

Кинетика и катализ, 2021, T. 62, № 2, стр. 263-276
Свойства многокомпонентного катализатора MoVSbNbCeOx/SiO2 в реакции окислительного дегидрирования этана в этилен
Г. А. Зенковец a, *, А. А. Шутилов a, В. М. Бондарева a, Л. С. Довлитова a, В. И. Соболев a, А. С. Марчук a, С. В. Цыбуля a, И. П. Просвирин a
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: zenk@catalysis.ru
Поступила в редакцию 30.07.2020
После доработки 25.09.2020
Принята к публикации 10.10.2020
Аннотация
Многокомпонентный оксидный катализатор MoVSbNbCeOx/SiO2, обладающий высокой каталитической активностью в реакции окислительного дегидрирования этана в этилен, приготовлен распылительной сушкой суспензии водных растворов исходных компонентов с последующей термообработкой в Не при 350 и 600°С. При температуре 400–450°С в широком интервале изменения конверсии этана катализатор характеризуется достаточно высокой активностью и селективностью по этилену. Максимальный выход этилена на нем достигает 74% (конверсия этана – 91%, селективность образования этилена – 81.5%), что значительно превышает выход в присутствии известных из литературных источников Sb-содержащих катализаторов. Катализатор длительное время устойчиво работает в условиях реакционной среды без изменения фазового состава и ухудшения каталитических характеристик. Показано, что каталитические свойства полученного катализатора сопоставимы со свойствами одного из лучших в данном процессе MoVTeNbOx-катализаторов. Однако использование Те-содержащего катализатора ограничивается высокой токсичностью и летучестью теллура в процессе приготовления и эксплуатации катализатора. По результатам рентгенофазового анализа основные компоненты катализатора – фазы М1 и М2, стабилизированные на поверхности SiO2. Данные ПЭМВР показывают, что структурной особенностью катализатора является наличие межфазной границы, образованной когерентно сросшимися между собой кристаллами фаз M1 и M2.
ВВЕДЕНИЕ
Каталитическое дегидрирование этана, один из распространенных промышленных методов производства этилена, является эндотермической и обратимой реакцией, которая достаточно эффективно протекает на Cr2O3/Al2O3 и Pt/Sn/Al2O3-катализаторах. Основная проблема процессов дегидрирования связана с протеканием реакции вблизи равновесия, поэтому даже при высоких температурах выход продукта небольшой. Кроме того, проведение процесса осложняет образование кокса [1–4].
Перспективный метод получения этилена – прямое окислительное дегидрирование этана в этилен (ОДЭ). Процесс осуществляется при достаточно низких температурах (400–500°С) в присутствии кислорода в реакционной смеси. В настоящее время в литературе наиболее подходящими для процесса ОДЭ считаются многокомпонентные катализаторы MoVTe(Sb)NbOx [3–16]. Известно, что Te-содержащие катализаторы MoTeVNbOx проявляют более высокую активность в ОДЭ по сравнению с Sb-содержащими катализаторами MoVSbNbOx. На одних из лучших MoVTeNbОx-катализаторах при температуре реакции 400°С достигается конверсия этана 88–90% при селективности образования этилена 81–87%, что обеспечивает выход этилена 72–76% [3, 5, 6, 9, 10, 13–16].
Несмотря на то, что известные MoVSbNbОx-катализаторы также активны в ОДЭ, они характеризуются более низкой активностью и селективностью по этилену, и выход этилена на них меньше по сравнению с Te-содержащими системами. Так, для одного из лучших катализаторов MoVSbNbCaOx при температуре 400°C максимальный выход этилена составляет 52% при конверсии этана 73% и селективности образования этилена 71% [17]. Несмотря на уже достигнутые результаты, исследования, направленные на улучшение каталитических показателей катализаторов MoVTe(Sb)NbОx в ОДЭ, не прекращаются.
В ряде работ [2, 6] было показано, что активные в парциальном окислении катализаторы MoVNbTe(Sb)Ox в основном содержат две оксидные кристаллические фазы, которые в литературе описаны как орторомбическая (M1) и гексагональная (M2), а также некоторое количество других примесных фаз типа Mo5O14 и бинарных оксидов MoV, SbV и MoTe. Структура фазы М1 характеризуется наличием сложных по мотиву плоских сеток из сопряженных по вершинам или по вершинам и по ребрам полиэдров, в центре которых находятся катионы молибдена и ванадия (MO6) и ниобия (MO7). В самих сетках в зависимости от расположения и вида полиэдров образуются шести- и семиугольные петли, в которых находятся структурные единицы Te–O. Сетки объединяются общими вершинами и, накладываясь трансляционно друг на друга в направлении [001], формируют каналы, проходящие через всю структуру фазы М1. Это обуславливает морфологию иглоподобных кристаллов, в которой плоскости [001] расположены перпендикулярно оси длины иглы. Было высказано предположение, что концевые плоскости [001] фазы M1 содержат наиболее активные и селективные участки поверхности для ОДЭ [6, 9, 12–31].
Кристаллическая структура фазы M2 близко соотносится с таковой для гексагональной бронзы. В катализаторах MoVNbTe(Sb)Ox фаза М2 обычно сосуществует с фазой M1. В ряде литературных источников [18, 32] сообщалось, что в реакции ОДЭ синергетический эффект между фазами M1 и M2 в Sb-содержащих катализаторах не наблюдается, и фаза M2 практически не активна. Утверждается, что чистая Sb-содержащая фаза M1, полученная после удаления фазы M2 путем eе специального растворения в пероксиде водорода, характеризуется более высокой селективностью образования этилена, чем их смесь. Отмечается, что при одних и тех же условиях реакции Tе- и Sb-содержащие чистые фазы M1 имеют практически одинаковую селективность по этилену в широком интервале изменения конверсии этана. В обоих катализаторах при конверсии этана 66% селективность в образовании этилена достигает 92%. Однако при одинаковых времeнах контакта скорость превращения этана на Te-содержащей фазе M1 значительно выше, чем на Sb-содержащей.
Основной недостаток при использовании катализаторов MoVNbTeOx – довольно высокая летучесть Te. Поэтому при их прокаливании в инертной атмосфере при высокой температуре и в условиях каталитической реакции происходит восстановление теллура до металлического состояния и его сублимация из катализатора [2, 33–35]. Эти процессы в свою очередь приводят к разложению фазы M1 с образованием структур типа Mo5O14 или MoO2 и к ухудшению каталитических свойств. Такие особенности катализатора являются существенным ограничением при его масштабировании до промышленного уровня.
В настоящее время в мире постоянно проводятся достаточно интенсивные исследования, направленные на уменьшение сублимации теллура из катализаторов MoVTeNbOx. В работах [36, 37] было показано, что промотирование MoVTeNbOx-катализатора добавками Bi или Ce повышает стабильность фазы М1 в процессе ОДЭ. Методами EXAFS-спектроскопии и РФА in situ продемонстрировано, что в MoVTeNbBiOx-катализаторах висмут стабилизируется в протяженных шестичленных каналах структуры фазы M1, в которых стабилизирован и Те. Включение Bi способствует ограничению подвижности Te, снижая его сублимацию и степень дезактивации катализатора. Введение Bi в состав фазы M1 также приводит к некоторому увеличению активности катализатора [38]. Однако модифицирование катализатора висмутом не полностью исключает сублимацию теллура из катализатора MoVTeNbBiOx.
В настоящее время Sb-содержащие катализаторы MoVNbSbOx рассматриваются как перспективная альтернатива для замены MoVNbTeOx-катализаторов в ОДЭ при сохранении высоких показателей процесса. Однако достоверные литературные сведения о высокой каталитической активности и стабильности MoVNbSbOx-катализаторов в настоящее время отсутствуют.
Целью представленной работы явилась разработка нового оксидного Sb-содержащего катализатора MoVNbSbCeOx/SiO2, характеризующегося высокой каталитической активностью в реакции окислительного дегидрирования этана.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Катализатор, в состав которого входит 50 мас. % Mo1V0.25Sb0.23Nb0.08Ce0.01Ox и 50 мас. % SiO2, приготовлен смешением водных растворов парамолибдата аммония, метаванадата аммония, триоксида сурьмы, нитрата церия, SiO2 и оксалата ниобия при температуре 50–80°C с последующей сушкой полученной суспензии на лабораторной распылительной сушилке Mini Spray Dryer B-290 (“Buchi Labortechnik AG”, Швейцария) при температуре 105°C. Полученный порошок таблетировали и прокаливали в токе Не при температуре 350°C в течение 2 ч, а затем при 600°C также в течение 2 ч [39]. Для приготовления катализатора использовали следующие исходные вещества: парамолибдат аммония (NH4)6Mo7O24 · 4H2O (“Вектон”, Россия), метаванадат аммония NH4VO3 (“Вектон”, Россия), Sb2O3 (“Реахим”, Россия), нитрат церия Ce(NO3)3 6H2O (“Aldrich”, США), пентахлорид ниобия NbCl5 (“Acros Organics”, Бельгия), SiO2 (“Nalco”, США). Оксалат ниобия был приготовлен из пентахлорида ниобия по методике, приведенной в [40, 41]. NbCl5 растворяли в дистиллированной воде, а затем из полученного раствора осаждали гидроксид ниобия 25%-ным водным раствором NH4OH при постоянном рH 7. Полученный осадок отфильтровывали, промывали дистиллированной водой до отсутствия ионов хлора в промывной воде, после чего растворяли в растворе щавелевой кислоты при соотношении ${{{\text{C}}}_{2}}{\text{H}}_{4}^{{2 - }}$ : Nb = 3.
Химический состав прокаленного катализатора определяли методом атомно-эмиссионной спектрометрии с индуктивно-связанной плазмой на спектрометре OPTIMA 4300 DV (“Perkin Elmer”, США).
Изучение пористой структуры катализатора проводили методом низкотемпературной адсорбции азота при 77.4 K на приборе DigiSorb-2600 (“Micromeritics”, США). Катализатор предварительно тренировали в вакууме при 200°C и давлении 10–4 мм рт. ст. в течение 5 ч. Распределение мезопор по размерам и преимущественный размер пор рассчитывали классическим методом Barrett–Joyner–Halenda (BJH).
Рентгенофазовый анализ осуществляли на дифрактометре Bruker D8 (“Bruker”, Германия), монохроматическое CuKα-излучение. Рентгенограммы снимали в режиме сканирования в области 2θ = 5°–70° с шагом 0.02° и накоплением в каждой точке 3 с. Для детектирования сигнала применяли многоканальный детектор LynxEye. Количественный фазовый анализ проводили методом Ритвельда с помощью программного пакета TOPAS v.4.2 [42], используя структурные параметры, приведенные в [3], и базы структурных данных ICSD [43]. Размер областей когерентного рассеяния (ОКР) рассчитывали по уравнению Шеррера.
Исследование методом просвечивающей электронной микроскопии высокого разрешения (ПЭМВР) выполняли на микроскопах JEM-2010 (“JEOL”, Япония, ускоряющее напряжение – 200 кВ, разрешающая способность – 1.4 Å) и JEM-2200FS (“JEOL”, Япония, ускоряющее напряжение – 200 кВ, разрешение по решетке – 1 Å). Образец наносили на медную подложку с помощью ультразвукового диспергатора. Цифровую обработку полученных электронно-микроскопических изображений с расчетом наблюдаемых межплоскостных расстояний по Фурье-анализу области проводили в программе Gatan DigitalMicrograph.
Исследования методом дифференцирующего растворения (ДР) осуществляли в проточном динамическом режиме растворения с использованием стехиографа на атомно-эмиссионном спектрофотометре ICP (“Baird”, США) [44]. Поток раствора, содержащий растворенный образец, с постоянной объемной скоростью (3.6 мл/мин) подавали в детектор-анализатор. По спектральным линиям молибдена (202.0 нм), ванадия (292.4 нм), сурьмы (217.5 нм), ниобия (316.1 нм) и кремния (288.1 нм) каждые 3 с устанавливали элементный состав. Чувствительность определения элементов составляла 0.001 мкг/мл. Погрешность определения стехиометрических коэффициентов в эмпирических формулах и количественного содержания фаз не превышает 5–10 отн. %. Поскольку в методе ДР возможность детектора прибора определять водород и кислород ограничена, получаемые при анализе эмпирические формулы фаз являются фрагментарными, отражающими стехиометрию только в отношении определяемых элементов [45].
Растворение образца катализатора (5 мг) начинали в водном растворе, после в режиме стехиографического титрования переходили к раствору соляной кислоты с концентрацией 1.2 и 3 М, а затем, для полного растворения, к раствору HF (3.6 M). Температуру растворителя повышали в интервале от 22 до 90°С. Постепенное увеличение концентрации растворителя и смена кислот приводит к возрастанию химического потенциала растворителя. Это позволяет проводить селективное растворение отдельных фаз катализатора. Для определения формульного состава растворенных фаз и их количества из полученных кинетических кривых рассчитывали стехиограммы, представляющие собой мольные соотношения скоростей растворения химических элементов от времени растворения [44, 45]. Количественную оценку содержания фаз в их смеси осуществляли по площади под кинетической кривой растворения, величину которой находили численным интегрированием.
Рентгеновские фотоэлектронные спектры (РФЭС) записывали на спектрометре SPECS (Германия) с анализатором PHOIBOS–150-MCD–9 и монохроматором FOCUS–500 (излучение AlKα, hν = 1486.74 эВ, 200 Вт). Шкала энергий связи была предварительно откалибрована с использованием уровней положений пиков Au4f7/2 (84.0 эВ) и Cu2p3/2 (932.67 эВ). Энергию связи калибровали по положению пика C1s (284.8 эВ), соответствующего углеводородным отложениям на поверхности образца [46]. Образец в виде порошка наносили на проводящий двухсторонний медный скотч. Обзорный спектр и индивидуальные спектры элементов регистрировали при энергии пропускания анализатора 20 эВ. Атомные отношения элементов рассчитывали из интегральных интенсивностей фотоэлектронных пиков, которые были скорректированы с помощью соответствующих факторов чувствительности на основе сечений фотоионизации Скофилда [47]. Обработку и анализ спектральной информации выполняли с помощью программного обеспечения XPS Peak 4.1 [48].
Исследование каталитических свойств образцов в реакции окислительного дегидрирования этана проводили в проточной установке с online хроматографическим анализом компонентов реакционной смеси на хроматографе Цвет-500 (Россия) с использованием детектора по теплопроводности (газ-носитель – гелий, поток газа-носителя – 3.2 л/ч, температура детектора – 250°С) и 2-х колонок (длина 2 м, внешний диаметр 4 мм), заполненных Porаpak Q и молекулярными ситами СаА. На первой колонке при комнатной температуре разделяли СО2, этан и этилен, на второй – кислород, азот и СО. Эксперименты осуществляли в неподвижном слое катализатора в трубчатом реакторе с коаксиально расположенным термопарным карманом, при атмосферном давлении, составе исходной реакционной смеси (об. %) C2H6 : O2 : N2 = 10 : 10 : 80 и температуре 400–450°C на фракции катализатора 0.25–0.50 мм. Скорость потока реакционной смеси (U, л/c) варьировали от 8.33 × 10–4 до 4.44 × 10–3 л/c, время контакта – от 0.5 до 15.0 с. Отсутствие гомогенного окисления в указанном температурном интервале подтверждено опытами с пустым реактором.
Конверсию этана (X, %) и селективность (Si, %) образования продуктов реакции (этилена и оксидов углерода) рассчитывали, как в работе [3]:
Выход этилена (В, %) вычисляли по уравнению:
Баланс по углероду во всех проведенных экспериментах составлял 98 ± 2%.
Для сравнения активности катализаторов, скорости превращения этана W1 и образования этилена W2 (моль м–2.с–1) при конверсии этана 10% рассчитывали по формулам:
В дополнение к вышеприведенным данным также были вычислены скорости реакции (W1 и W2), отнесенные к 1 г катализатора и 1 г активного компонента.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
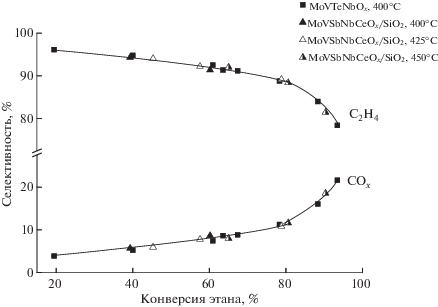
По данным химического анализа состав полученного катализатора соответствует расчетному: 50 мас. % (Mo1V0.25Sb0.23Nb0.08Ce0.01Ox)/50 мас. % SiO2. Как показывают результаты исследования каталитических свойств синтезированного образца MoVSbNbCeOx/SiO2 в реакции ОДЭ, продуктами превращения этана являются только этилен и оксиды углерода CO и CO2. На рис. 1 приведены зависимости селективности образования этилена и оксидов углерода от конверсии этана для приготовленного в настоящей работе MoVSbNbCeOx/SiO2 и, в качестве сравнения, для одного из лучших Те-содержащих катализаторов Mo1V0.3Te0.23Nb0.12Ox [3, 16, 31]. Как видно из рисунка в изученном температурном интервале селективности по этилену и продуктам полного окисления для этих катализаторов одинаковы. Так, при конверсии этана, равной 60%, селективность по этилену в присутствии обоих катализаторов составляет 92%. Следует отметить, что, как и для MoVTeNbOx-катализатора [49], зависимости селективностей по продуктам реакции от конверсии этана на катализаторе MoVSbNbCeOx/ SiO2 не изменяются при варьировании температуры в интервале 400–450°С. На обоих катализаторах, максимальный выход этилена достигает 74% при конверсии этана 91% и селективности по этилену 81.5%.
Рис. 1.
Зависимость селективностей образования этилена (C2H4) и оксидов углерода (COx) от конверсии этана для катализаторов MoVSbNbCeOx/SiO2 (треугольники) и MoVTeNbOx (квадраты) при температурах реакции 400–450°C.
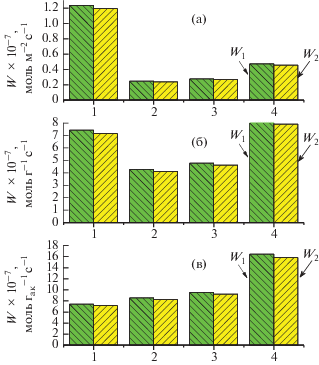
На рис. 2. для исследуемого образца MoVSbNbCeOx/SiO2 приведены зависимости скоростей расходования этана (W1) и образования этилена (W2) при температурах 400, 425 и 450°C. Для сравнения представлены аналогичные характеристики для MoVTeNbOx-катализатора. Видно, что в случае MoVSbNbCeOx/SiO2 скорости W1 и W2, рассчитанные на 1 м2 поверхности катализатора, значительно меньше таковых для MoVTeNbOx-системы даже при температуре 450°C. В то же время скорости W1 и W2, отнесенные к 1 г катализатора, для полученного в настоящей работе Sb-содержащего образца при 450°C даже несколько выше по сравнению с соответствующими показателями для Te-содержащего катализатора при 400°C. В интервале температур 400–450°C скорости общего превращения этана и образования этилена в расчете на 1 г активного компонента на MoVSbNbCeOx/ SiO2-катализаторе выше по сравнению с таковыми на MoVTeNbOx-катализаторе.
Рис. 2.
Скорости расходования этана (W1) и образования этилена (W2) в расчете на 1 м2 поверхности катализатора (а), 1 г катализатора (б) и 1 г активного компонента (в) для катализаторов MoVTeNbOx (1) и MoVSbNbCeOx/SiO2 (2, 3, 4) при конверсии этана 10% и температуре реакции 400 (1, 2), 425 (3) и 450°C (4).
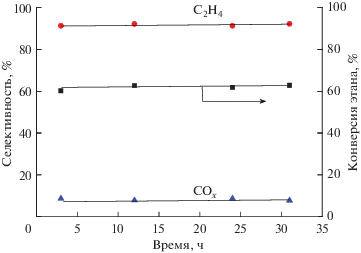
На рис. 3 показаны зависимости конверсии этана и селективностей образования продуктов реакции при 450°C в присутствии катализатора MoVSbNbCeOx/SiO2 при его длительной работе. Видно, что каталитические свойства не ухудшаются в течение 36 ч, что свидетельствует о достаточно хорошей стабильности катализатора в условиях реакционной смеси.
Рис. 3.
Влияние длительности испытания катализатора MoVSbNbCeOx/SiO2 при температуре 400°C на конверсию этана и селективность образования этилена и оксидов углерода.
Из рис. 1 видно, что с увеличением конверсии этана селективность по этилену постепенно снижается, а селективность по СО и СО2 возрастает. Такой вид зависимости позволяет описать процесс окислительного дегидрирования этана на катализаторе MoVSbNbCeOx/SiO2 последовательно-параллельной схемой (схема 1 ) [16, 31, 50]:
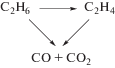
Схема 1 .
Согласно этой схеме этилен и оксиды углерода образуются как непосредственно из этана, так и при доокислении этилена. Экстраполяция кривой зависимости селективности по этилену в точку нулевой конверсии этана позволяет оценить долю этилена, образующегося из этана. На катализаторе MoVSbNbCeOx/SiO2 эта величина составляет 96.0%.
Таким образом, сравнение каталитических свойств Sb-содержащего катализатора 50 мас. % Mo1V0.24Sb0.23Nb0.08Ce0.01Ox/50 мас. % SiO2 со свойствами известного Te-содержащего катализатора Mo1V0.3Te0.23Nb0.12Ox при температурах 400–450°С показывает, что полученный в настоящей работе катализатор характеризуется достаточно высокой каталитической активностью в реакции окислительного дегидрирования этана в этилен.
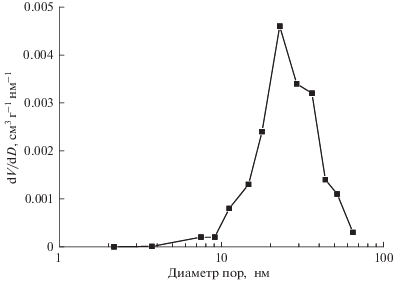
Синтезированный катализатор MoVSbNbCeOx/ SiO2 имеет достаточно развитую пористую структуру. Его удельная поверхность составляет 15.7 м2/г, что существенно больше, чем поверхность MoVTeNbOx-катализатора, величина которой не более 5–6 м2/г [16, 31, 36]. Объем мезопор для MoVSbNbCeOx/SiO2-катализатора равен 0.125 см3/г, катализатор характеризуется довольно широким распределением пор по размерам с максимумом в области 20–30 нм (рис. 4), значительно превышающим таковой для MoVТеNbOx-катализатора [14]. Это может быть обусловлено присутствием в MoVSbNbCeOx/SiO2 достаточно большого количества диоксида кремния.
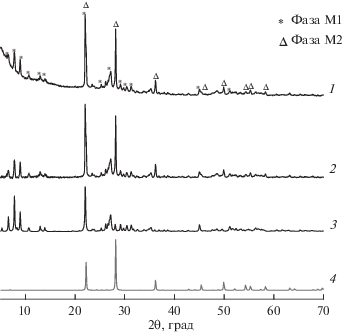
На рис. 5 представлена рентгенограмма свежеприготовленного катализатора MoVSbNbCeOx/ SiO2, прокаленного при температуре 600°С. На рентгенограмме наряду с линиями присутствующих в образце фаз, в области 2θ = 10°–32° четко наблюдается гало SiO2. Поэтому ниже на том же рисунке показана рентгенограмма этого катализатора после вычета гало, а также рентгенограммы фаз М1 и М2, рассчитанные метом Ритвельда из структурных данных, приведенных в [22, 26]. На рентгенограмме исследуемого образца четко виден интенсивный пик в области 2θ = 22.20°, соответствующий фазе M1, и пик в области 2θ = = 36.1°, относящийся к фазе M2, а также другие пики, характерные для этих фаз. Пиков, принадлежащих к иным кристаллическим фазам, на рентгенограмме не регистрируется. Следовательно, синтезированный катализатор MoVSbNbCeOx/SiO2 по фазовому составу представляет собой смесь фаз M1 и М2.
Рис. 5.
Дифрактограммы образцов исходного реального катализатора MoVSbNbCeOx/SiO2 (1), этого же катализатора после вычета фона и гало SiO2 (2) и модельных фаз M1 (3) и M2 (4).
В табл. 1 представлены результаты определения методом Ритвельда содержания фаз M1 и M2 в образце MoVSbNbCeOx/SiO2 и размер их кристаллов (ОКР). Содержание фаз M1 и M2 в активном компоненте равно 71.7 и 28.3% соответственно. Учитывая, что в состав полученного катализатора входит всего 50% активного компонента, а остальная часть представляет собой SiO2, содержание фаз M1 и M2 в катализаторе MoVSbNbCeOx/ SiO2 составляет 35.85 и 14.15% соответственно, что существенно ниже значений этих показателей для массивного MoVTeNbOx-катализатора [16, 31]. Из данных, представленных в табл. 1, следует, что размер ОКР фазы M1 (35.8 нм) в катализаторе MoVSbNbCeOx/SiO2 гораздо меньше, чем таковой для фазы M2 (66.5 нм). Это может быть связано непосредственно с присутствием SiO2 в катализаторе. По-видимому, SiO2 уменьшает агломерацию и спекание кристаллов фазы M1 при термообработке в большей степени, чем фазы M2.
Таблица 1.
Структурные параметры фаз M1 и M2 исходного катализатора и катализатора после работы в реакционной смеси
| Параметры | Катализатор | |||
|---|---|---|---|---|
| исходный | после реакции | |||
| М1 | М2 | М1 | М2 | |
| Пространственная группа | Pba2 | Pmm2 | Pba2 | Pmm2 |
| Параметры решетки, Å | a = 21.1026 b = 26.5995 c = 4.0191 |
a = 12.6736 b = 7.2942 c = 3.9900 |
a = 21.1298 b = 26.6350 c = 4.0013 |
a = 12.6056 b = 7.3096 c = 4.0250 |
| Содержание фазы, % | 71.7 | 28.3 | 80.4 | 19.8 |
| Размер кристалла (ОКР), нм | 35.8 | 66.6 | 29.4 | 70.3 |
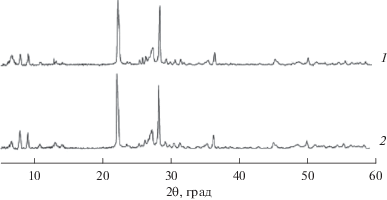
На рис. 6 приведено сравнение рентгенограмм исходного катализатора и катализатора после длительной работы в реакционной смеси. Видно, что рентгенограммы практически идентичны, новых кристаллических фаз не появляется. В то же время согласно данным табл. 1 содержание фазы М1 повышается до 80%, а фазы М2 снижается. После длительных каталитических испытаний в фазе М1 параметры кристаллической решетки a и b несколько увеличиваются, а параметр c уменьшается; для фазы М2 наблюдается незначительное уменьшение параметра a и рост параметров b и c. Размеры ОКР М1 и M2 фаз под воздействием реакционной смеси значительно не изменяются. Из сопоставления полученных данных следует, что реакционная среда не оказывает существенного влияния на фазовый состав катализатора: основные кристаллические фазы М1 и М2 сохраняются, происходят лишь некоторые изменения параметров кристаллических ячеек, что требует дальнейшего более глубокого рентгенографического исследования их структуры.
Рис. 6.
Дифрактограммы исходного катализатора MoVSbNbCeOx/SiO2 (1) и этого же катализатора после длительной работы в реакционной смеси (2).
Для уточнения фазового состава катализатора и определения химического состава фаз исходный катализатор был исследован методом ДР. На рис. 7а представлены кинетические кривые растворения Mo, V, Sb и Nb и стехиограммы V/Mo, Sb/Mo, Nb/Mo от времени растворения пробы. Видно, что на стехиограмме V/Mo присутствуют два линейных участка, переходящих один в другой, с постоянными мольными отношениями V/Mo = = 0.29 ± 0.01 и V/Mo = 0.24 ± 0.02. Эти данные свидетельствуют о наличие в катализаторе двух фаз, содержащих фрагменты V0.29Mo1 и V0.24Mo1. В области этих участков регистрируется еще два линейных участка с постоянными мольными отношениями Sb/Mo = 0.31 ± 0.02 и Sb/Mo = 0.09 ± ± 0.02. Это позволяет добавить к формулам фрагментов V0.29Mo1 и V0.24Mo1 сурьму в соответствующих количествах: Mo1V0.29Sb0.31 и Mo1V0.24Sb0.09. Стехиограмма Nb/Mo в интервале времени растворения 47–70 мин (рис. 7а) имеет линейный участок, характеризующийся мольным соотношением Nb/Mo = 0.11 ± 0.01, что доказывает наличие в составе фрагмента Mo1V0.24Sb0.09 соответствующего количества ниобия – Mo1V0.24Sb0.09Nb0.11. На рис. 7б показано растворение обнаруженных в катализаторе фаз постоянного состава: Mo1V0.29Sb0.31, обозначенной как Ф1, и Mo1V0.24Sb0.09Nb0.11, обозначенной как Ф2. Видно, что растворение фазы Ф1 происходит одновременно с кремнием в соляной кислоте, а Ф2 фаза растворяется одновременно с кремнием в НF. Это может говорить о стабилизации фаз Ф1 и Ф2 на поверхности диоксида кремния.
Рис. 7.
Кинетические кривые растворения Mo, V, Sb, Nb и стехиограммы V/Mo, Sb/Mo, Nb/Mo (a); кинетические кривые растворения Mo1Sb0.49, фаз Ф1 (Mo1V0.29Sb0.31) и Ф2 (Mo1V0.24Sb0.09Nb0.11) и соединений Sb и Si (б).
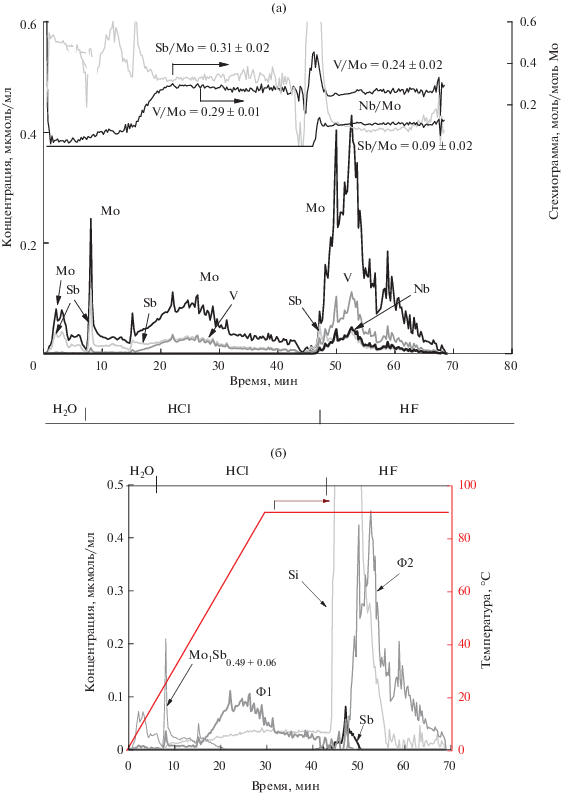
Кроме того, анализ полученных результатов свидетельствует, что после вычитания сурьмы, принадлежащей фазам Ф1 и Ф2, на кинетической кривой (рис. 7б) остаются еще две формы сурьмы. Одна форма (1.8 отн. %) растворяется в HF и, вероятнее всего, относится к оксиду сурьмы, другая растворяется в соляной кислоте вместе с молибденом. Стехиограмма Sb/Mo переменна во времени (Sb/Mo = 0.49 ± 0.06), что в соответствии с принципами ДР свидетельствует об отсутствии стехиометрического соединения между Mo и Sb. Тот факт, что оба эти элемента начинают и заканчивают растворение одновременно, позволяет предположить присутствие пространственно неоднородной фазы (7.4 отн. %) переменного состава Mo1Sb0.49 ± 0.06.
К сожалению, содержание церия в катализаторе определить не удалось. Церий и ниобий анализируются в монохроматоре поочередно, и сложность связана с выбором подходящей спектральной линии церия, свободной от спектральных помех, создаваемых ниобием. Чувствительные линии церия, в основном, располагаются в области спектра 340–440 нм, которая также сильно насыщена и линиями ниобия, поэтому можно ожидать спектральное наложение линий Nb на линию Се, которая может быть использована при его определении.
Таким образом, по данным ДР в катализаторе обнаруживается присутствие двух основных фаз Ф1 и Ф2. Сопоставление полученных результатов с данными РФА и литературными сведениями позволяет отнести фазу Ф1 к фазе М2, а фазу Ф2 – к фазе M1. Следовательно, химический состав фазы M1 (без учета содержания Ce) соответствует Mo1V0.24Sb0.09Nb0.11, а состав фазы M2 – Mo1V0.29Sb0.31. Соединения Mo1Sb0.49 ± 0.06 и небольшое количество оксида Sb рентгенографически не определяются.
Анализ обзорного РФЭ-спектра Sb-содержащего катализатора MoVSbNbCeOx/SiO2 показал, что поверхностный состав образца соответствует заданному, никаких дополнительных химических элементов не обнаружено. Для уточнения химического состояния элементов в катализаторе и их поверхностных концентраций были проанализированы узкие спектральные регионы элементов поверхности исходного образца и образца после реакции. В исходном катализаторе (рис. 8а) в спектре Mo3d значение энергии связи Mo3d5/2 составляет 232.8 ± 0.1 эВ, что соответствует состоянию Mo6+ [51, 52]. В спектре V2p пик V2p3/2 описывается двумя компонентами с энергиями связи 516.2 ± 0.1 и 517.2 ± 0.1 эВ, которые относятся к состояниям V4+ и V5+ [53–55]. Проведенные оценки показывают, что соотношение V5+/V4+ на поверхности исходного катализатора равно ~0.6. В спектре Nb3d энергия связи пика Nb3d5/2 составляет 207.1 ± ± 0.1 эВ и соответствует состоянию Nb5+ [56, 57]. На рис. 8а также представлен спектральный регион Sb3d + O1s. В связи с тем, что более интенсивный пик Sb3d5/2 перекрывается с линией O1s, идентификацию химического состояния сурьмы проводили по пику Sb3d3/2. Значение энергии связи этого пика равно 539.9 ± 0.1 эВ, что характерно для состояния Sb3+ [58, 59]. Анализ спектрального региона Ce3d показывает наличие пиков с энергией связи 886.0 эВ (Ce3+3d5/2) и 901.7 эВ (Ce3+3d3/2), типичных для состояния Ce3+. Пики с энергией связи, характерной для состояния Ce4+ (916.6 ± ± 0.2 эВ) отсутствуют. Это указывает на то, что церий находится преимущественно в состоянии Ce3+ [58, 60]. Энергия связи пика Si2p (103.5 эВ) характерна для SiO2 [46].
Рис. 8.
РФЭ-спектры элементов на поверхности исходного катализатора VMoSbNbCeOx/SiO2 (а) и этого же катализатора после длительной работы в реакционной смеси (б).
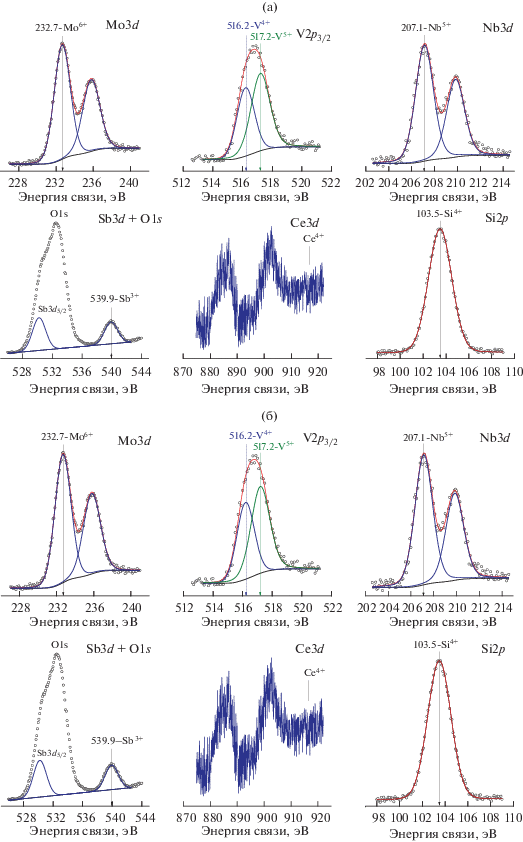
Анализ РФЭ-спектров образца, подвергнутого испытаниям в реакционной смеси (рис. 8б) свидетельствует о том, что в катализаторе не происходит изменения валентного состояния элементов Mo6+, Nb5+, Sb3+, Ce3+, V5+/V4+, но при этом увеличивается концентрация пятивалентного ванадия: соотношение V5+/V4+ возрастает от 0.6 до 1.2.
На электронно-микроскопическом снимке высокого разрешения Sb-содержащего катализатора (рис. 9) отчетливо видно два типа частиц (A и B) с хорошо упорядоченной кристаллической структурой. Согласно Фурье-дифрактограмме, полученной с этого изображения, кристаллическая структура частицы А соответствует фазе М1, а кристаллическая структура частицы В достоверно может быть отнесена к фазе М2 [26]. На электронно-микроскопическом снимке также четко регистрируется образование когерентной межфазной границы между микрокристаллами фаз M1 и M2. Можно предположить, что Ce в небольших количествах может быть стабилизирован в области межфазной границы, образованной микрокристаллами фаз M1 и M2. Отметим, что данный тип межфазной границы является основной микроструктурной особенностью структурного устройства синтезированного катализатора MoVSbNbCeOx/SiO2, и ранее в катализаторах такого типа она не наблюдалась. Вопрос о структурной модели границы будет изучен в наших дальнейших работах.

ЗАКЛЮЧЕНИЕ
Таким образом, на основании полученных данных можно заключить, что использование Sb вместо Te позволяет получать достаточно эффективные и стабильные катализаторы для процесса окислительного дегидрирования этана в этилен. Так, на катализаторе 50 мас. % Mo1V0.24Sb0.23Nb0.08Ce0.01Ox/50 мас. % SiO2, приготовленном в настоящей работе, выход этилена составляет 74% (селективность по этилену − 81.5% и при конверсии этана − 91.0%). На сегодняшний день это лучший из известных в литературе показателей для Sb-содержащих катализаторов в ОДЭ. Катализатор длительное время устойчиво работает в условиях реакционной среды без изменения фазового состава и ухудшения каталитических свойств.
Катализатор может быть приготовлен смешением водных растворов парамолибдата аммония, метаванадата аммония, нитрата церия, Sb2O3, SiO2 и оксалата ниобия при 50–80°C с последующей распылительной сушкой суспензии и прокаливанием в токе гелия при 350 и 600°C в течение 2 ч.
В основном катализатор представляет собой смесь оксидных фаз с орторомбической М1 и гексагональной M2 кристаллической структурой в количестве 71.7 и 28.3%. По данным метода ДР химический состав фазы M1 соответствует Mo1V0.24Sb0.09Nb0.11, а состав фазы M2 − Mo1V0.29Sb0.31. Кроме того в катализаторе присутствует небольшое количество аморфного молибден-сурьмяного соединения. По данным РФЭС основные элементы в катализаторе находятся в валентных состояниях Mo6+, Nb5+, Sb3+, Ce3+, V5+/V4+, которые не меняются под воздействием реакционной среды. Наблюдается лишь повышение концентрации пятивалентного ванадия: соотношение V5+/V4+ увеличивается от 0.6 до 1.2.
Характерной особенностью Sb-содержащего катализатора является формирование межфазной границы между кристаллами M1 и M2, выявленное методом ПЭМВР. Предположительно, это может привести к образованию активного состояния в катализаторе, обеспечивающего улучшенные каталитические свойства. Окислительно-восстановительные свойства катализатора в области межфазной границы могут отличаться от таковых для чистых фаз M1 и M2.
Список литературы
Baroi G., Gaffney A.M., Fushimi R. // Catal. Today. 2017. V. 298. P. 138.
Valente J.S., Armendariz-Herrera H., Quintana-Solorzano R., Angel P., Nava N., Masso A., Lopez Nieto J.M. // ACS Catal. 2014. V. 4. P. 1292.
Бондарева В.М., Кардаш Т.Ю., Ищенко Е.В., Соболев В.И. // Катализ в промышленности. 2014. № 6. С. 38. (Bondareva V.M., Kardash T.Yu., Ishchenko E.V., Sobolev V.I. // Catal. Ind. 2015. V. 7. P. 104.)
Bhasin M.M., McCain J.H., Vora B.V., Imai T., Pujado P.R. //Appl. Catal. A: Gen. 2001. V. 221. P. 397.
Cavani F., Trifirò F. // Catal. Today. 1995. V. 24. P. 307.
Xie Q., Chen L., Weng W., Wan H. // J. Mol. Catal. A: Chem. 2005. V. 240. P. 191.
Valente J.S., Quintana-Solorzano R., Armendariz-Herrera H., Barragan-Rodríguez G., Lopez Nieto J.M. // Ind. Eng. Chem. Res. 2014. V. 53. P. 1775.
Sanfilipp D., Miracca I. // Catal. Today. 2006. V. 111. P. 133.
Lopez Nieto J.M., Botella P., Vazquez M.I., Dejoz A. // Chem. Commun. 2002. P. 1906.
Lopez Nieto J.M., Botella P., Vazquez M.I., Dejoz A. Pat. 03/064035 A1 WO, 2003.
Lopez Nieto J.M., Botella P., Vzquez M.I., Dejoz A. Pat. 7319179 B2 US, 2008.
Solsona B., Vazquez M.I., Ivars F., Dejoz A., Concepcion P., Lopez Nieto J.M. // J. Catal. 2007. V. 252. P. 271.
Nguyen T.T., Burel L., Nguyen D.L., Pham-Huu C., Millet J.M. // Appl. Catal. A: Gen. 2012. V. 433. P. 41.
Nguyen T.T., Aouine M., Millet J.M.M. // Catal. Commun. 2012. V. 21. P. 22.
Botella P., Garcıa-Gonzalez E., Dejoz A., Lopez Nieto J.M., Vazquez M., Gonzalez-Calbet J. // J. Catal. 2004. V. 225. P. 428.
Ishchenko E.V., Kardash T.Y., Gulyaev R.V., Ishchenko A.V., Sobolev V.I., Bondareva V.M. // Appl. Catal. A: Gen. 2016. V. 514. P. 1.
McCain J.H. Pat. 5524236 US, 1985.
Nguyen T.T., Deniau B., Delichere P., Millet J.M.M. // Top. Catal. 2014. V. 57. P. 1152.
Baca M., Millet J.M.M. // Appl. Catal. A: Gen. 2005. V. 279. P. 67.
Murayama H., Vitry D., Ueda W., Fuchs G., Anne M., Dubois J.L. // Appl. Catal. A: Gen. 2007. V. 318. P. 137.
DeSanto P., Buttrey D.J., Grasselli R.K., Lugmair C.G., Volpe A.F., Toby B.H., Vogt T. // Top. Catal. 2003. V. 23. P. 23.
DeSanto P.J., Buttrey D.J., Grasselli R.K., Lugmair C.G., Volpe A.F., Toby B.H., Vogt T. // Z. Crystallogr. 2004. V. 219. P. 152.
Sadakane M., Yamagata K., Kodato K., Endo K., Toriumi K., Ozawa Y., Ozeki T., Nagai T., Matsui Y., Sakaguchi N., Pyrz W.D., Buttrey D.J., Blom D.A., Vogt T., Ueda W. // Angew. Chem. 2009. V. 48. № 21. P. 3782.
Aouine M., Dubois J.L., Millet J.M.M. // ACS Catal. 2016. V. 6. P. 4775.
Deniau B., Millet J.M.M., Loridanta S., Christin N., Dubois J.L. // J. Catal. 2008. V. 260. P. 30.
Li X., Buttrey D.J., Blom D.A., Vogt T. // Top. Catal. 2011. V. 54. P. 614.
Wagner J.B., Timpe O., Hamid F.A., Trunschke A., Wild U., Su D.S, Widi R.K, Hamid S.B.A, Schlögl R. // Top. Catal. 2006. V. 38. P. 51.
Hävecker M., Wrabetz S., Kröhnert J., Csepei L.-I., Naumann R., Kolen’ko Y.V., Girgsdies F., Schlögl R., Trunschke A. // J. Catal. 2012. V. 285. P. 48.
Lopez Nieto J.M., Botella P., Concepción P., Dejoz A., Vázquez M.I. // Catal. Today. 2004. V. 91−92. P. 241.
Oliver J.M., López Nieto J.M., Botella P., Mifsud A. // Appl. Catal. A: Gen. 2004. V. 257. P. 67.
Kardash T.Yu., Lazareva E.V., Svintsitskiy D.A., Ishenko A.V., Bondareva V.M., Neder R.B. // RSC Adv. 2018. V. 8. P. 35 903.
Deniau B., Nguyen T.T., Delichere P., Safonova O., Millet J.M.M. // Top. Catal. 2013. V. 56. P. 1952.
Ivars F., Solsona B., Hernandez S., Lopez-Nieto J.M. // Catal. Today. 2010. V. 149. P. 260.
Mishanin I.I., Kalenchuk A.N., Maslakov K.I., Lunin V.V., Koklin A.E., Finashina E.D., Bogdan V.I. // Russ. J. Phys. Chem. 2016. V. 90. P. 1132.
Chu B., Truter L., Nijhuis T.A., Cheng Y. // Appl. Catal. A: Gen. 2015. V. 498. P. 99.
Ishchenko E.V., Gulyaev R.V., Kardash T.Yu., Ishchenko A.V., Gerasimov E.Yu., Sobolev V.I., Bondareva V.M. // Appl. Catal. A: Gen. 2017. V. 534. P. 58.
Yun Y.S., Lee M., Sung J., Yun D., Kim T.Y., Park H., Lee K.R., Song C.K., Kim Y., Lee J., Seo Y.J., Song I.K., Yi J. // Appl. Catal. B: Env. 2018. V. 237. P. 554.
Svintsitskiy D.A., Kardash T.Yu, Lazareva E.V., Saraev A.A., Derevyannikova E.A., Vorokhta M., Šmíd B., Bondareva V.M. // Appl. Catal. A: Gen. 2019. V. 579. P. 141.
Zenkovets G.A., Bondareva V.M., Shutilov A.A., Ivanova G.G., Shadrina L.A., Sobolev V.I. Пaт. 2 552 984 C1, Ru, 2015.
Andrushkevich T.V., Popova G.Y., Chesalov Y.A., Ischenko E.V., Kramov M.I, Kaichev V.V. // Appl. Catal. A. Gen. 2015. V. 506. P. 109.
Ischenko E.V., Andrushkevich T.V., Popova G.Y., Chesalov Y.A., Plyasova L.M., Ischenko A.V., Kardash T.Yu., Dovlitova L.S. // Cat. Ind. 2010. № 2. P. 291.
Topas 4.3. Bruker AXS. Germany. 2015.
Database: Inorganic Crystal Structure Database, ICSD. In Release 2008. Fashinformationszentrum Karsruhe D #8211 1754 Eggenstein #8211 Leopoldshafen, Germany, 2008.
Малахов В.В, Васильева И.Г. // Успехи химии. 2008. Т. 77. № 4. С. 370.
Малахов В.В., Болдырева Н.Н., Власов А.А., Довлитова Л.С. // Журн. аналит. химии. 2011. Т. 66. № 5. С. 473. (Malakhov V.V., Boldy’reva N.N., Vlasov A.A., Dovlitova L.S. // J. Analyt. Chem. 2011. V. 66. № 5. P. 458).
Moudler J., Stickle W., Sobol P., Bomben K. Handbook of X-ray Photoelectron Spectroscopy. Perkin-Elmer Corp.: Eden. Prairie, MN, 1992.
Scofield J.H. // J. Electron Spectrosc. Relat. Phenom. 1976. V. 8. P. 129. https://doi.org/10.1016/0368-2048(76)80015-1
http://xpspeak.software.informer.com/4.1/
Бондарева В.М., Ищенко Е.В., Шадрина Л.А., Соболев В.И. // Катализ в промышленности. 2015. Т. 15. № 6. С. 36. (Bondareva V.M., Ishchenko E.V., Shadrina L.A., Sobolev V.I. // Catal. Ind. 2016. V. 8. P. 112.)
Gärtner C.A., Van Veen A.C., Lercher J.A. // ChemCatChem. 2013. V. 5. P. 3196.
Sanders A.F.H., De Jong A.M., De Beer V.H.J., Van Veen C.A., Niemantsverdriet J.W. // Appl. Surf. Sci. 1999. V. 144−145. P. 380.
Saih Y., Segawa K. // Appl. Catal. A: Gen. 2009. V. 353. P. 258.
Demeter M., Neumann M., Reichelt W. // Surf. Sci. 2000. V. 454−456. P. 41.
Silversmit G., Depla D., Poelman H., Marin G.B., De Gryse R. // J. Electron Spectrosc. Relat. Phenom. 2004. V. 135. P. 167.
Sawatzky G.A., Post D. // Phys. Rev. B. 1979. V. 20. P. 1546.
Farahani M.D., Dasireddy V.D.B.C., Friedrich H.B. // ChemCatChem. 2018. V. 10. P. 2059.
Xue J., Wang R., Zhang Z., Qiu S. // Dalton Trans. 2016. V. 45. P. 16519.
He G.-H., Liang C.-J., Ou Y.-D., Liu D.-N., Fang Y.-P., Xu Y.H. // Mater. Res. Bull. 2013. V. 48. P. 2244.
Li L., Zhang Y.X., Fang X.S., Zhai T.Y., Liao M.Y., Wang H.Q., Li G.H., Koide Y., Bando Y., Golberg D. // Nanotechnol. 2011. V. 22. P. 165 704.
Xu J., Overbury S.H. // J. Catal. 2004. V. 222. P. 167.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ