07.2020
После доработки 15.10.2020
Принята к публикации 09.11.2020
Аннотация
Совокупностью кинетических методов и электронной микроскопии дискриминированы гипотезы о причинах антибатной зависимости частоты оборотов гидрирования ненасыщенных соединений (алкин, алкинол, олефин) от концентрации катализатора: диссоциация поликристаллических Pd–P-частиц, смещение равновесия “стабилизированный кластер – кластер + стабилизатор” и агрегирование–дезагрегирование Pd–P-частиц. Установлено, что основной причиной влияния концентрации катализатора на кажущуюся частоту оборотов в диапазоне 0.125–1 ммоль/л является агрегирование–дезагрегирование Pd–P-частиц в растворе. В зависимости от природы субстрата действие этого фактора на активность различно. На примере гидрирования стирола показано соответствие предложенной кинетической модели экспериментальным данным и определены константы скоростей отдельных стадий.
ВВЕДЕНИЕ
Катализаторы на основе палладия находят широкое применение в различных областях катализа, в том числе и в промышленно важных процессах – селективном гидрировании алкинов до алкенов [1–4], алкинолов до аллиловых спиртов [5–7], предотвращая их полное гидрирование до алканов или алканолов соответственно. Повышение селективности палладиевых катализаторов в хемоселективном гидрировании ацетиленовых соединений достигается различными способами. К ним относятся промотирование палладия элементами, образующими с ним сплавы (Ag, Ni, Co, Ti, Si [1, 4, 6]), добавление органических или неорганических веществ, конкурирующих с субстратом за активный центр [3] или повышение электронной плотности на Pd, облегчающее десорбцию алкенов [1], например, в результате “сильного взаимодействия металл–носитель” [8]. Рост селективности в этих случаях обычно сопровождается снижением каталитической активности. В связи с новыми стандартами экологической безопасности ставится задача создания высокоселективных и более экологически чистых катализаторов, которые отвечают стратегии устойчивого развития и характеризуются разумным соотношением активности и селективности [9].
Ранее нами было показано, что применение фосфорного модификатора позволяет получать высокоэффективные в гидрировании алкинолов палладиевые катализаторы [7]. Pd–P-частицы, сохраняя высокую селективность при 96–98% конверсии субстрата, по активности в гидрировании 2-метил-3-бутин-2-ола (TOF = 2380 мин–1) превосходили не только близкие по размерам нанокластеры палладия в форме куба (TOF = 918 мин–1), октаэдра (TOF = 1050 мин–1) и кубооктаэдра (TOF = = 876 мин–1) [9], но и системы циглеровского типа (TOF = 794 мин–1) [7]. При выборе оптимальной концентрации катализатора было установлено, что частота оборотов растет с ее уменьшением [7]. Антибатный характер зависимости между кажущейся частотой оборотов и концентрацией прекурсора ранее был описан в гидрировании алкенов под действием циглеровских систем [10, 11] и в реакции Хека [12].
Нелинейный вид зависимости скорости реакции от концентрации катализатора чаще всего связывают с ассоциацией активных комплексов переходных металлов [13] или агрегированием частиц активного компонента [11, 14]. Однако причины так называемого “гомеопатического” катализа могут быть иными [10]. В частности, авторы [12] объясняли антибатную зависимость в области низких концентраций прекурсора образованием более реакционноспособных в реакции С–С-сочетания малых кластеров Pd с незавершенной внешней оболочкой. Смещение равновесия “стабилизированный кластер – кластер + стабилизатор” при уменьшении концентрации прекурсора, в соответствии с правилом Ле-Шателье–Брауна, в сторону десорбции стабилизатора с поверхности кластера [10] или гидролиз молекул AlR3, выполняющих роль стабилизатора и каталитического яда, следами воды в растворителе [11], приводят к росту числа активных центров, и, как следствие этого, антибатной зависимости. Экспериментальное обоснование причин антибатной зависимости между кажущейся частотой оборотов и концентрацией катализатора на основе коллоидных растворов Pd–P-частиц в гидрировании ненасыщенных соединений представлено в настоящей работе.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Растворители (бензол, N,N-диметилформамид) очищали по стандартным методикам [15]. N,N-диметилформамид (ДМФА) выдерживали над безводным сульфатом меди до получения раствора зеленого цвета с целью обезвоживания и удаления примесей аминов и дважды подвергали вакуумной разгонке (8 мм рт. ст.) при температуре не выше 42°С [15].
Для более глубокой осушки бензол дополнительно перегоняли над LiAlH4 с использованием ректификационной колонки. Растворители хранили в атмосфере аргона в запаянных ампулах над молекулярными ситами 4А.
Белый фосфор непосредственно перед применением механически очищали от поверхностных продуктов окисления и промывали в безводном бензоле. Раствор белого фосфора в бензоле готовили и хранили в инертной атмосфере в сосуде Шленка типа “палец”, предварительно вакуумированном и заполненным аргоном.
Бис-(ацетилацетонат) палладия Pd(acac)2 получали согласно методике, приведенной в патенте [16]. Образующийся осадок желтого цвета перекристаллизовывали из ацетона.
2-Метил-3-бутин-2-ол (MBY) фирмы “Sigma–Aldrich” (содержание более 98%) использовали без дополнительной очистки.
Фенилацетилен (PA) выдерживали в течение 2 недель над предварительно прокаленным CaCl2 и перегоняли в вакууме (40°С/10 мм рт. ст.) [15].
Стирол очищали путем встряхивания его с 5%-ным раствором щелочи до тех пор, пока порция щелочи не становилась бесцветной. Промывали дистиллированной водой, сушили над безводным хлоридом кальция и перегоняли в вакууме (32°С/ 10 мм рт. ст.) [15].
Примеры проведения экспериментов
Гидрирование ненасыщенных соединений проводили в стеклянном термостатируемом сосуде типа “утка” при 30°C и начальном давлении водорода 2 атм в присутствии коллоидного раствора Pd–P-частиц, который получали следующим образом. К раствору Pd(аcac)2 (0.00608 г, 2 × 10–5 моль) в 19 мл ДМФА, помещенному в термостатируемый сосуд “утка”, в потоке водорода добавляли по каплям 1 мл раствора фосфора в бензоле (0.6 × 10–5 моль в расчете на атомную форму фосфора) и перемешивали в течение 7 мин при комнатной температуре. Затем температуру поднимали до 80°C и перемешивали реакционную смесь в водороде в течение 40–45 мин до количественного превращения Pd(аcac)2 при давлении 2 атм. Восстановление Pd(аcac)2 контролировали методом УФ-спектроскопии. Коллоидный раствор черно-коричневого цвета охлаждали до 30°C. Для исследования свойств Pd–P-частиц в гидрировании ненасыщенных соединений при концентрациях катализатора 0.125, 0.25, 0.5 ммоль л–1 проводили разбавление предварительно сформированной каталитической системы $\left( {{{C}_{{{\text{Pd(acac}}{{{\text{)}}}_{{\text{2}}}}}}} = {\text{1}}\,\,{{{\text{ммоль}}} \mathord{\left/ {\vphantom {{{\text{ммоль}}} {\text{л}}}} \right. \kern-0em} {\text{л}}}} \right){\text{.}}$ Для осуществления гидрирования создавали давление водорода 2 атм. Шприцем через тефлоновую пробку с резиновой прокладкой вводили субстрат – 2-метил-3-бутин-2-ол, фенилацетилен или стирол. Гидрирование вели при интенсивном перемешивании, исключающем протекание реакции в диффузионной области, периодически отбирая аликвоты реакционной смеси для ГЖХ-анализа.
Гидрирование ацетиленовых соединений протекает в две стадии: на первой стадии преимущественно гидрируется тройная связь, на второй стадии реакции образующийся 2-метил-3-бутен-2-ол (MBE) (при гидрировании MBY) или стирол (при гидрировании фенилацетилена) восстанавливаются до насыщенного спирта или этилбензола соответственно. Количество продукта полного гидрирования (2-метилбутан-2-ола или этилбензола), образующегося на первой стадии гидрирования, не превышало 4–5%. Учитывая этот факт, скорость реакции r (моль H2/мин) на каждой стадии определяли из кинетической кривой следующим образом. Скорость гидрирования MBY (или PA) до 2-метил-3-бутен-2-ола (или стирола) рассчитывали по тангенсу угла наклона прямых участков кинетических кривых в диапазоне поглощения водорода 0.1–0.3 моль H2 на моль MBY (PA). Скорость гидрирования MBE (или стирола) до насыщенного спирта (или этилбензола) вычисляли по тангенсу угла наклона прямых участков кинетических кривых в диапазоне поглощения водорода 1.1–1.3 моль H2 на моль MBY (PA).
Расчет частоты оборотов (TOF) Pd–P-частиц проводили по формулам (1)–(3):
где А – каталитическая активность в расчете на весь палладий (мольсубстрата/(мольPd общий мин)); DПЭМ – дисперсность, определенная из данных просвечивающей электронной микроскопии. где MPd и APd – атомная масса Pd (г/моль) и площадь поверхности атома Pd (м2Pd поверх/атомPd поверх) соответственно, ρ – плотность палладия, NA – число Авогадро, dПЭМ – среднеповерхностный диаметр частиц. где ni – число частиц с диаметром di.Методы исследования
УФ-спектры растворов на стадии формирования катализатора снимали на спектрофотометре СФ-2000 (Россия) в кварцевых кюветах с толщиной поглощающего слоя 0.1 см. Контроль за превращением Pd(аcac)2 проводили по полосе поглощения 330 нм (ε330 = 10 630 л см–1 моль–1).
ГЖХ-анализ продуктов гидрирования выполняли на хроматографе Кристалл 5000 (“Хроматэк”, Россия), снабженном капиллярной колонкой длиной 30 м (фаза – 5% дифенил, 95% диметилполисилфениленсилоксан) и пламенно-ионизационным детектором (ДИП) по методу внутреннего стандарта, используя температурное программирование: 100°C (1.5 мин), 270°C (10 мин), скорость нагрева 30°/мин. Давление газа-носителя (азот) – 20 кПа.
Снимки ПЭМ получали на электронном микроскопе LEO 906E (“Сarl Zeiss”, Германия) с ускоряющим напряжением 120 кВ. Каплю полученного реакционного раствора наносили на науглероженную медную сеточку, высушивали в боксе в инертной атмосфере при комнатной температуре. Для нахождения среднего размера обрабатывали участок, содержащий не менее 150–200 частиц.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
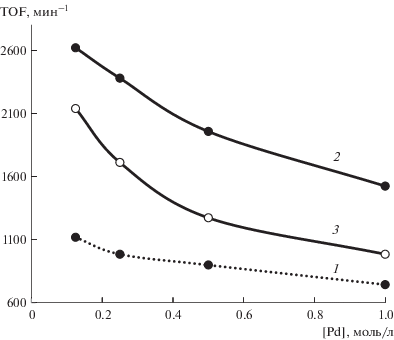
Зависимость частоты оборотов от концентрации Pd–P-частиц изучена на примере гидрирования трех модельных субстратов: фенилацетилена, 2-метил-3-бутин-2-ола и стирола. При разбавлении коллоидного раствора катализатора от 1 до 0.125 ммоль/л частота оборотов (TOF) в гидрировании фенилацетилена возрастала в 1.5 раза (рис. 1, кривая 1), а в гидрировании 2-метил-3-бутин-2-ола – в 1.7 раза (рис. 1, кривая 2). Наибольшие изменения частоты оборотов от концентрации Pd–P-катализатора для рассматриваемых субстратов наблюдался в гидрировании стирола (рис. 1, кривая 3) – рост TOF в этом же диапазоне концентраций Pd–P-частиц (0.125–1 ммоль/л) достигал двукратной величины (рис. 1, кривая 3). Изменение TOF выходит за рамки ошибки экспериментов, относительное отклонение которых не превышает 5–6%.
Рис. 1.
Зависимость частоты оборотов гидрирования фенилацетилена (1), 2-метил-3-бутин-2-ола (2) и стирола (3) от концентрации палладий-фосфорного катализатора. Условия реакции: Р : Pd = 0.3, [PA] = 0.68 моль/л, [MBY] = 0.65 моль/л, [стирол] = 0.79 моль/л, Т = 30°С, Р(Н2) = 2 атм, растворитель – ДМФА, 10 мл.
Исходя из основных положений теории о гетерогенно-каталитических процессах, истинная частота оборотов, рассчитанная на долю активных центров, должна оставаться постоянной величиной и не зависеть от концентрации катализатора [17, 18]. Причины так называемого “гомеопатического” катализа могут быть различны [10]. Для небольшого числа процессов, объединенных в группу размерно-чувствительных реакций, к которым относится гидрирование терминальных алкинов [19, 20] и алкинолов [9], наблюдается зависимость частоты оборотов, рассчитанной на все поверхностные атомы, от размера частиц активного компонента. Но в рассматриваемом случае антибатная зависимость частоты оборотов от концентрации катализатора не связана с размерным эффектом. Во-первых, для гидрирования терминальных алкинов характерен отрицательный размерный эффект [3]. Во-вторых, гидрирование стирола не относится к размерно-чувствительным процессам [21].
При протекании реакции в кинетической области рост частоты оборотов реакции с уменьшением концентрации катализатора указывает на увеличение числа активных центров или на формирование более реакционноспособных частиц. Для экспериментального обоснования зависимости частоты оборотов реакции от концентрации Pd–P-частиц методом ПЭМ были определены размеры Pd–P-частиц при различных концентрациях (рис. 2).
Рис. 2.
ПЭМ-снимки Pd–P-частиц (P : Pd = 0.3), полученных при концентрации Pd(acac)2 равной 1 (а), 0.5 (б), 0.25 (в) и 0.125 (г) ммоль/л.

Средний диаметр Pd–P-частиц составляет 5.9 ± ± 1.1 нм ([Pd] = 1 ммоль/л), 6.23 ± 1.12 нм ([Pd] = = 0.5 ммоль/л), 6.17 ± 1.16 нм ([Pd] = 0.25 ммоль/л) и 6.29 ± 0.86 нм ([Pd] = 0.125 ммоль/л). Близкие значения среднего размера Pd–P-частиц в диапазоне концентраций от 1 до 0.125 ммоль/л позволяют исключить из рассмотрения диссоциацию поликристаллических Pd–P-частиц на более мелкие фрагменты или переход палладия с поверхности частиц в раствор при разбавлении катализатора растворителем (ДМФА).
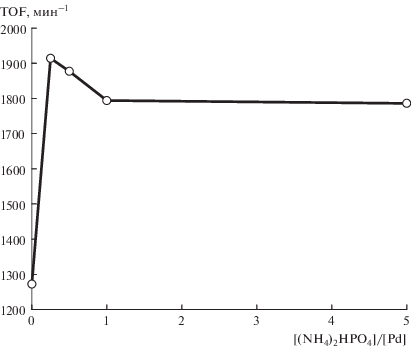
Ранее нами было показано [22], что Pd–P-наночастицы стабилизированы аммонийными солями фосфорной или фосфористой кислот, которые образуются на стадии формирования катализатора. В соответствии с теорией ДЛФО (теория Дерягина, Ландау, Фервея, Овербека) [23] в качестве потенциалопределяющих ионов выступают анионы фосфорной и фосфористой кислот, а противоионами являются катионы диметиламмония. Поэтому из общего числа причин антибатной зависимости нельзя исключать увеличение числа активных центров при разбавлении раствора катализатора в результате смещения равновесия “катализатор–стабилизатор” в соответствии с правилом Ле-Шателье–Брауна. В случае сильного стабилизатора его конкуренция с субстратом за активный центр должна ингибировать протекание реакции гидрирования с ростом концентрации стабилизатора. Для проверки данной гипотезы проведены каталитические эксперименты на примере гидрирования стирола в присутствии различных количеств (NH4)2HPO4. Оказалось, что введение небольших количеств гидрофосфата аммония не только не уменьшает, но наоборот, увеличивает частоту оборотов реакции гидрирования стирола (рис. 3). В то же время в присутствии эквимольных количеств хлорида аммония наблюдалось практически полное подавление каталитической активности. Следовательно, анионы фосфорных кислот, в отличие от хлорид-аниона, не относятся к сильным стабилизаторам (каталитическим ядам) Pd–P-наночастиц. Этот вывод согласуется с результатами авторов [23], согласно которым по стабилизирующей способности нанокластеров иридия анионы располагаются в следующий ряд:
Рис. 3.
Влияние добавок (NH4)2HPO4 на частоту оборотов гидрирования стирола в присутствии Pd–P-частиц (Р : Pd = 0.3). Условия реакции: [Pd] = 0.5 ммоль/л, [стирол] = 0.79 моль/л, Т = 30°С; Р(Н2) = 2 атм, растворитель – ДМФА.
Таким образом, смещение равновесия “стабилизированный кластер – кластер + стабилизатор” в соответствии с правилом Ле-Шателье–Брауна можно исключить из числа основных рассматриваемых причин роста кажущейся частоты оборотов при уменьшении концентрации коллоидного раствора Pd–P-частиц. Наблюдаемые на ПЭМ-снимках рыхлые ветвистые структуры, образующиеся в результате агрегирования Pd–P-частиц, также согласуются со слабой стабилизирующей способностью гидрофосфат-анионов (рис. 2). Однако нельзя однозначно сказать, когда образовались эти структуры: в растворе при формировании Pd–P-частиц или на стадии пробоподготовки (высушивания) к анализу методом электронной микроскопии. Поэтому влияние ассоциации Pd–P-частиц в растворе с ростом их концентрации на частоту оборотов гидрирования ненасыщенных соединений было рассмотрено с использованием кинетического подхода на примере следующей кинетической модели:
Ранее такая кинетическая модель была предложена для гидрирования фенилацетилена под действием полиядерных комплексов палладия с фосфидными и фосфиниденовыми лигандами [24]. Возможность описания гидрирования MBY под действием Pd–P-частиц в рамках механизма Ленгмюра–Хиншельвуда показана в работе [7].
Скорость реакции (r) и общая концентрация палладия (Pd0) в каталитической системе могут быть представлены уравнениями (4) и (5) соответственно:
Исходя из принципа квазиравновесия и квазистационарных концентраций, концентрации палладия в виде ассоциатов [Pdn], в свободном виде [Pd] и промежуточного соединения [Pd(S)] будут равны:
(7)
$[Pd(S)] = \frac{{{{k}_{2}}[Pd][S]}}{{{{k}_{{ - 2}}} + k[{{H}_{2}}]}} = \frac{r}{{k[{{H}_{2}}]}},$(9)
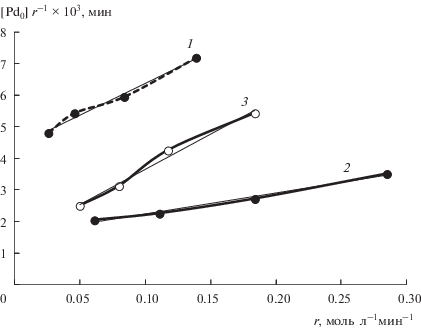
$\begin{gathered} \frac{{[P{{d}_{0}}]}}{r} = \frac{{({{k}_{{ - 2}}} + k[{{H}_{2}}]) + {{k}_{2}}[S]}}{{{{k}_{2}}k[S][{{H}_{2}}]}} + \\ + \,\,n{{K}_{1}}{{\left( {\frac{{{{k}_{{ - 2}}} + k[{{H}_{2}}]}}{{{{k}_{2}}k[S][{{H}_{2}}]}}} \right)}^{n}}{{r}^{{n - 1}}}. \\ \end{gathered} $Для проверки гипотезы – проявление антибатной зависимости между частотой оборотов и концентрацией катализатора как результата дезагрегирования Pd–P-частиц – были построены зависимости $\frac{{[P{{d}_{0}}]}}{r} = f\left( {{{r}^{{n - 1}}}} \right){\text{.}}$ Зависимости $\frac{{[P{{d}_{0}}]}}{r} = f\left( {{{r}^{{n - 1}}}} \right)$ для всех трех субстратов (стирол, фенилацетилен, 2-метил-3-бутин-2-ол) линеаризуются при n = = 2 в диапазоне концентраций катализатора 0.125–1 ммоль/л c коэффициентами корреляции, равными 0.9939, 0.9874 и 0.9884 соответственно (рис. 4).
Рис. 4.
Линеаризация зависимости $\frac{{[P{{d}_{0}}]}}{r} = f\left( {{{r}^{{n - 1}}}} \right)$ в случае гидрирования фенилацетилена (1), 2-метил-3-бутин-2-ола (2) и стирола (3) при n = 2.
Дополнительная проверка адекватности предложенной кинетической модели проведена на примере гидрирования стирола. На рис. 5 представлены кинетические кривые, полученные при варьировании концентрации стирола, катализатора и давления водорода. Экспериментальные данные линеаризуются в координатах $\frac{{[P{{d}_{0}}]}}{r} = f\left( {{{r}^{{n - 1}}}} \right)$ при n = 2 (табл. 1). Линейная зависимость ${{a}_{{i(S)}}} = f\left( {{1 \mathord{\left/ {\vphantom {1 {[S]}}} \right. \kern-0em} {[S]}}} \right)$ и ${{a}_{{i({{H}_{2}})}}} = f\left( {{1 \mathord{\left/ {\vphantom {1 {[{{H}_{2}}]}}} \right. \kern-0em} {[{{H}_{2}}]}}} \right)$ свидетельствует о формальном соответствии предложенной схемы наблюдаемым кинетическим закономерностям и позволяет определить константы скоростей отдельных стадий (рис. 6). Из значения отрезка, отсекаемого прямой ${{a}_{{i(S)}}} = f({1 \mathord{\left/ {\vphantom {1 {[S]}}} \right. \kern-0em} {[S]}})$ на оси ординат, рассчитана константа скорости третьей стадии (k), которая составила 1.75 × 105 л моль–1 мин–1:
Рис. 5.
Зависимость скорости гидрирования стирола в присутствии Pd–P-частиц от концентрации стирола (а) и водорода (б). Условия реакции: Т = 30°С; Р(Н2) = 2 атм (а); Т = 30°С; [S] = 0.79 моль/л (б). [Pd] = 0.125 (1), 0.25 (2), 0.5 (3) и 1.0 (4) ммоль/л; растворитель – ДМФА, 10 мл.
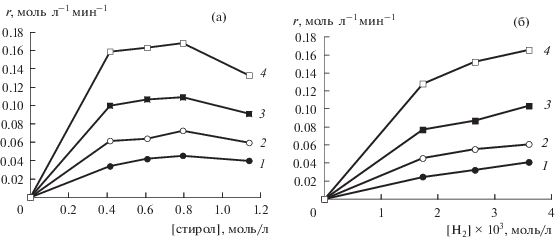
Таблица 1.
Параметры прямой а и b уравнения (9)
| Условия экспериментов | а × 103 | b × 103 | η | |
|---|---|---|---|---|
| Р(Н2), МПа | [стирол], моль/л | |||
| 0.202 | 0.4156 | 2.780 | 29.235 | 0.9924 |
| 0.202 | 0.6089 | 2.4231 | 24.795 | 0.9948 |
| 0.202 | 0.7934 | 2.2327 | 21.144 | 0.9965 |
| 0.202 | 1.138 | 1.9131 | 32.742 | 0.9635 |
| 0.165 | 0.7934 | 3.2718 | 23.026 | 0.9272 |
| 0.125 | 0.7934 | 4.4766 | 23.493 | 0.9996 |
Рис. 6.
Зависимость параметра а прямой $\frac{{[P{{d}_{0}}]}}{r} = f\left( {{{r}^{{n - 1}}}} \right)$ от 1/[S] (a) и 1/[H2] (б). Условия реакции: [H2] = 3.6 ммоль/л (а); [S] = 0.79 моль/л (б).

Отрезок, отсекаемый прямой ${{a}_{{i({{H}_{2}})}}} = f({1 \mathord{\left/ {\vphantom {1 {[{{H}_{2}}]}}} \right. \kern-0em} {[{{H}_{2}}]}})$ на оси ординат, позволяет рассчитать константу скорости второй стадии (k2), а тангенс угла наклона – константу скорости k–2:
Константы скорости k2 и k–2 равны 3.76 × × 103 л моль–1 мин–1 и 6.18 × 102 мин–1 соответственно. Константа равновесия первой стадии K1 (константа агрегирования), рассчитанная из уравнения
Кинетическое описание гидрирования стирола в присутствии каталитической системы на основе Pd–P-частиц подтверждает ассоциацию активных в гидрировании частиц палладия с образованием менее активных в гидрировании форм с ростом концентрации катализатора и согласуется с предложенной причиной антибатной зависимости.
ЗАКЛЮЧЕНИЕ
Таким образом, в отличие от систем циглеровского типа, в которых сокатализатор (AlEt3) выступает в роли восстановителя, стабилизатора и сильного каталитического яда [10, 11, 25], основной причиной зависимости кажущейся частоты оборотов от концентрации катализатора является диссоциация агрегатов Pd–P-частиц при разбавлении в результате адсорбции молекул растворителя и/или субстратов. В зависимости от природы субстрата действие этого фактора на активность различно.
Список литературы
Николаев С.А., Занавескин Л.Н., Смирнов В.В., Аверьянов В.А., Занавескин К.Л. // Успехи химии. 2009. Т. 78. № 3. С. 248. (Nikolaev S.A., Zanaveskin L.N., Smirnov V.V., Averyanov V.A., Zanaveskin K.L. // Russ. Chem. Rev. 2009. V. 78. P. 231.)
Stolarov I.P., Yakushev I.A., Churakov A.V., Cherkashina N.V., Smirnova N.S., Khramov E.V., Zubavichus Y.V., Khrustalev V.N., Markov A.A., Klyagina A.P., Kornev A.B., Martynenko V.M., Gekhman A.E., Vargaftik M.N., Moiseev I.I. // Inorg. Chem. 2018. V. 57. № 18. P. 11 482.
Vile G., Albani D., Almora-Barrios N., Lopez N., Perez-Ramirez J. // ChemCatChem. 2016. V. 8. P. 21.
McCue A.J., Anderson J.A. // Front. Chem. Sci. Eng. 2015. V. 9. № 2. P. 142.
Nikoshvili L., Bykov A., Khudyakova T., LaGrange T., Héroguel F., Luterbacher J.S., Matveeva V. G., Sulman E.M., Dyson P.J., Kiwi-Minsker L. // Ind. Eng. Chem. Res. 2017. V. 56. P. 45.
Johnston S.K., Cherkasov N., Pérez-Barrado E., Aho A., Murzin D.Y., Ibhadon A.O., Francesconi M.G. // Appl. Catal. A: Gen. 2017. V. 544. P. 40.
Скрипов Н.И., Белых Л.Б., Стеренчук Т.П., Гвоздовская К.Л., Жердев В.В., Дашабылова Т.М., Шмидт Ф.К. // Кинетика и катализ. 2020. Т. 61. № 4. С. 526.
Gong T., Huang Yu, Qin L., Zhang W., Li J., Hui L., Feng H. // Appl. Surf. Sci. 2019. V. 495. P. 143 495.
Crespo-Quesada M., Yarulin A., Jin M., Xia Y., Kiwi-Minsker L. // J. Am. Chem. Soc. 2011. V. 133. P. 12 787.
Crooks A.B., Yih K.-H., Li L., Yang J.C., Ozkar S., Finke R.G. // ACS Catal. 2015. V. 5. № 6. P. 3342.
Shmidt F.K., Nindakova L.O., Shainyan B.A., Saraev V.V., Chipanina N.N., Umanetz V.A. // J. Mol. Catal. A: Chem. 2005. V. 235. № 1–2. P. 161.
Schmidt A.F., Smirnov V.V. // Top. Catal. 2005. V. 32. № 1–2. P. 71.
Sanchez-Delgado R.A., Andriollo A., Puga J., Martin G. // Inorg. Chem. 1987. V. 26. № 12. P. 1867.
Widegren J.A., Finke R.G. // J. Mol. Catal. A: Chem. 2003. V. 198. № 1–2. P. 317.
Гордон А., Форд Р. Спутник химика. Москва: Мир, 1976. 572 с. (Gordon A.J., Ford R.A. The Chemist’s Companion; Wiley & Sons: NY, 1972.)
Matthews J.C., Nashua N.H., Wood L.L. USA, Patent 3 474 464, 1969.
Crespo-Quesada M., Yoon S., Jin M., Xia Y., Weidenkaff A., Kiwi-Minsker L. // ChemCatChem. 2014. V. 6. № 3. P. 767.
Markov P.V., Mashkovsky I.S., Bragina G.O., Warna J., Gerasimov E.Yu., Bukhtiyarov V.I., Stakheev A.Yu., Murzin D.Yu. // Chem. Eng. J. 2019. V. 358. P. 520.
Ярулин А.Э., Креспо-Кесада М.P., Егорова Е.В., Киви-Минскер Л.Л. // Кинетика и катализ. 2012. Т. 53. № 2. С. 263. (Yarulin A.E., Crespo-Quesada R.M., Egorova E.V., Kiwi-Minsker L.L. // Kinet. Catal. 2012. V. 53. № 2. P. 253.)
Ruta M., Semagina N., Kiwi-Minsker L. // J. Phys. Chem. C. 2008. V. 112. № 35. P. 13 635.
Hu J., Zhou Z., Zhang R., Li L., Cheng Z. // J. Mol. Catal. A: Chem. 2014. V. 381. P. 61.
Белых Л.Б., Скрипов Н.И., Стеренчук Т.П., Акимов В.В., Таусон В.Л., Шмидт Ф.К. // Журн. общей химии. 2016. Т. 86. № 9. С. 1454. (Belykh L.B., Skripov N.I., Sterenchuk T.P., Akimov V.V., Tauson V.L., Schmidt F.K. // Russ. J. Gen. Chem. 2016. V. 86. № 9. P. 2022.)
Ott L.S., Finke R.G. // Coord. Chem. Rev. 2007. V. 251. № 9–10. P. 1075.
Миронова Л.В., Белых Л.Б., Усова И.В., Шмидт Ф.К. // Кинетика и катализ. 1985. Т. 26. № 2. С. 469. (Mironova L.V., Belykh L.B., Usova I.V., Shmidt F.K. // Kinet. Catal. 1985. V. 26. № 2. P. 413.)
Шмидт Ф.К., Титова Ю.Ю., Белых Л.Б. // Кинетика и катализ. 2015. Т. 56. № 5. С. 582. (Shmidt F.K., Titova Yu.Yu., Belykh L.B. // Kinet. Catal. 2015. V. 56. № 5. P. 574.)
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ