

Кинетика и катализ, 2021, T. 62, № 2, стр. 135-174
Катализаторы на основе высокованадиевых растворов молибдованадофосфорных гетерополикислот: достижения, проблемы, перспективы
Е. Г. Жижина a, Л. Л. Гогин a, *, Ю. А. Родикова a, В. И. Бухтияров a
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: gogin@catalysis.ru
Поступила в редакцию 16.11.2020
После доработки 04.12.2020
Принята к публикации 04.12.2020
Аннотация
Ретроспективный обзор, основанный на работах авторов, посвящен рассмотрению гомогенных каталитических систем на основе водных растворов Мо–V–P-гетерополикислот (ГПК-х, х – число атомов ванадия), впервые предложенных в Институте катализа СО РАН для осуществления окислительных реакций. Рассмотрены свойства и методы синтеза растворов ГПК-х, наиболее перспективные процессы, протекающие в их присутствии, а также проблемы, возникающие при использовании растворов ГПК-х, и пути их устранения. Показаны перспективы создания промышленных процессов на основе модифицированных высокованадиевых растворов ГПК-х некеггиновских составов.
СОДЕРЖАНИЕ
Введение
1. Окисление субстратов в растворах Мо−V−P-ГПК-х
2. Состав растворов ГПК-х
3. Получение растворов ГПК-х
4. Свойства растворов ГПК-х
4.1. Кислотные свойства растворов
4.2. Окислительные свойства растворов ГПК-х
4.2.1. Редокс-потенциалы водных растворов ГПК-х
4.2.2. Термохимия восстановления и окисления ГПК-х в водных растворах
4.3. Физико-химические свойства растворов ГПК-х
4.3.1. Плотность и вязкость растворов ГПК-х
4.3.2. Электропроводность растворов ГПК-х
4.3.3. Термостойкость растворов ГПК-х
4.3.4. Коррозионная активность растворов ГПК-х
5. Каталитические процессы в присутствии растворов ГПК-х
5.1. Низкотемпературное окисление СО в СО2 в присутствии гомогенного двухкомпонентного катализатора Pd(II) + ГПК-х. Гетерогенный вариант системы
5.2. Окисление низших олефинов (С2−С4) в карбонильные соединения в присутствии гомогенной двухкомпонентной каталитической системы Pd(II) + + ГПК-х. МЭК-процесс
5.3. Окисление алкилфенолов бензольного и нафталинового рядов в соответствующие пара-хиноны в присутствии растворов ГПК-х (однокомпонентные каталитические системы)
5.3.1. Окисление 2-метилнафтола-1 в 2-метил-1,4-нафтохинон (витамин К3). Разработка промышленной технологии “Викасиб” синтеза витаминов группы К
5.3.2. Окисление 2,3,6-триметилфенола в 2,3,5-триметил-1,4-бензохинон – ключевой полупродукт синтеза витамина Е
5.3.3. Окисление диалкилфенолов в соответствующие пара-бензохиноны
5.4. Каталитическое окисление 5-гидроксиметилфурфурола в 2,5-диформилфуран в присутствии ГПК-х
5.5. Диеновый синтез. One-pot процессы
6. Регенерация растворов ГПК-х кислородом
Заключение
ВВЕДЕНИЕ
Катализ полиоксометаллатами (гетерополисоединениями, ГПС) – интенсивно развивающееся направление в современной науке о катализе. Исследования в этой области ведутся уже около 50 лет. О высоком интересе к указанному классу катализаторов свидетельствует большое число научных работ по катализу ГПС, рассмотренных в многочисленных обзорах [1–15]. Накопленные за эти годы данные позволили существенно продвинуться в понимании механизма каталитического действия ГПС на молекулярном уровне и приступить к реализации промышленных процессов с их участием.
Впервые использовать ГПС – гетерополикислоты (ГПК) и их соли – в составе катализаторов предложил К.И. Матвеев (Институт катализа СО РАН, ИК СО РАН, г. Новосибирск), разработавший с сотрудниками двухкомпонентный гомогенный катализатор Pd + ГПК для окисления этилена в ацетальдегид кислородом [16], в котором ГПК играла роль обратимо действующего окислителя. Отсутствие Cl–-ионов было главным преимуществом нового катализатора по сравнению с “хлоридным” катализатором PdCl2 + CuCl2, используемым в промышленном Вакер-процессе [17]. Именно работами Матвеева было положено начало развитию нового направления катализа – катализа ГПС. Всесторонние исследования ГПС с целью их применения в катализе, начатые в ИК СО РАН, вызвали неподдельный интерес и очень быстро распространились по всему миру. Уникальность свойств ГПС и многообразие составов позволили с успехом использовать их в кислотном и окислительном катализе, в качестве однокомпонентных катализаторов или в составе многокомпонентных, в гомогенном или гетерогенном варианте. Мировым лидером в коммерциализации каталитических процессов на основе ГПС стала Япония [4, 5, 7, 9, 10].
Большое число опубликованных научных работ и внедренных крупнотоннажных каталитических процессов органического синтеза показывают, что в первую очередь ГПС применяются как кислотные (гомогенные или гетерогенные) катализаторы [9, 10, 18]. Что касается промышленных процессов окисления, которых значительно меньше, то в их ряду можно отметить, в частности, парофазное окисление метакролеина в метакриловую кислоту на гетерогенном катализаторе Мо–V–P-ГПК (процесс реализован в 1982 г.) [4] и окисление этилена в уксусную кислоту на гетерогенном катализаторе Рd + SiW12 (1997 г.) [19]. Несмотря на быстрый рост внедрения каталитических процессов с участием ГПС в последние десятилетия, промышленные гомогенно-каталитические процессы окисления в присутствии ГПС до сих пор не реализованы. Именно поэтому в настоящем обзоре мы сконцентрировались в основном на гомогенном окислительном катализе с участием растворов Мо−V−P-ГПК-х (х – число атомов ванадия в составе ГПК). Здесь рассмотрены свойства этих растворов и способы их синтеза, окислительные процессы в присутствии ГПК-х, а также возникающие при их реализации проблемы и возможные пути устранения этих препятствий.
Следует отметить, что использование ГПК и их солей очень перспективно в качестве катализаторов процессов тонкого органического синтеза, включая асимметрический синтез. Благодаря высокой цене продуктов выигрыш в селективности за счет введения более эффективного катализатора (каким часто оказывается ГПК) может многократно перекрывать затраты на сам катализатор. Поэтому в последнее время выполнено много работ по применению ГПС в синтезе антиоксидантов, лекарственных препаратов, витаминов, биологически активных веществ [4, 9, 10, 20–22]. Кроме того, важным направлением катализа является использование бифункциональных свойств растворов ГПК-х, способных одновременно осуществлять кислотные и окислительные превращения органических молекул в жидкой фазе.
Несмотря на то, что изучением свойств ГПС занимаются сейчас во всем мире, и за последние годы создано немало новых каталитических процессов на их основе, ИК СО РАН продолжает оставаться одним из лидеров фундаментальных исследований свойств этих соединений и механизмов каталитических процессов с их участием. Настоящая работа, посвященная поиску перспективных каталитических реакций окисления с участием растворов Mo–V–Р ГПК-х и изучению их механизмов, нацелена на то, чтобы высокоэффективные гомогенные каталитические процессы окисления с участием ГПК-х были в ближайшей перспективе реализованы в промышленности.
1. ОКИСЛЕНИЕ СУБСТРАТОВ В РАСТВОРАХ ГПК-x
ГПК – это свободная протонная форма гетерополианиона (ГПА), в нашем случае ГПА-х. Водные растворы ГПК-х являются сильными бренстедовскими кислотами. В то же время, за счет присутствия V(V) в их составе они могут быть довольно сильными окислителями [1, 2, 23]. В окислительном катализе часто используют растворы ГПК-х структуры Кеггина с общей формулой Hx+ 3PMo12– xVxO40. Эти растворы обладают исключительно важным для катализа свойством – обратимой окисляемостью. Оно заключается в том, что восстановленные формы ГПК-х способны окисляться молекулярным кислородом, поэтому в их присутствии можно в 2 стадии (методом нестационарного катализа) осуществить каталитическое окисление кислородом различных органических субстратов (Su). Например, окисление Su кислородом по реакции (III) можно представить суммой реакций (I) и (II):
В таких процессах ГПК-х, являясь обратимо действующим окислителем, играет роль катализатора суммарной реакции (III). Важно отметить, что редокс-превращения в растворах ГПК-х претерпевают только атомы ванадия: V(V) → V(IV) → → V(V). Стадии (I) и (II) каталитической реакции (III) часто проводят раздельно в разных условиях. При этом удается достичь максимальной селективности в целевой реакции (I) и к тому же предотвратить взаимодействие Su с O2 в тех случаях, когда возможно образование взрывоопасных смесей или О2 инициирует побочные реакции. Стадия (II) регенерации катализатора кислородом – общая стадия всех окислительных процессов с участием ГПК-х. Эта стадия, как правило, ключевая, поскольку именно возможность быстрой регенерации растворов ГПК-х определяет технологичность окислительных процессов (III) с их участием.
Растворы ГПК-х, являющиеся многоэлектронными окислителями [10, 24, 25], весьма перспективны в качестве катализаторов окисления субстратов разных классов. Тем не менее, создать промышленные окислительные процессы с их участием оказалось довольно трудно. Главные причины – неполная регенерация катализаторов кислородом по реакции (II) и недостаточная гидролитическая устойчивость растворов ГПК-х. Для применения новых катализаторов окисления в промышленности необходимо было решить вышеуказанные проблемы. В этой связи весьма актуальными стали исследования каталитических и физико-химических свойств растворов ГПК-х в процессах окисления различных органических субстратов на основе данных о составе этих растворов и исследования механизмов отдельных стадий каталитических превращений.
2. СОСТАВ РАСТВОРОВ ГПК-х
Следует отметить, что смешанные Мо−V−Р-ГПК-х состава H3 +хPMo12– хVхO40, на которых будет сосредоточено наше внимание, образуются при замещении части ионов Мо(VI) на V(V) в кеггиновской структуре гетерополианиона ${\text{РМ}}{{{\text{о}}}_{{12}}}{\text{О}}_{{40}}^{{3 - }}$ (рис. 1).
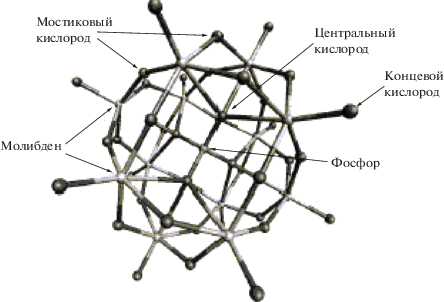
Такие соединения исследовали разные группы ученых [24, 26–33], однако бóльшая часть основополагающих работ была выполнена в ИК СО РАН с использованием методов ЯМР, ЭПР, рентгеноструктурного анализа и колебательной спектроскопии [34–47]. Этому способствовало то, что в 70-е годы ХХ века именно в ИК СО РАН начало интенсивно развиваться направление катализа ГПС.
Особенности строения ГПК-х при изменении числа ионов V(V) в их составе детально изучались в работах [24, 30, 31, 39–41]. Для всех ГПК-х, независимо от значения х, характерна структура Кеггина, в которой ионы ванадия распределены статистически. Ионы V (Mo) расположены в центре искаженных октаэдров МО6 с наиболее коротким расстоянием до концевого атома кислорода и наиболее длинным расстоянием до атома Отранс, общего для трех М-октаэдров и одного Р-тетраэдра.
Характер анион-катионного взаимодействия в ГПА-х закономерно изменяется с увеличением х. При замещении двух и трех ионов Мо(VI) на V(V) рост отрицательного заряда гетерополианиона (ГПА) может быть скомпенсирован увеличением числа протонов. При х > 3 кислоты неустойчивы: выделены только кислые и средние соли щелочных металлов [41]. В кристаллах этих солей катионы щелочных металлов наиболее прочно связаны с концевыми атомами кислорода. При х > 6 реализуется еще один путь компенсации заряда ГПА – присоединение катионов VO3+ через четыре мостиковых атома кислорода. Так, в [42] рассмотрена структура Na4H4(VO)[PMo4V8O40] ⋅ 20H2O. В работе [48] показано, что высокий заряд ГПА состава ${\text{Р}}{{{\text{V}}}_{{12}}}{\text{O}}_{{40}}^{{15 - }}$ компенсируется присоединением двух катионов VO3+ с образованием ${\text{Р}}{{{\text{V}}}_{{14}}}{\text{O}}_{{42}}^{{9 - }}.$ По спектрам ЯМР 17О и 51V установлено, что этот анион сохраняет свою структуру и в водных растворах [43].
Mo−V−Р-ГПА имеют ИК-спектры, характерные для структуры Кеггина. Они содержат полосы, относящиеся к колебаниям связей РО4 (в области 1050 см–1), М=О (в области 950 см–1) и М−О−М двух типов (около 870 и 780 см–1) [42]. Сохранение структуры Кеггина в водных растворах ГПА-х подтверждают также спектры КРС [49].
Растворы ГПК-х, используемые нами в качестве катализаторов в двухстадийных процессах (III) окисления Su кислородом, претерпевают поочередные редокс-превращения V(V) → V(IV) → V(V), поэтому в их растворах обычно содержатся как ионы V(V), так и V(IV). Следовательно, брутто-состав частично восстановленной ГПК-х должен быть записан как ${{{\text{Н}}}_{{3 + х + }}}_{m}{\text{PV}}_{m}^{{{\text{IV}}}}{\text{V}}_{{х - m}}^{{\text{V}}}{\text{M}}{{{\text{o}}}_{{12 - х}}}{{{\text{O}}}_{{40}}}$ (HmГПК-х). Здесь m – это степень восстановления ГПК-х, равная числу ионов ванадия(IV) в ее составе или числу принятых ею электронов [20, 25].
Состав растворов ГПК-х сложен, что подтверждено методом ЯМР на разных ядрах (31Р, 51V) и методом ЭПР [34, 36, 50–54]. Надежно доказано, что в процессе редокс-превращений в растворе ГПК-х структура Кеггина сохраняется [34], однако соотношение интенсивностей сигналов изменяется в зависимости от рН и числа атомов ванадия (х). Показано, что в спектрах растворов ГПК-х, в частности ГПК-2 [36], имеются группы линий, относящиеся к ГПА с 1, 2, 3 и более атомами ванадия. При этом число линий в спектрах ГПК-х возрастает с ростом х [34, 36]. Кроме того, могут наблюдаться линии Mo−V-изополианионов и катионов ${\text{VO}}_{2}^{ + }.$ Внутри каждой группы линий ЯМР 31Р и 51V для ГПК одного состава видна тонкая структура, обусловленная присутствием стереоизомеров [36, 50]. Изомеры с изолированными и соседними атомами ванадия удалось зафиксировать по ЭПР-спектрам частично восстановленных растворов ГПА-х [55–57]. Сложный состав растворов ГПК-х в сильнокислой области (рН < 1) можно наблюдать по спектрам ЯМР 31Р и 51V благодаря различающейся способности к протонированию у разных ГПА. Плавное же изменение положения линий ГПА с изменением кислотности объясняется протонным обменом между ГПА [36]. В индивидуальном виде существует только ГПК-1 (H4PMo11VO40).
На основе многочисленных исследований для растворов ГПК-х доказано существование нескольких типов равновесий. Во-первых, имеет место равновесие деструктивной диссоциации ГПА под действием протонов с отщеплением оксокатионов ванадия. Так, невосстановленные молекулы ГПК-х с х ≥ 2 под действием Н+-ионов претерпевают обратимую диссоциацию с отщеплением катиона ${\text{VO}}_{2}^{ + }$ и образованием ГПК-(х – 1) с меньшим числом атомов ванадия по уравнению (IV) [3, 37, 54, 58–60]:
(IV)
$\begin{gathered} (13--х){{{\text{H}}}_{{3 + х}}}{\text{P}}{{{\text{V}}}_{х}}{\text{M}}{{{\text{o}}}_{{12 - х}}}{{{\text{O}}}_{{40}}} + 12{{{\text{H}}}^{ + }}~\overset {{{K}_{4}}} \leftrightarrows \\ \rightleftarrows (12--х){{{\text{H}}}_{{2 + х}}}{\text{P}}{{{\text{V}}}_{{х - 1}}}{\text{M}}{{{\text{o}}}_{{13 - х}}}{{{\text{O}}}_{{40}}} + 12{\text{VO}}_{2}^{ + } + \\ + \,\,{{{\text{H}}}_{{\text{3}}}}{\text{P}}{{{\text{O}}}_{4}} + 12{{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $Полностью восстановленные ГПК-х диссоциируют в кислой среде с отщеплением катиона VO2+ по реакции (V) [51–53, 56, 61]:
(V)
$\begin{gathered} (13--х){{\left[ {{{{\text{H}}}_{{2х - 1}}}{\text{PV}}_{х}^{{{\text{IV}}}}{\text{M}}{{{\text{o}}}_{{12 - х}}}{{{\text{O}}}_{{40}}}} \right]}^{{4 - }}} + 28{{{\text{H}}}^{ + }}\overset {{{K}_{5}}} \leftrightarrows \\ \rightleftarrows (12--х){{\left[ {{{{\text{H}}}_{{2х - 3}}}{\text{PV}}_{{х - 1}}^{{{\text{IV}}}}{\text{M}}{{{\text{o}}}_{{13 - х}}}{{{\text{O}}}_{{40}}}} \right]}^{{4 - }}} + 12{\text{V}}{{{\text{O}}}^{{2 + }}} + \\ + \,\,{{{\text{H}}}_{{\text{3}}}}{\text{P}}{{{\text{O}}}_{4}} + 24{{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $В частично же восстановленных растворах ГПК-х в кислой среде от ГПА будут отщепляться как катионы VO2+, так и ${\text{VO}}_{2}^{ + }{\text{.}}$ Эти многоступенчатые процессы для частично восстановленных ГПК-х можно представить уравнением (VI) [25, 51, 61, 62]:
(VI)
$\begin{gathered} \left( {13--x} \right){{\left[ {{{{\text{H}}}_{{x + m - 1}}}{\text{PV}}_{m}^{{{\text{IV}}}}{\text{V}}_{{x - m}}^{{\text{V}}}{\text{M}}{{{\text{o}}}_{{12 - x}}}{{{\text{O}}}_{{40}}}} \right]}^{{4 - }}} + \\ + \,\,\left( {28 + x--m} \right){{{\text{H}}}^{{ + ~}}}\overset {{{K}_{6}}} \leftrightarrows ~ \\ \rightleftarrows \left( {12--x} \right){{\left[ {{{{\text{H}}}_{{x + m - 3}}}{\text{PV}}_{{m - 1}}^{{{\text{IV}}}}{\text{V}}_{{x - m}}^{{\text{V}}}{\text{M}}{{{\text{o}}}_{{13 - x}}}{{{\text{O}}}_{{40}}}} \right]}^{{4 - }}} + \\ + \,\,(x--m){\text{V}}{{{\text{O}}}^{{2 + }}} + (12--x + m){\text{VO}}_{2}^{ + } + {{{\text{H}}}_{{\text{3}}}}{\text{P}}{{{\text{O}}}_{4}} + \\ + \,\,(24 + x--m){{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $Оценки констант некоторых равновесий распада ГПА-х с отщеплением катионов ${\text{VO}}_{2}^{ + }$ при температурах 25–77°С проведены в работах [54, 60].
Кроме равновесий деструктивной диссоциации под действием протонов (IV)−(VI) в растворах ГПК-х имеют место равновесия диспропорционирования (VII) по числу атомов ванадия в молекуле, в результате которых образуются ГПК-х с меньшим и бóльшим числом атомов ванадия [2, 25, 60], например:
(VII)
$\begin{gathered} {\text{2}}{{{\text{H}}}_{{{\text{3 + }}x}}}{\text{P}}{{{\text{V}}}_{x}}{\text{M}}{{{\text{o}}}_{{{\text{12}} - x}}}{{{\text{O}}}_{{{\text{40}}}}}\overset {{{K}_{7}}} \leftrightarrows \\ \rightleftarrows {{{\text{H}}}_{{{\text{2 + }}x}}}{\text{P}}{{{\text{V}}}_{{x - {\text{1}}}}}{\text{M}}{{{\text{o}}}_{{{\text{12}} - {\text{(}}x - {\text{1)}}}}}{{{\text{O}}}_{{{\text{40}}}}} + \\ + \,\,{{{\text{H}}}_{{{\text{4 + }}x}}}{\text{P}}{{{\text{V}}}_{{x{\text{ + 1}}}}}{\text{M}}{{{\text{o}}}_{{{\text{12}} - {\text{(}}x{\text{ + 1)}}}}}{{{\text{O}}}_{{{\text{40}}}}}{\text{.}} \\ \end{gathered} $В этих же растворах существует также равновесие (VIII) диспропорционирования ГПА по степени восстановления m, при котором общее число атомов ванадия в ГПА-х остается постоянным, а изменяется соотношение числа ионов V(IV) и V(V) [25, 53]:
(VIII)
$\begin{gathered} \mathop {~2{{{\left[ {{{{\text{H}}}_{x}}{\text{P}}{{{\text{V}}}^{{{\text{IV}}}}}{\text{V}}_{{x - 1}}^{{\text{V}}}{\text{M}}{{{\text{o}}}_{{12 - x}}}{{{\text{O}}}_{{40}}}} \right]}}^{{4 - }}}}\limits_{(m = 1)} ~\overset {{{K}_{8}}} \leftrightarrows \\ \rightleftarrows \mathop {{{{\left[ {{{H}_{{x - 1}}}{\text{PV}}_{x}^{{\text{V}}}{\text{M}}{{{\text{o}}}_{{12 - x}}}{{{\text{O}}}_{{40}}}} \right]}}^{{4 - }}}}\limits_{(m = 0)} + \\ + \,\,\mathop {{{{\left[ {{{{\text{H}}}_{{x + m - 1}}}{\text{PV}}_{{\text{2}}}^{{{\text{IV}}}}{\text{V}}_{{x - 2}}^{{\text{V}}}{\text{M}}{{{\text{o}}}_{{12 - x}}}{{{\text{O}}}_{{40}}}} \right]}}^{{4 - }}}}\limits_{(m = 2)} . \\ \end{gathered} $Таким образом, из сказанного выше следует, что растворы ГПК-х, отвечающие брутто-формуле ${{{\text{Н}}}_{{3 + х + m}}}{\text{PV}}_{m}^{{{\text{IV}}}}{\text{V}}_{{х - m}}^{{\text{V}}}{\text{M}}{{{\text{o}}}_{{12 - х}}}{{{\text{O}}}_{{40}}}{\text{,}}$ представляют собой сложные равновесные смеси, содержащие ГПА с разным числом ионов V(V) и V(IV), протоны, катионы ${\text{VO}}_{2}^{ + }$ и VO2+, свободную фосфорную кислоту.
Количественные характеристики равновесий (IV)–(VIII) были получены при исследовании состава растворов методами ЯМР и ЭПР. В соответствии с данными ЯМР 31Р в растворе 0.1−0.3 М ГПК-4 при рН 3−4 всегда присутствует 10−15% ГПК-5 и столько же ГПК-3. Их содержание зависит от некоторых особенностей получения раствора ГПК-4, например, от рН в ходе синтеза [60]. Следует отметить, что в растворе ГПК-2 имеются также значительные концентрации продуктов диспропорционирования и гидролиза, особенно при низких значениях рН. Так, раствор ГПК-2, имеющий концентрацию 0.1 М и рН 0.53, содержит 5.7% ГПК-3; перекристаллизация из воды не позволяет от нее избавиться. В качестве примера в табл. 1 представлен состав раствора ГПК-2 в зависимости от концентрации и рН (по данным работы [54]).
Таблица 1.
Изменение состава раствора, отвечающего брутто-формуле Н5РМо10V2О40 (ГПК-2), от рН и концентрации раствора (по данным работы [54], получено разрешение издателя на использование материалов)
| Начальная концентрация ГПК-2, М | рН | ГПК-2, % | ГПК-1, % | ГПК-3, % | Н3РО4,% |
|---|---|---|---|---|---|
| 0.1 | 1.31 | 91.2 | 0 | 8.8 | 0 |
| 0.53 | 68.1 | 26.2 | 5.7 | 0 | |
| 0.50 | 62.3 | 28.7 | 7.4 | 1.6 | |
| 0.17 | 48.4 | 38.1 | 7.9 | 5.6 | |
| 0.03 | 1.55 | 100 | 0 | 0 | 0 |
| 0.49 | 53.5 | 42.3 | 0 | 4.2 | |
| 0.01 | 1.45 | 85.1 | 14.9 | 0 | 0 |
Данные этой таблицы показывают, что изменения рН и концентрации ГПК-2 влекут за собой сдвиг всех равновесий (IV)–(VIII), приводящий, в свою очередь, к изменению состава раствора.
За изменением состояния растворов ГПК-х при восстановлении следили методом ЭПР [51]. Оказалось, что при рН 1 в полностью восстановленном растворе ГПК-1 (m = 1) около половины всего V(IV) находится в виде катиона VO2+, а остальной входит в состав ГПА. В растворе же ГПК-2 (m = 1) бóльшая часть V(IV) присутствует в виде VO2+. В спектрах ЭПР раствора ГПК-3 (m = 1) уже весь V(IV) наблюдается в виде VO2+. При полном восстановлении раствора ГПК-3 (m = 3) 60% V(IV) содержится в виде VO2+, а остальной − в составе ГПА [51].
По спектрам ЭПР в работе [52] определена зависимость доли свободного катиона VO2+ от рН растворов ГПК-х (табл. 2). Видно, что в полностью восстановленных растворах доля свободного VO2+ убывает с увеличением рН вследствие сдвига влево уравнения (V). Аналогичные выводы сделаны в работах [45, 56], где при изучении состава растворов восстановленных ГПК-1 и ГПК-2 методом ЭПР было обнаружено, что существует область рН, в которой V(IV) сохраняется в составе восстановленной ГПК-х, но уменьшение рН приводит к отщеплению части V(IV).
Таблица 2.
Зависимость доли свободного катиона VO2+ от рН в растворах ГПК-х при комнатной температуре (по данным работы [52], получено разрешение издателя на использование материалов)
| ГПК-х | рН | VO2+, % |
|---|---|---|
| 0.1 MГПК-2(m = 2) | 1.25 | 47 |
| 1.65 | 20.5 | |
| 1.75 | 19 | |
| 2.25 | 11.5 | |
| 2.45 | 8 | |
| 3.20 | 0 | |
| 3.60 | 0 | |
| 0.05 M | 1.85 | 48 |
| ГПК-6 | 1.95 | 33 |
| (m = 6) | 2.80 | 27 |
Важно отметить, что изменения спектров ГПК-х при изменении рН растворов происходят мгновенно и обратимо [63]. Показано, что при рН 3–4 растворы ГПК-х стабильны и при постоянном значении рН сохраняют свой состав длительное время [60].
Таким образом, методами колебательной и радиоспектроскопии доказано, что растворы Мо–V–Р-ГПК представляют собой сложные равновесные смеси большого числа различных частиц. Сложность растворов ГПК-х, безусловно, затрудняет исследование механизмов каталитических реакций в их присутствии. В первую очередь это относится к стадии регенерации катализатора молекулярным кислородом по реакции (II) – ключевой стадии каталитических окислительных процессов с использованием растворов ГПК-х. В ходе этой реакции возрастает концентрация Н+-ионов, и все равновесия (IV)–(VIII) смещаются, непрерывно изменяя состав раствора ГПК-х, от которого очень сильно зависит скорость реакции (II).
3. ПОЛУЧЕНИЕ РАСТВОРОВ Мо−V−Р-ГПК-х
Наиболее распространенным способом синтеза ГПК-х является метод подкисления [24, 64]. Как правило, полиоксометаллаты формируются в растворах из простых оксоанионов и необходимого гетероатома путем самосборки по уравнению (IX) при подкислении, например:
(IX)
$\begin{gathered} 11{\text{MoO}}_{4}^{{2 - }} + {\text{VO}}_{3}^{ - } + {\text{HPO}}_{4}^{{2 - }} + 21{{{\text{H}}}^{{ + ~}}} \to \\ \to {{\left[ {{\text{PM}}{{{\text{o}}}_{{11}}}{\text{V}}{{{\text{O}}}_{{40}}}} \right]}^{{4 - }}} + 11{{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $Подкисление обычно достигается добавлением минеральных кислот, таких как HCl, H2SO4, HClO4 или HNO3 [10]. Часто при синтезе ГПК-х сначала получают их кислые соли [65]. Реакцию (X) при этом ведут, используя растворы натриевых солей фосфатов, молибдатов и ванадатов, которые подкисляют серной кислотой:
(X)
$\begin{gathered} {\text{N}}{{{\text{a}}}_{{\text{2}}}}{\text{HP}}{{{\text{O}}}_{{{\text{4(aq}})}}} + (12 - x){\text{N}}{{{\text{a}}}_{{\text{2}}}}{\text{Mo}}{{{\text{O}}}_{{4({\text{aq}})}}} + \\ + \,\,x{\text{NaV}}{{{\text{O}}}_{{3({\text{aq}})}}} + {\text{избыток}}\,\,{{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{4}} \to \\ \to {{\{ {\text{N}}{{{\text{a}}}_{y}}{{{\text{H}}}_{{(3 + x - y)}}}{\text{PM}}{{{\text{o}}}_{{(12 - x)}}}{{{\text{V}}}_{x}}{{{\text{O}}}_{{40}}}\} }_{{{\text{(aq)}}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + \\ + \,\,{\text{избыток}}\,\,{\text{N}}{{{\text{a}}}_{z}}{{{\text{H}}}_{{(2 - z)}}}{\text{S}}{{{\text{O}}}_{{{\text{4(aq)}}}}}. \\ \end{gathered} $В результате получают раствор натриевой соли Mo–V–фосфата, содержащий значительные количества сульфата и бисульфата натрия, загрязняющих раствор [32]. Это является существенным недостатком такого метода синтеза.
Выделение ГПА из воды может быть проведено путем добавления соответствующего катиона: аммония, тетраалкиламмония и т.д. Например, тетрабутиламмониевая соль обычно нерастворима в воде, поэтому ее можно отделить и перекристаллизовать из таких растворителей, как ацетонитрил и ацетон [10].
Существует классический эфирный метод, основанный на экстракции ГПК-х диэтиловым эфиром из сильно подкисленных водных растворов [66, 67]. При этом в виде нижнего слоя образуется тяжелый масляный эфират ГПК. Эфирный слой отделяют и эфират разлагают при добавлении воды под током воздуха. Затем продукт синтеза кристаллизуют из воды [10, 24]. Однако, как показано в [68], не все ГПК-х этим методом можно экстрагировать количественно. Следовательно, способ подкисления с помощью минеральных кислот имеет серьезные недостатки: продукт загрязнен кислотами, использование эфирной экстракции неприменимо для крупнотоннажных процессов и проблематично даже для лабораторного использования [32].
Описаны и другие способы подкисления при синтезе ГПК-х, например, подкисление катионообменными смолами [67], однако этот метод сопряжен с большим количеством вредных отходов, возникающих на этапе регенерации катионита [64, 67]. Кроме того, таким путем получают очень разбавленные растворы ГПК-х, которые необходимо концентрировать выпариванием или высаливанием.
Для подкисления при синтезе ГПК-х может быть использован также электрохимический способ, суть которого заключается в удалении катионов из раствора и компенсации их протонами. Процесс проводят в электродиализаторах (в анодной камере находится раствор исходных компонентов, а в катодной – раствор щелочи; камеры разделены мембраной) [69]. Производительность электрохимического процесса невелика.
Существует также метод подкисления (XI), заключающийся в разложении солей под действием сильных кислот [64]:
(XI)
${\text{B}}{{{\text{a}}}_{{\text{3}}}}{\text{ГПА}} + 3{{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{4}}~ \to 3{\text{BaS}}{{{\text{O}}}_{{\text{4}}}} + {{{\text{H}}}_{{\text{6}}}}{\text{ГПА}}.$Сильного подкисления можно избежать, если проводить синтез путем взаимодействия простых веществ: оксидов и кислот. Свободные ГПК-х могут быть приготовлены растворением в воде МоО3 и V2O5 со стехиометрическим количеством фосфорной кислоты по уравнению (XII) [32]:
(XII)
$\begin{gathered} {{{\text{H}}}_{{\text{3}}}}{\text{P}}{{{\text{O}}}_{4}} + (12--x){\text{Мо}}{{{\text{О}}}_{3}} + \raise.5ex\hbox{$\scriptstyle 1$}\kern-.1em/ \kern-.15em\lower.25ex\hbox{$\scriptstyle 2$} x{{{\text{V}}}_{{\text{2}}}}{{{\text{O}}}_{5}} + \raise.5ex\hbox{$\scriptstyle 1$}\kern-.1em/ \kern-.15em\lower.25ex\hbox{$\scriptstyle 2$} x{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to {{{\text{H}}}_{{(3 + x)}}}{\text{PM}}{{{\text{o}}}_{{(12 - x)}}}{{{\text{V}}}_{x}}{{{\text{O}}}_{{40}}}. \\ \end{gathered} $Этим методом в раствор вводят только нужные компоненты, однако так получаются растворы с очень низким содержанием ванадия. Растворение V2O5 в растворе полиоксометаллата протекает довольно медленно и требует больших времен реакции, обычно около недели. Синтез 0.3 М ГПК-1 проходит с полным введением V2O5, но при приготовлении 0.3 М ГПК-2 не удается ввести весь V2O5 в состав раствора даже при длительном времени реакции. Так, авторы работы [32] через семь дней синтеза отфильтровали оставшийся V2O5 и аналитически определили состав раствора. В итоге при синтезе 0.3 М ГПК-2 реальный состав был H4.9PMo10.1V1.9O40, а для 0.3 М ГПК-3 − H5.4PMo9.8V2.4O40. Полное введение V2O5 становится более трудным с повышением требуемого содержания ванадия, поэтому данный метод становится бесполезным для получения растворов ГПК-х с х > 2.
Описанный способ, однако, был адаптирован для приготовления использующихся в катализе растворов кислых солей ГПК-х (х > 2) [32]. Не растворившийся в ходе синтеза V2O5 отфильтровывали, отдельно растворяли в воде с минимальным количеством щелочного карбоната и возвращали в раствор ГПК-х. Этим методом синтезируют, например, Na1.2H4.8PMo9V3O40 и Li1.15H5.85PMo8V4O40. В том случае, когда нужно синтезировать соль ГПК-х, растворение МоО3 в водном растворе ванадата натрия со стехиометрическим количеством фосфорной кислоты позволяет прямо получить нужный раствор соли по уравнению (XIII):
(XIII)
$\begin{gathered} {{{\text{H}}}_{{\text{3}}}}{\text{P}}{{{\text{O}}}_{4}} + (12 - x){\text{Мо}}{{{\text{О}}}_{3}} + {\text{NaV}}{{{\text{O}}}_{3}} \to \\ \to {\text{N}}{{{\text{a}}}_{x}}{{{\text{H}}}_{{\text{3}}}}{\text{PM}}{{{\text{o}}}_{{(12 - x)}}}{{{\text{V}}}_{x}}{{{\text{O}}}_{{40}}}. \\ \end{gathered} $Образование раствора Мо–V–фосфата этим методом протекает без добавления других минеральных солей. Ванадат натрия может быть получен из V2O5 путем добавления карбоната натрия. Это позволяет в одном реакторе масштабно синтезировать кислые соли ГПК-х из оксидов металлов. Раствор натриевой соли ГПК-4 можно приготовить в лабораторных условиях довольно быстро – за 5–6 ч [32]. Однако из-за низкой растворимости и реакционной способности реагентов метод имеет ограничения. Для некоторых ГПК, например ГПК-1, требуется до 20 сут реакции [32, 68].
Используют и другой способ, по которому нужные ГПК получают через ГПА других ненасыщенных ГПК [24, 64]. Схематично реакцию (XIV) можно представить так:
(XIV)
${{\left( {{\text{P}}{{{\text{M}}}_{{11}}}} \right)}^{{n - }}} + {{{\text{Z}}}^{{x + }}} \to {{\left[ {{\text{P}}{{{\text{M}}}_{{{\text{11}}}}}{\text{Z}}} \right]}^{{x - n}}}.$Лучший, на наш взгляд, метод синтеза растворов свободных ГПК-х (х = 1–6), не содержащих посторонних ионов, описан в работах [68, 70], где получение ГПК-х осуществляют по уравнению (XV) с использованием декаванадиевой кислоты H6V10O28:
(XV)
$\begin{gathered} {{{\text{H}}}_{{\text{3}}}}{\text{P}}{{{\text{O}}}_{{4~}}} + \left( {12 - х} \right){\text{Mo}}{{{\text{O}}}_{{3~}}} + 0.1х{{{\text{H}}}_{{\text{6}}}}{{{\text{V}}}_{{{\text{10}}}}}{{{\text{O}}}_{{28~}}} + \\ + \,\,0.2х{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {{{\text{H}}}_{{3 + х}}}{\text{PM}}{{{\text{o}}}_{{12 - х}}}{{{\text{V}}}_{х}}{{{\text{O}}}_{{40}}}. \\ \end{gathered} $Декаванадиевую кислоту готовят путем растворения V2O5 в холодной (3−5°С) H2O2 c последующим разложением промежуточно образующейся “надванадиевой” кислоты HVO4. Раствор декаванадиевой кислоты вводят в кипящую взвесь MoO3 + H3PO4 и кипятят до полного растворения. Такой способ позволяет получить растворы ГПК-х c х = 2–4 за 4.5–7.5 ч, с х = 5–6 – за 8–10 ч, c х = 1 – за 19 ч [68].
Таким образом, растворы ГПК-х и их солей можно синтезировать растворением реагентов, взятых в нужных пропорциях, либо при подкислении раствора различными способами, либо без подкисления. Второй вариант дает возможность готовить чистые растворы ГПК-х без примесей минеральных кислот. Лучший способ получения чистых кислот, разработанный в ИК СО РАН [68, 70], позволяет провести синтез ГПК-х нужного состава с полным введением всего ванадия в состав раствора. Отметим, что растворы ГПК-х, использованные нами при разработке описанных ниже каталитических процессов, были приготовлены именно этим способом.
Как будет показано ниже, наиболее эффективными в окислительном катализе оказались разработанные нами растворы высокованадиевых ГПК-х некеггиновских (модифицированных) составов [70–74]. Их синтезы были освоены при создании технологии производства метилэтилкетона путем гомогенного окисления н-бутиленов в присутствии катализатора (Pd + ГПК-х) не только в лабораторном, но и в опытно-промышленном масштабе [71–73] (раздел 5.2). Для осуществления пилотных испытаний было наработано 1350 л 0.25 М раствора брутто-состава H12P3Mo18V7O85 (ГПК-7) [73]. В отличие от кеггиновских растворов, в модифицированных ГПК-х концентрации ванадия, отвечающего за редокс-превращения, может быть в 2−3 раза выше. При этом они обладают значительно большей гидролитической и термической устойчивостью (вплоть до 180°С), позволяющей проводить эффективную регенерацию катализатора при более высокой температуре, чем это допустимо в случае с кеггиновскими растворами.
4. СВОЙСТВА РАСТВОРОВ ГПК-х
4.1. Кислотные свойства
ГПК различного состава, в том числе ГПК-х, находят широкое применение как катализаторы кислотно-каталитических реакций. Высокая эффективность ГПК в качестве кислотных катализаторов определяется рядом причин:
а) ГПК – это многоосновные бренстедовские кислоты, которые значительно сильнее обычных минеральных кислот, таких как H2SO4, HBr, HCl, HNO3, HClO4 [2, 75]. Наиболее просто различие в силе ГПК и обычных кислот может быть объяснено в рамках электростатической теории с учетом размеров анионов и количества атомов кислорода, доступных для делокализации заряда аниона. Поскольку ГПА значительно превосходят по размеру анионы обычных кислот, энергия связи протона в ГПК должна быть ниже, а их константы кислотной диссоциации – выше по сравнению с обычными кислотами. Кроме того, чем более делокализован заряд аниона, тем меньше эффективный отрицательный заряд на его отдельных оснόвных центрах, акцептирующих протоны, и потому слабее притяжение протона к аниону кислоты. Формальные отрицательные заряды гетерополианионов [PM12O40]3– и [SiM12O40]4– совпадают с зарядами анионов соответствующих простых кислот [PO4]3– и [SiO4]4–, однако заряд, приходящийся на один внешний атом кислорода в ГПА, в девять раз меньше, чем в фосфат- и силикат-ионах. Благодаря этому кислоты H8 –z[Xz–M12O40] значительно сильнее, чем H8 –z[Xz–O4] [23].
б) ГПК хорошо растворимы в воде и кислородсодержащих растворителях. Например, растворимость кислоты H4SiMо12O40 ⋅ 8H2O при 25°С составляет 88 мас. % в воде, 86 мас. % в этилацетате, 85 мас. % в диэтиловом эфире. В таких растворителях, как бензол, петролейный эфир, хлороформ, эта ГПК не растворяется [23].
в) ГПК обладают достаточно высокой термостабильностью. Для твердых кислот со структурой Кеггина она возрастает в ряду H4SiMо12O40 < < H3PMо12O40 < H4SiW12O40 < H3PW12O40. Разложение последней происходит при Т > 400°С. Важно отметить, что соли ГПК более стабильны, чем сами кислоты [1].
г) ГПК, как правило, не вступают в побочные реакции с органическими реагентами, что характерно для обычных минеральных кислот (сульфирование, хлорирование, нитрование и т.д.) [8, 10, 24].
Органические реакции в присутствии ГПК и минеральных кислот в растворах обычно следуют одним и тем же закономерностям [23], но механизмы их каталитического действия различаются. Прежде всего, это проявляется в сильном солевом эффекте (при полной диссоциации H4SiMо12O40 ионная сила в десять раз больше, чем при диссоциации 1–1 электролита – HNO3). Для реакции гидратации изобутилена порядок по катализатору в случае минеральных кислот равен единице, а в случае ГПК он переменный и выше первого. Рост порядка с ростом концентрации ГПК обусловлен солевым эффектом [20].
Надо отметить, что в кислотном катализе, как правило, используются вольфрамовые ГПК [75], поскольку они более сильные кислоты, чем Mo–V–фосфорные. Однако и последние обладают достаточной кислотностью: значения рН 0.3–0.2 М растворов ГПК-х лежат в диапазоне 0–1.5 [25].
Следует учитывать, что константы кислотной диссоциации ГПК-х, в отличие от вольфрамовых ГПК, уменьшаются при их восстановлении. Снижение кислотности ГПК-х в ходе восстановления происходит из-за протонирования аниона по уравнению (XVI) [1]:
(XVI)
${\text{Su}} + {\text{ГПА - }}х + m{{{\text{H}}}^{ + }} \to {\text{P}} + {{{\text{Н}}}_{m}}{\text{ГПА - }}х.$Таким образом, кислотно-каталитическая активность ГПК-х должна зависеть от восстановительной способности реакционной среды [23, 64].
Большое число опубликованных научных работ демонстрирует, что в первую очередь ГПК используются как кислотные катализаторы (в гомогенном и гетерогенном варианте). Тем не менее, имеющиеся на сегодняшний день данные о кислотности растворов ГПК часто противоречивы. Это происходит вследствие того, что применяемый классический индикаторный метод определения кислотности (когда в раствор кислоты вводится основание-индикатор, а затем осуществляется спектрофотометрическая индикация [64, 75]) в случае ГПК встречает большие трудности. Они заключаются в том, что многие ГПК окрашены, значительная часть из них полностью диссоциирована в водных растворах, поэтому константы их протонной диссоциации измеряют в неводных растворителях, где растворимость многих из них ограничена (например, в уксусной кислоте). Кроме того, ГПК в растворах взаимодействуют с полярными органическими молекулами, даже образуют ассоциаты, которые сохраняются, например, при экстракции. Можно полагать, что противоречивость данных о кислотности ГПК сдерживает их более широкое применение в кислотном катализе.
Не так давно появились работы [76, 77], авторы которых предприняли попытку разработать другой способ определения кислотности ряда растворов ГПК. Для этой цели использовали метод ЯМР на ядрах 13С, 14N, 17О, индикаторами кислотности были о-нитроанилин (традиционный индикатор Гаммета) и 2,6-диметил-γ-пирон. Был сделан вывод, что метод ЯМР 17О можно применять в качестве альтернативного для оценки кислотности водных растворов ГПК.
Коротко рассмотрим наиболее важные примеры кислотных реакций, катализируемых ГПК. Среди них промышленно важные реакции гидратации олефинов С3−С4. Например, гидратация пропилена – первый промышленный каталитический процесс на основе ГПК (1972 г.) [78]. Применение в этом процессе вместо минеральных кислот гомогенного катализатора H4SiW12O40 позволило получить более высокую селективность и уменьшить количество побочных продуктов [8]. В 1984 г. запущен процесс жидкофазной гидратации изобутена в изобутиловый спирт, а в 1989 г. – процесс гидратации 2-бутена во вторичный бутиловый спирт. Гомогенный катализатор этих процессов – Н3РМо12О40 [10]. Более эффективно, чем H2SO4 и HClO4, ГПК катализирует реакцию гидратации фенилацетилена [79].
Циклоалкены гидратируются в соответствующие спирты с селективностью 99% в присутствии катализатора, содержащего концентрированный раствор арилсульфокислоты и вольфрамовую ГПК [8].
Применение ГПК перспективно также в реакциях этерификации при синтезе лекарственных и парфюмерных препаратов, в производстве стабилизаторов, ингибиторов полимеризации и т.д. Например, этерификация пара-нитробензойной кислоты – стадия синтеза анестезирующих препаратов [79]. В присутствии H3PW12O40 выход продукта при 75°С составляет 99%. Изначально гомогенная реакционная смесь становится двухфазной в конце реакции. Верхняя фаза содержит продукт, а нижняя – концентрированный раствор ГПК.
Существует возможность использования ГПК в качестве катализаторов реакций конденсации. Так, в присутствии растворов H3PW12O40 при температурах 45–80°С происходит конденсация ацетона с образованием окиси мезитила и мезитилена [80]. H3PW12O40 и H4SiW12O40 являются эффективными катализаторами конденсации изофитола c 1-ацетокси-4-гидрокси-2-метилнафталином, а также с 2,3,5-триметилгидрохиноном. Эти реакции – важные стадии в синтезах витаминов К1 и Е соответственно. Процессы конденсации протекают с выходами выше 95% при температурах 90−120°С [81].
Следует отметить эффективность ГПК и в реакциях деалкилирования. Так, массивные и нанесенные вольфрамовые ГПК (0.5–7%) − это универсальные катализаторы полного транс-де-трет-бутилирования фенолов. Реакции протекают при температурах 100–140°С, а выход продукта за 0.2–4 ч достигает 92–98% [81].
Важный промышленный процесс полимеризации тетрагидрофурана (1985 г.) – одна из стадий синтеза полиуретанов. Процесс ведут при 60°С в двухфазной системе; катализатором служит H3PW12O40/H2O [4].
Эффективность ГПК отмечена и в реакциях циклоприсоединения. Авторы работы [82] впервые показали, что Н3PW12O40/SiO2 катализирует реакцию Дильса−Альдера, в которой взаимодействуют 1,4-нафтохинон и 2,3-диметилбутадиен. Полная конверсия исходного хинона наблюдалась за 30 мин, выход продукта составил 75%. Эта работа открыла перспективы для создания новых способов синтеза пара-хинонов, в которых в качестве бифункциональных катализаторов используются растворы ГПК-х (см. раздел 5.4).
Можно отметить также, что ГПК катализируют изомеризацию алканов [83], конверсию МеОН в олефины [84], алкилирование парафинов [85], олигомеризацию алкенов [86], реакции Фриделя–Крафтса [87] и др. Круг реакций, в которых ГПК применяются как кислотные катализаторы, очень широк.
4.2. Окислительные свойства растворов ГПК-х
Как было сказано выше, растворы Мо–V–P-ГПК-х за счет присутствия V(V) являются довольно сильными окислителями с редокс-потенциалом Е ~ 1 В (НВЭ) [1, 2, 23]. Поэтому растворы ГПК-х структуры Кеггина (Hx+ 3PMo12– xVxO40) очень часто используют в окислительном катализе. Обратимая окисляемость этих растворов [2, 6, 7, 10], позволяющая замкнуть каталитический двухстадийный цикл окисления субстрата кислородом (III) по реакциям (I) + (II), исключительно важна. При этом надо понимать, что окисление Su в растворе ГПК-х на стадии (I) происходит за счет кислорода молекул воды.
Следует заметить, что ГПК-х катализируют реакции окисления, где в качестве окислителя применяется не только кислород, но и пероксид водорода, алкилгидропероксиды, реже другие окислители. Оценку их перспективности для каталитических процессов с участием ГПК-х сделал C.L. Hill в обзоре [6]. На основе анализа большого числа разнообразных окислителей автор заключил, что с экологической и коммерческой точек зрения наиболее привлекательным является О2.
4.2.1. Редокс-потенциалы водных растворов ГПК-х. Первые данные о зависимости скорости окисления Su от окислительных свойств растворов ГПК-х были получены при изучении реакции окисления этилена в ацетальдегид в присутствии водных растворов ГПК-х + Pd(II) [88]. Именно в этой работе была найдена корреляция между скоростью реакции и значениями Е растворов ГПК-х. Позже было установлено, что Е раствора ГПК-х влияет не только на скорость, но и на селективность окислительного процесса. Например, такие данные имеются для окисления 2-метилнафтола-1 в 2-метил-1,4-нафтохинон [22, 89, 90] и 2,3,6-триметилфенола в 2,3,5-триметил-1,4-бензохинон [21] в растворах ГПК-х. Отсюда следует, что активность и селективность гомогенных катализаторов на основе ГПК-х можно регулировать, изменяя значения Е их растворов.
Для эффективного использования растворов ГПК-х в качестве гомогенных катализаторов окисления необходимо знать величину Е в условиях каталитической реакции. В работах [62, 91, 92] представлены количественные данные о редокс-потенциалах растворов ГПК-х, однако они получены в присутствии посторонних солей (сульфатов), которые не участвуют в каталитических реакциях, но влияют на состав растворов ГПК-х. Выше было показано, что в кислых частично восстановленных растворах ГПК-х находятся оксокатионы ${\text{VO}}_{2}^{ + }$ и VO2+. Оба этих катиона образуют комплексы с анионом ${\text{SO}}_{{\text{4}}}^{{{\text{2}} - }}$ [93]. Несомненно, это отражается на составе частиц в растворах ГПК-х и измеренных в работах [62, 91, 92] значениях Е. Ранее при исследовании Е такое комплексообразование не учитывалось, и для обеспечения постоянной величины рН в растворы ГПК-х обычно добавляли H2SO4 [62, 94]. Кроме того, некоторое количество H2SO4 попадало в растворы ГПК-х при их синтезе по устаревшей методике с использованием экстракции эфиром [66].
В стандартных условиях ([H+] = 1 M или рН 0 [95]) равновесия деструктивной диссоциации ГПК-х типа (IV)−(VI) очень сильно сдвинуты вправо. В таких условиях ГПК-х в окислительном катализе, как правило, не используются, поскольку стадия (II) регенерации раствора ГПК-х кислородом при низких рН протекает очень медленно [96]. При применении ГПК-х в качестве катализаторов (0.2 < pH < 2) равновесия (IV)−(VI) смещены вправо в значительно меньшей степени. При таких нестандартных условиях мы исследовали редокс-потенциалы систем ГПК-х/HmГПК-х.
Восстановление частично протонированных ГП-анионов представляет собой одноэлектронные процессы, протекающие ступенчато. В ряде случаев это подтверждено экспериментально. Так, методом ЭПР доказано существование частично восстановленного аниона HаPVIVVVMo10${\text{O}}_{{{\text{40}}}}^{{{\text{(6}} - {\text{a)}} - }}$ [38]. На ступенчатое одноэлектронное восстановление ГПК-х указывают также кинетические данные [97].
При восстановлении растворов ГПК-х с х ≥ 2 их рН возрастает [90]. Это объясняется тем, что между отщепляемыми катионами ${\text{VO}}_{2}^{ + }$ и VO2+ существует равновесие (XVII) с участием Н+-ионов:
(XVII)
${\text{VO}}_{{2{\text{aq}}}}^{ + } + 2{{{\text{H}}}^{ + }} + {{{\text{e}}}^{ - }} \rightleftarrows {\text{VO}}_{{{\text{aq}}}}^{{2 + }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Редокс-система ${{{\text{VO}}_{2}^{ + }} \mathord{\left/ {\vphantom {{{\text{VO}}_{2}^{ + }} {{\text{V}}{{{\text{O}}}^{{{\text{2 + }}}}}}}} \right. \kern-0em} {{\text{V}}{{{\text{O}}}^{{{\text{2 + }}}}}}}$ (XVII) хорошо исследована как в стандартных, так и нестандартных условиях [98]. Для нее легко учесть влияние рН на значение Е, поэтому она была выбрана нами для сопоставления с исследуемыми каталитическими системами ГПК-х/HmГПК-х. В нашем случае по величине [V(IV)]∑ и известной концентрации ГПК-х можно легко вычислить степень восстановления раствора m = [V(IV)]∑/[ГПК-х] (может принимать целые и дробные значения в интервале 0 < m < x). Зная величины m и х, нетрудно определить соотношение окисленной и восстановленной форм ГПК-х ([V(V)]/[V(IV)]∑), равное (х – m)/m. Следовательно, значение Е раствора ГПК-х может быть выражено уравнением (1) через равновесные концентрации катионов ${\text{VO}}_{2}^{ + }$ и VO2+. Главной окисляющей частицей в растворах ГПК-х являются именно катионы ${\text{VO}}_{2}^{ + }.$
(1)
${{E}_{V}} = E_{V}^{0} + \frac{{RT}}{F}ln\frac{{[VO_{2}^{ + }]{{{[{{H}^{ + }}]}}^{2}}}}{{[V{{O}^{{2 + }}}]}},$Величина $E_{V}^{0}$ (стандартный потенциал) для реакции (XVII) составляет 1.00 В [95]. Редокс-потенциал пары катионов ${\text{VO}}_{2}^{ + }$/VO2+ равен редокс-потенциалу каждой пары ГП-анионов ГПА-хm/ ГПА-хm + 1, поскольку все они связаны между собой равновесиями (IV)−(VI). Наличие этих равновесий сглаживает отдельные ступени одноэлектронного восстановления ГПА, поэтому значение Е раствора ГПК-х снижается плавно.
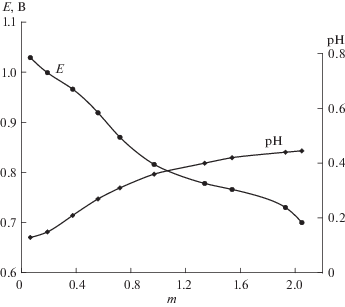
В работе [25] представлены результаты измерений Е и рН растворов HmГПК-х (х = 1–4) разных концентраций в зависимости от m. Показано, что для всех этих растворов значения Е снижается с увеличением m (рН возрастает). При этом чем меньше х, тем сильнее меняется −dE/dm. При m = х достигается полное восстановление всего V(V) в V(IV) в растворе ГПК-х.
Мы доказали, что зависимости Е = f(m) с успехом используются в качестве калибровочных кривых, по которым, измеряя Е раствора НmГПК-х, можно отслеживать состояние катализатора в ходе редокс-превращения [99]. В качестве примера на рис. 2 представлены зависимости Е и рН от m для кеггиновского раствора ГПК-2 [25]. Эти зависимости сохраняются также для регенерированных растворов кеггиновских и модифицированных ГПК-х. Во всех исследованных нами процессах (раздел 5) за состоянием катализаторов (т.е. за степенью их восстановления, m) следили с помощью зависимости Е = f(m).
4.2.2. Термохимия восстановления и окисления ГПК-х в водных растворах. Выше отмечено, что каталитические реакции типа (III) в присутствии растворов ГПК-х (например, окисление низших олефинов в карбонильные соединения [71], получение витамина К3 окислением 2-метилнафтола-1 [22, 90] и др.) из практических соображений нередко осуществляют раздельно по стадиям (I) и (II). Таким образом исключается прямой контакт окисляемого Su с кислородом в том случае, если О2 образует с ним (например, с C2H4) взрывоопасные смеси. Это необходимо также, если О2 инициирует различные побочные реакции – тогда стадии (I) и (II) также должны проводиться раздельно (см., например, [100]). Технология таких процессов, в свою очередь, требует знания тепловых эффектов и энтальпий каждой стадии: ΔrH1 = –q1/nSu и ΔrH2 = ${{ - {{q}_{2}}} \mathord{\left/ {\vphantom {{ - {{q}_{2}}} {{{n}_{{{{{\text{O}}}_{2}}}}}}}} \right. \kern-0em} {{{n}_{{{{{\text{O}}}_{2}}}}}}},$ где n − число молей, q1 и q2 – теплоты, а ΔrH1 и ΔrH2 − соответствующие энтальпии реакций (I) и (II).
Энтальпия суммарной реакции (III) легко находится как разность табличных энтальпий образования продукта SuO и субстрата Su: ∆rH3 = = ∆fHSuO – ∆fHSu. Однако энтальпии стадий (I) и (II) остаются неизвестными, поскольку в литературе нет надежных данных об энтальпиях редокс-превращений растворов ГПК-х. Детальные сведения об энтальпиях этих стадий нужны и для теории регулирования каталитической активности ионов ГПА-x, поскольку с изменениями энтальпий валентных переходов V(V) $ \rightleftharpoons $ V(IV) в ионах ГПА-x связаны изменения реакционной способности ГПК-x как в отношении субстрата, так и кислорода. Энтальпии зависят от значения m, а также от природы растворителя, влияющего на степень диссоциации ГПК-x и HmГПК-x по реакциям (IV)–(VI). Именно поэтому термохимические данные, представленные в этом разделе, внесли серьезный вклад в понимание механизмов редокс-превращений в растворах ГПК-х и возможности регулирования их каталитической активности. Остановимся на этом более подробно.
В расчетах тепловых эффектов реакций (I) и (II) достаточно знать разность энтальпий ∆fHHmГПК-х – ∆fHГПК-х, которую можно найти экспериментально, используя какую-либо модельную реакцию. Для каталитических реакций типа (III) стадия (II) является общей, но прямое измерение ее теплового эффекта затруднено из-за переменной и недостаточно высокой для таких измерений скорости реакции (II) [101]. Более надежным оказалось определение дифференциального теплового эффекта (q18i = −∆rH18) реакции (XVIII) селективного окисления гидразин-гидрата (ГГ) в водных растворах ГПК-х при ступенчатом наращивании m (т.е. при мгновенных значениях mi):
(XVIII)
$\begin{gathered} {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{4}} + {4 \mathord{\left/ {\vphantom {4 m}} \right. \kern-0em} m}\,\,{\text{ГПК - }}x \to {{{\text{N}}}_{2}}{\kern 1pt} \uparrow + \\ + \,\,{4 \mathord{\left/ {\vphantom {4 m}} \right. \kern-0em} m}\,\,{{{\text{H}}}_{m}}{\text{ГПК - }}x. \\ \end{gathered} $Из величин ∆rH18 и ∆rH19 достаточно легко вычислить ∆rH2 по уравнению (XX). Сумма реакций (XVIII) и (II) соответствует реакции (XIX) полного окисления ГГ:
(XIX)
${{{\text{N}}}_{{\text{2}}}}{{{\text{Н}}}_{4}} \cdot {{{\text{H}}}_{{\text{2}}}}{\text{O}} + {{{\text{O}}}_{2}}~ \to {{{\text{N}}}_{2}}{\kern 1pt} \uparrow + {\text{ }}3{{{\text{H}}}_{{\text{2}}}}{\text{O}}.$(XX)
${{\Delta }_{{\text{r}}}}{{{\text{H}}}_{2}} = {{\Delta }_{{\text{r}}}}{{{\text{H}}}_{{19}}} - {{\Delta }_{{\text{r}}}}{{{\text{H}}}_{{18}}} = - 606 - {{\Delta }_{{\text{r}}}}{{{\text{H}}}_{{18}}}.$Надо упомянуть, что в выполненном до нас термохимическом исследовании реакции (XVIII) [103] растворы ГПК-x содержали значительные концентрации анионов ${\text{SO}}_{4}^{{2 - }}{\text{.}}$ Это внесло большие неопределенности в состав частиц растворов, поскольку катионы ${\text{VO}}_{2}^{ + }$ и VO2+ образуют комплексы с ${\text{SO}}_{4}^{{2 - }}$ [93, 104]. Энтальпии образования сульфатных комплексов различны и по-разному зависят от рН растворов, на значения которых, в свою очередь, влияет m. Это, несомненно, отражалось на составе частиц в растворах и измеренных в [103] тепловых эффектах. Кроме того, в указанной работе восстановление растворов ГПК-x при 26°С протекало слишком медленно (до 80 мин), что влекло за собой неконтролируемые потери тепла вследствие рассеяния.
В нашем же исследовании [105, 106] растворы ГПК-х не содержали посторонних анионов-комплексообразователей, а реакция (XVIII) при 70°С заканчивалась за 10 мин, что сводило к минимуму влияние неконтролируемых факторов на результаты. В ходе опыта в раствор ГПК-х, находившийся в термостатированном реакторе-калориметре типа Тиана−Кальве [107] со стеклянной мешалкой и бюреткой для измерения объема N2, выделявшегося по реакции (XVIII), дробно, через резиновую мембрану водили шприцем малые порции раствора ГГ. Это позволяло ступенчато наращивать m и находить дифференциальные теплоты реакции (XVIII), q18i, зависящие от мгновенных значений mi.
Установлено, что дифференциальные энтальпии –∆rH18i при восстановлении растворов ГПК-х снижались по-разному. Так, для низкованадиевых растворов ГПК-1 и ГПК-2 величины –∆rH18i с ростом m уменьшались монотонно (рис. 3, кривые 1 и 2), причем перед полным восстановлением V(V) они падали особенно резко. Для раствора ГПК-3 (кривая 3) наблюдался максимум –∆rH18i при m ≈ 0.65, после которого значение –∆rH18i уменьшалось [105].
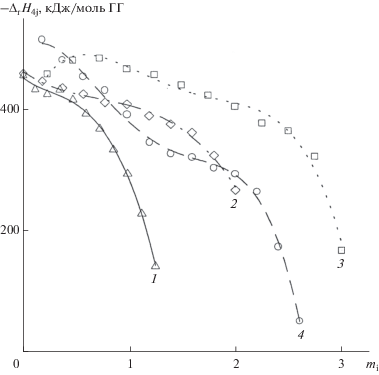
При восстановлении растворов двузамещенных Na+-, K+- и Mg2+-солей ГПК-3 тепловыделение было одинаковым (кривая 4), т.е. природа катиона не влияла на величину –∆rH18i. В то же время для солей ГПК-3 значение –∆rH18i было ниже, чем для свободной ГПК-3. Такое различие объясняется разными рН0 соответствующих 0.2 M растворов: для ГПК-3 pH0 ~ 0.4, а для ее двузамещенных солей pH0 ~ 0.6. При равных значениях рН одинаковыми оказались и величины тепловыделения, т.е. действительно, природа катиона не оказывает влияния на –∆rH18i.
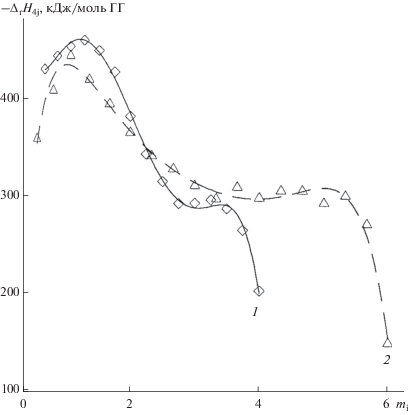
Для ГПК-x c x ≥ 4, как и для ГПК-3, зависимости –∆rH18i = f(mi) имели максимум в области малых значений mi (рис. 4). Оказалось, что у высокованадиевых ГПК-4 и ГПК-6 этот максимум более острый и сильнее сдвинут в сторону высоких значений m. Так, для раствора ГПК-3 он наблюдался при m ≈ 0.65 (рис. 3, кривая 3), а для ГПК-4 и ГПК-6 − при m ≈ 1.2. Для двух последних растворов после максимума можно выделить область m, где величины −∆rH18i практически не менялись и составляли 300 (±7) кДж/моль. Для ГПК-4 это область 2.5 ≤ m ≤ 3.5; для ГПК-6 – область ~3 ≤ m ≤ 5.8 [105].
Важно отметить, что для растворов ГПК-х, регенерированных кислородом по реакции (II), зависимости −∆rH18i = f(mi) точно совпали с аналогичными зависимостями для свежеприготовленных растворов ГПК-х. Это доказывает полную обратимость составов и свойств растворов ГПК-х в ходе редокс-реакций.
Степень диссоциации ГПК-x с отщеплением иона ${\text{VO}}_{2}^{ + }$ по реакции (IV) в кислых растворах возрастает с увеличением x и для 0.2 М раствора ГПК-3 достигает ~17% [68]. Поэтому при интерпретации величин –∆rH18i, относящихся к растворам ГПК-x, необходимо знать их значения для свободного иона ${\text{VO}}_{2}^{ + }$ в растворе.
Тепловыделение при восстановлении 0.1 М раствора VO2ClO4 в 1 М HClO4 почти не зависит от доли восстановленного ${\text{VO}}_{2}^{ + }$ и составляет ~435 (±6) кДж/моль ГГ. Эта величина близка к начальным значениям –∆rH18i при восстановлении ГПК-1 (431 ± 11 кДж/моль ГГ) и ГПК-2 (440 ± 14 кДж моль ГГ) (рис. 3). Однако в 4 М HClO4 наблюдается четкий максимум −∆rHi при [${\text{VO}}_{2}^{ + }$] ≈ [VO2+], отсутствующий в 1 М HClO4. Наличие такого максимума можно объяснить образованием из ${\text{VO}}_{2}^{ + }$ и VO2+ смешанно-валентного катиона ${{{\text{V}}}_{2}}{\text{O}}_{3}^{{3 + }}.$ В работе [108] спектрофотометрически доказано, что в ≥ 3 М HClO4 такой катион действительно существует.
Частично восстановленные 0.2 М растворы ГПК-х имеют значения pH в пределах 0.4 ≤ рН ≤ ≤ 1.5 [90]. В этой области катион ${\text{VO}}_{{2{\kern 1pt} {\text{aq}}}}^{ + }$ очень легко гидролизуется с появлением малорастворимой кислоты состава H2V12O31 ⋅ xH2O [109]; ее растворимость в 0.2 М HClO4 равна ~0.047 г-атом V/л. Между тем равновесная концентрация ${\text{VO}}_{2}^{ + }$ в 0.2 М растворе ГПК-3 составляет ~0.1 М, в 0.2 М растворе ГПК-4 − ~0.16 M, а в 0.2 М растворе ГПК-6 − ~0.4 М (определено в [68] методом ЯМР 51V). Это значительно выше, чем равновесная концентрация VO2+ в отсутствие ГПА. Отсюда следует вывод, что в растворах ГПК-х катион VO2+не является свободным, он находится во внешней координационной сфере ГПА. Таким образом, большой сольватированный ГПА в растворе (диаметр >1.3 нм [110]) можно уподобить кислой частице, на которой “адсорбированы” менее кислые катионы ${\text{VO}}_{2}^{ + }$ и VO2+, размеры которых не превышают 0.3 нм [111]. Следовательно, взаимодействие катионов VO2+ и VO2+ происходит не только в 4 М HClO4, но и во внешней координационной сфере ГПК-х с 3 ≤ x ≤ 6. Это вытекает из аналогичного положения максимумов рассматриваемой зависимости. Менее резкий максимум на кривых −∆rH18i = f(mi) для ГПК-х, по-видимому, объясняется меньшей энтальпией этой реакции во внешней координационной сфере ГПА.
Отсутствие максимума −∆rH18i = f(mi) для 0.2 М растворов ГПК-1, ГПК-2 и солей ГПК-3 вызвано малой равновесной концентрацией ${\text{VO}}_{2}^{ + }$ [68]. Согласно спектрам ЯМР 51V концентрация ${\text{VO}}_{2}^{ + }$ равна нулю в 0.2 М растворе ГПК-1, менее 0.01 М в 0.2 М растворе ГПК-2, но достигает ~0.1 М в 0.2 М растворе ГПК-3. С увеличением рН равновесная концентрация ${\text{VO}}_{2}^{ + }$ в растворах Мо–V–фосфатов падает [112], поэтому в растворах солей ГПК-3 она ниже, чем в самой ГПК-3.
Термохимические исследования [105, 106] позволили сделать ряд весьма важных выводов о механизмах редокс-превращений в растворах ГПК-х. Доказана определенная последовательность восстановления различных форм V(V) в этих растворах. В первую очередь (при малых m) восстанавливается свободный (равновесный) катион ${\text{VO}}_{2}^{ + },$ для которого −∆rH18i ≈ 435 кДж/моль. Для ГПК-2 восстановление этого квазисвободного иона ${\text{VO}}_{2}^{ + }$ имеет место при m ≤ 0.5. Следует отметить, что нормальный окислительный потенциал пары ${{{\text{VO}}_{{2{\text{aq}}}}^{ + }} \mathord{\left/ {\vphantom {{{\text{VO}}_{{2{\text{aq}}}}^{ + }} {{\text{VO}}_{{{\text{aq}}}}^{{2 + }}}}} \right. \kern-0em} {{\text{VO}}_{{{\text{aq}}}}^{{2 + }}}}$ (1.00 В [98]) выше, чем формальные окислительные потенциалы ГПК-х (0.68–0.715 В [91]). Поэтому в первую очередь, вероятнее всего, восстанавливается ${\text{VO}}_{2}^{ + }$ во внешней сфере ГПА. Для ГПК-x с 3 ≤ x ≤ 6 это происходит в области начальных и максимальных значений –∆rH18i. Во вторую очередь восстанавливается V(V) во внутренней координационной сфере ГПА. Это соответствует участкам кривых –∆rH18i = f(mi) с относительно малым углом наклона для ГПК-2 и ГПК-3 и приблизительно постоянным значением –∆rH18i ≈ 300 кДж/моль для ГПК-4 и ГПК-6 (рис. 4). Считаем, что на этих участках кривых частично восстановленная ГПК-х диссоциирует с отщеплением катиона VO2+. На последнем этапе (при m близких к х) –∆rH18i резко падает. Здесь происходит восстановление V(V) во внутренней координационной сфере ГПА без отщепления катиона VO2+.
Проведены расчеты энтальпий –∆rH2i реакции (II) окисления растворов HmГПК-x кислородом [105, 106]. Из уравнения (XX) следует, что изменения –∆rH2i и –∆rH18i носят противоположный характер. При уменьшении mi величины –ΔrH2i для исследованных ГПК-x (х = 1–6) падают, и для всех ГПК-х с х ≥ 3 имеется минимум ‒ΔrH2i при mi < 1. Обнаружено [101], что при уменьшении mi снижаются и скорости реакции (II), т.е. для этой реакции наблюдаются симбатные изменения ее скорости и –ΔrH2i.
Последовательность окисления кислородом различных форм V(IV) в растворе HmГПК-x обратна последовательности восстановления V(V). В первую очередь (при больших m) с высокими, но постепенно снижающимися энтальпиями –ΔrH2i окисляется V(IV) внутри координационной сферы ГПА. При средних значениях m происходит окисление внешнесферного VO2+, слабо связанного с ГПА, а образующийся V(V) входит во внутреннюю сферу ГПА. В последнюю очередь (с наименьшими –ΔrH2i, равными 120–180 кДж/моль O2) окисляется внешнесферный VO2+. Образовавшийся катион ${\text{VO}}_{2}^{ + }$ остается во внешней сфере ГПА. Внешнесферные катионы ${\text{VO}}_{2}^{ + }$ и VO2+ обратимо взаимодействуют между собой с превращением в катион ${{{\text{V}}}^{{\text{V}}}}{{{\text{V}}}^{{{\text{IV}}}}}{\text{O}}_{3}^{{3 + }}.$
Таким образом, термохимическое исследование редокс-превращений растворов ГПК-x не только подтвердило участие катионов ${\text{VO}}_{2}^{ + }$ и VO2+ в этих процессах, но и показало, что они могут взаимодействовать между собой во внешней координационной сфере ГПА, а также доказало полную обратимость изменений состава ГПК-х в ходе редокс-превращений в водных растворах по реакциям (I) и (II).
4.3. Физико-химические свойства растворов ГПК-х
В разделе 2 было продемонстрировано, что окисленные растворы ГПК-х являются сложными равновесными смесями. Состав же частично восстановленных растворов еще более сложен, поскольку в них появляются ГПА с разной степенью восстановления (НmГПА) и катионы VO2+ [25]. Это серьезно затрудняет исследование изменений физико-химических свойств растворов ГПК-x в редокс-процессах. Между тем для разработки технологий двухстадийных каталитических процессов окисления (III) с участием растворов ГПК-х необходимо знать, насколько сильно в ходе реакций (I) и (II) эти свойства меняются. Детальное изучение физико-химических свойств растворов ГПК-х проведено нами на примере одного из наиболее перспективных с технологической точки зрения высокованадиевых растворов брутто-состава Н12P3Mo18V7O85 (ГПК-7), результаты представлены в работах [113, 114].
4.3.1. Плотность и вязкость растворов ГПК-х. Катализаторы окисления органических соединений кислородом по схеме (I) + (II) на основе ряда модифицированных высокованадиевых растворов ГПК-х обладают достаточной термической устойчивостью, высокой окислительной емкостью и производительностью [115]. Физико-химические свойства таких катализаторов непрерывно меняются в ходе реакций (I) и (II). В работе [114] исследовано изменение свойств раствора (Pd(II) + + 0.25 М ГПК-7), используемого в качестве катализатора двухстадийного окисления кислородом бутена-1 в бутанон (метилэтилкетон) [71]. Установлено, что наибольшие значения вязкости (η), плотности (ρ) и pH этого катализатора достигаются после целевой реакции (I) при 60–90°С. Наименьшие значения этих параметров имеют место после реакции (II) при 150–160°С. Значение E, наоборот, максимально после регенерации по реакции (II) и минимально после реакции (I).
Известно, что катионы ${\text{VO}}_{2}^{ + }$ весьма устойчивы в сильнокислых растворах, поэтому из них не выпадают ванадийсодержащие осадки (V2O5 · xH2O). Это подтверждается известными диаграммами ‒pV = f(pH) [116, 117]. В ходе каталитических превращений катионы ${\text{VO}}_{2}^{ + }$ восстанавливаются по реакции (I) до катионов VO2+, которые образуют димерные катионы ${{{\text{V}}}^{{{\text{IV}}}}}{{{\text{V}}}^{{\text{V}}}}{\text{O}}_{{\text{3}}}^{{{\text{3 + }}}},$ также устойчивые в сильнокислой среде [118, 119]. Таким образом, именно высокая кислотность каталитической системы (Pd + ГПК-х) позволяет сохранять ее гомогенность в ходе редокс-превращений (I) + (II).
Плотности и вязкости исходных и восстановленных растворов ГПК-7 приведены в табл. 3 и 4 [113, 114].
Таблица 3.
Плотность и вязкость исходного раствора ГПК-7 при различных концентрациях и температурах (m = 0.35)
| Концентрация ГПК-7, моль/л | Показатели | Температура, °С | ||||
|---|---|---|---|---|---|---|
| 50 | 60 | 70 | 80 | 90 | ||
| 0.10 | Плотность, г/см3 Вязкость, сП |
1.277 0.785 |
1.271 0.664 |
1.264 0.575 |
1.257 0.501 |
1.252 0.439 |
| 0.25 | Плотность, г/см3 Вязкость, сП |
1.682 1.794 |
1.675 1.384 |
1.668 1.115 |
1.662 0.928 |
1.659 0.787 |
| 0.30 | Плотность, г/см3 Вязкость, сП |
1.797 2.610 |
1.791 1.969 |
1.784 1.548 |
1.780 1.230 |
1.779 1.030 |
Таблица 4.
Плотность и вязкость 0.25 M раствора ГПК-7 при различных значениях m и температурах
| m | Показатели | Температура, °С | ||||
|---|---|---|---|---|---|---|
| 50 | 60 | 70 | 80 | 90 | ||
| 0.35 | Плотность, г/см3 Вязкость, сП |
1.682 1.794 |
1.675 1.384 |
1.668 1.115 |
1.662 0.928 |
1.659 0.787 |
| 2.12 | Плотность, г/см3 Вязкость, сП |
1.684 1.966 |
1.677 1.502 |
1.670 1.202 |
1.665 0.987 |
1.661 0.879 |
| 4.22 | Плотность, г/см3 Вязкость, сП |
1.685 2.254 |
1.679 1.744 |
1.671 1.393 |
1.666 1.143 |
1.662 0.949 |
| 6.10 | Плотность, г/см3 Вязкость, сП |
1.686 2.311 |
1.680 1.760 |
1.673 1.404 |
1.667 1.161 |
1.663 0.979 |
Видно (табл. 4), что в исследованном интервале температур (50–90°С) величина ρ слабо увеличивается с ростом m и незначительно снижается с повышением Т. Вязкость же исходных и восстановленных растворов ГПК-7 возрастает с увеличением концентрации ГПК-7 (табл. 3) и резко уменьшается с ростом Т. В то же время с ростом m вязкость этих растворов становится заметно больше (табл. 4). Это объясняется присоединением H+-ионов к восстановленным ГПА [24], в результате чего в растворе число водородных связей становится больше. Заряды же ГПА при восстановлении в общем случае не возрастают.
Поскольку регенерацию катализатора по реакции (II) обычно проводят при 150–160°С [71], то именно для этих температур необходимо было знать значения ρ и η. Экстраполяция значений ρ−1 0.25 М раствора ГПК-7 в координатах ρ−1–T−1 дает нужные величины ρ150 = 1.626 г/см3 и ρ160 = = 1.621 г/см3 [114]. Температурная зависимость эффективной вязкости описывается как экспоненциальная функция (уравнение (2)) [120]:
(2)
$\eta = k {{e}^{{{{{{E}_{\eta }}} \mathord{\left/ {\vphantom {{{{E}_{\eta }}} {RT}}} \right. \kern-0em} {RT}}}}},$Это уравнение позволяет провести линейную экстраполяцию в координатах lg η–T −1. В итоге для исходного 0.25 М раствора ГПК-7 получили η150 ≈ 0.30 сП и η160 ≈ 0.26 сП, а для раствора, восстановленного до m = 6.10, η150 ≈ 0.36 сП и η160 ≈ 0.31 сП (рис. 5 [113]). Таким образом, при повышении температуры с 50 до 160°C вязкость исследованных растворов снижается в ~7 раз. Величина Eη, согласно нашему расчету, равна 20.7 ± ± 0.7 кДж/моль. Это заметно выше, чем величина Eη для растворов обычных электролитов [120].
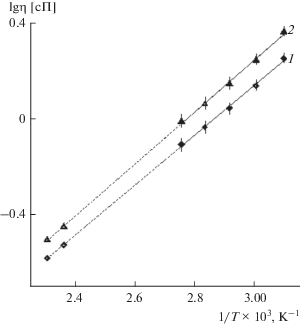
Катализаторы на основе растворов ГПК-х (ГПК-7 в частности) в процессах (I) + (II) используются как при низких, так и при повышенных температурах, а сами процессы стартуют с различных значений m. Поэтому при разработке технологий каталитических процессов (III), связанных с движением, перемешиванием, выпариванием и разбавлением гомогенных катализаторов, необходимо учитывать изменения вязкости растворов ГПК-х, определяемые параметром Eηуравнения (2). Важно понимать, что вышеприведенное экспериментальное значение Eη для 0.25 M раствора ГПК-7 отражает не только вклад уменьшения вязкости растворителя (воды) с повышением температуры, но и изменение констант равновесий в растворе [25].
После рассмотрения данных о существенных изменениях физико-химических свойств растворов ГПК-х в двухстадийных каталитических редокс-процессах (III) очень важно отметить, что высокованадиевые модифицированные растворы ГПК-х [70, 115] доказали свою устойчивость в циклических процессах по стадиям (I) + (II). В качестве примера в [113, 114] приведены результаты многоцикловых испытаний катализатора (Pd + + 0.25 M ГПК-7) в процессе окисления бутена-1 в метилэтилкетон, МЭК (табл. 5). Отметим, что низкие концентрации палладия не оказывали практически никакого влияния на физико-химические свойства раствора катализатора.
Таблица 5.
Изменения физико-химических свойств гомогенного катализатора (Pd + ГПК-7) в ходе многоциклового тестирования в процессе окисления бутена-1 кислородом в МЭК по схеме (I) + (II)
| Статус катализатора | [Pd] × 103, моль/л | ρ50, г/см3* | η50, сП* | Е25,В* |
|---|---|---|---|---|
| Перед испытаниями | 4.02 | 1.682 | 1.794 | 1.080 |
| После 1 цикла | 3.99 | 1.680 | 1.852 | 1.041 |
| После 2 цикла | 4.02 | 1.678 | 1.850 | 1.043 |
| После 5 цикла | 3.97 | 1.681 | 1.856 | 1.038 |
| После 10 цикла | 3.99 | 1.679 | 1.851 | 1.041 |
| После 15 цикла | 3.99 | 1.683 | 1.849 | 1.043 |
Примечание. Условия: Катализатор: V0 = 100 мл; [Pd]0 = 4 × 10–3 M; [ГПК-7] = 0.25 M. Реакция (I): T = 60°С, mвосст = 6.9–6.95. Реакция (II): Т = 160°С, ${{P}_{{{{{\text{O}}}_{2}}}}}$ = 0.4 МПа, τ = 20 мин [121]; mокисл = 0.97–1.03. * Стартовые значения ρ, η и Е измерены при m = 0.35, последующие – при m ≈ 1.
Многоцикловое тестирование в реакциях (I) + (II) подтвердило, что состав катализатора сохраняется, а его физико-химические свойства полностью обратимы [113, 114]. В ходе этих испытаний органические примеси, остающиеся в растворе катализатора после отделения МЭК, полностью окисляются при регенерации катализатора. Сохранение гомогенности катализатора в течение 15 циклов свидетельствует о достаточной термостойкости 0.25 M раствора ГПК-7 при температурах до 160°С (см. раздел 4.3.3).
4.3.2. Электропроводность растворов ГПК-х. При проектировании опытных и промышленных установок для редокс-процессов с использованием растворов ГПК-х (например, для пилотной установки синтеза МЭК [72, 121]) очень важно знать, как по ходу процесса изменяются не только рассмотренные выше физико-химические свойства этих растворов (ρ, η, рН, Е), но также и удельная электропроводность (χ). Однако в литературе не было данных о значениях χ для высокованадиевых растворов ГПК-х. В работе [122] на примере ГПК-7 мы впервые продемонстрировали, как изменяется χ раствора в ходе редокс-превращений (I) + (II). Были найдены величины χ для растворов [ГПК-7] при разных температурах и степенях восстановления m, а также обнаружена сильная зависимость χ от m (рис. 6) и показана связь χ с равновесным составом раствора ГПК-7.
Рис. 6.
Зависимость χ от степени восстановления m для 0.25 М раствора ГПК-7 при 60°С. Рисунок 6 адаптирован из работы [122] с разрешения издателя Pleiades Publishing.
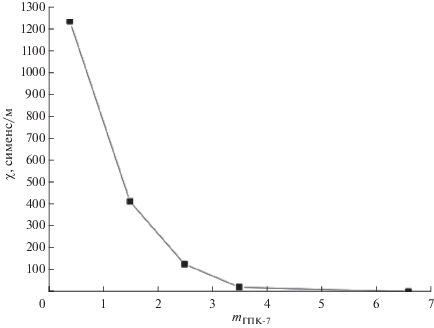
Подтверждено, что регенерированные растворы ГПК-7 полностью восстанавливают свою электропроводность, как и другие физико-химические свойства, доказывая тем самым высокую устойчивость модифицированных ГПК-х в качестве гомогенных катализаторов окислительных процессов и перспективность их практического использования.
4.3.3. Термостойкость растворов ГПК-х. Наши исследования продемонстрировали, что все высокованадиевые растворы кеггиновских ГПК-х (х > 2) имеют низкую термическую устойчивость при повышенных температурах реакции (II) [121]. Например, 0.2 M раствор ГПК-4 стабилен лишь при Т < 135°C. При бόльших температурах он начинает выделять ванадийсодержащие осадки. Недостаточная термостойкость растворов кеггиновских ГПК-х не позволяет проводить их быструю регенерацию при повышенных температурах [123] и поэтому является существенным недостатком для использования этих растворов в катализе. Именно поэтому на их основе не удалось создать высокотехнологичные промышленные катализаторы для окисления органических соединений кислородом по схеме (I) + (II).
В работе [115], посвященной синтезу модифицированных растворов ГПК-х некеггиновских составов, была детально исследована и их термостойкость. Представлены многоцикловые испытания 0.25 М растворов ГПК-х в редокс-превращениях по схеме (I) + (II), где на стадии (I) ГПК-х окисляла гидразин-гидрат при 100°С, а на стадии (II) происходило ее окисление молекулярным кислородом при температурах 130–190°C (табл. 6). Тут же для сравнения приведены данные для кеггиновского 0.2 М раствора ГПК-4.
Таблица 6.
Исследование термостойкости растворов модифицированных ГПК-7 и ГПК-10, а также кеггиновской ГПК-4 в циклической работе по реакциям (I) + (II) (по данным работы [115], получено разрешение на использование данных)
| Концентрация и брутто-состав раствора ГПК-х | Номер цикла | Количество осадка, выпавшего из раствора ГПК-х, г, при различных температурах регенерации кислородом по реакции (II), °С | ||||||
|---|---|---|---|---|---|---|---|---|
| 130 | 140 | 150 | 160 | 170 | 180 | 190 | ||
| 0.25 M H10P3Mo18V7O84 |
1 | Нет | Нет | Нет | Нет | Нет | Нет | Нет |
| 5 | Нет | Нет | Нет | Нет | Нет | Нет | Нет | |
| 10 | Нет | Нет | Нет | Нет | Нет | Нет | 0.24 | |
| 20 | Нет | Нет | Нет | Нет | Нет | Нет | 0.52 | |
| Σ, г a | Нет | Нет | Нет | Нет | Нет | Нет | 0.76 | |
| α, % b | 0 | 0 | 0 | 0 | 0 | 0 | 0.43 | |
| 0.25 M H11P3Mo16V10O86 |
1 | Нет | Нет | Нет | Нет | Нет | Нет | 0.38 |
| 5 | Нет | Нет | Нет | Нет | Нет | Нет | 0.95 | |
| 10 | Нет | Нет | Нет | Нет | Нет | 0.27 | 0.98 | |
| Σ, г a | Нет | Нет | Нет | Нет | Нет | 0.27 | 2.31 | |
| α, % b | 0 | 0 | 0 | 0 | 0 | 0.11 | 0.92 | |
| 0.20 M H7PMo8V4O40 |
1 | Нет | Нет | 0.24 | 0.96 | Опыты при этих температурах не проводили | ||
| 5 | Нет | 0.04 | 0.88 | 3.13 | ||||
| 10 | Нет | 0.14 | 0.97 | 3.38 | ||||
| Σ, г a | Нет | 0.18 | 2.09 | 5.07 | ||||
| α, % b | 0 | 0.23 | 2.61 | 9.34 | ||||
Примечание. Условия: Vкат = 50 мл; Т(I) = 100°C; время регенерации τ(II) = 30 мин при ${{P}_{{{{{\text{O}}}_{2}}}}}$ = 0.4 МПа [123]. a Σ – общее количество V-содержащих осадков во всех циклах. b α – доля ванадия, потерянного раствором ГПК-х в виде осадка.
Из данных табл. 6 следует, что растворы ГПК-10 и ГПК-7 устойчивы до 170 и 180°C соответственно. Это означает, что 0.25 M раствор ГПК-7 можно успешно многократно регенерировать кислородом при 170°C с сохранением его гомогенности. В то же время 0.2 M раствор ГПК-4 уже при 140°C начинает выделять ванадийсодержащие осадки. Следовательно, большее, чем 1/12, атомное отношение P : (Mo + V) обеспечивает повышенную термостойкость растворов ГПК-x.
Нетрудно оценить и эффективность модифицированных растворов ГПК-х в качестве окислителя. Так, по нашим данным, при 20-минутном окислении полностью восстановленного 0.25 M раствора ГПК‑7 при 170°C (${{P}_{{{{{\text{O}}}_{2}}}}}$ = 0.4 МПа) концентрация V(IV) падает с 1.75 до 0.20 M, в то время как при 130°C – лишь до 0.45 M. Легко подсчитать, что производительность этого раствора после его регенерации при 170°C на 15% выше в каждом цикле (I) + (II), чем после его регенерации при 130°C. Поэтому при разработке эффективных окислительных процессов по схеме (I) + + (II) регенерацию катализатора необходимо проводить при максимально возможной высокой температуре.
Таким образом, модифицированные растворы ГПК-х, устойчивые до 170°C, можно достаточно быстро регенерировать. Это открывают благоприятные перспективы создания экологичных малоотходных технологий каталитического синтеза органических соединений путем эффективного окисления различных субстратов кислородом в присутствии модифицированных растворов ГПК-х.
4.3.4. Коррозионная активность растворов ГПК-х. Используемые в процессах (I) + (II) водные растворы кеггиновских ГПК-х, во-первых, имеют сильнокислую реакцию (pH0 ~ 0.5) и, во-вторых, являются довольно сильными окислителями (E0 ≈ ≈ 1.0 В) [25, 99]. Еще более сильные кислоты (pH0 < 0) и окислители (E0 ≈ 1.10 В) – модифицированные растворы ГПК-х. При столь высокой кислотности и окислительной способности от этих растворов следует ожидать значительной коррозионной активности.
Очевидно, что все этапы двухстадийного процесса (III) следует проводить в коррозионноустойчивой аппаратуре. При этом из-за высокой температуры (около 160–170°С) наибольшая коррозионная нагрузка приходится на реактор для регенерации катализатора по реакции (II). Идеальным материалом для различных аппаратов процессов (III) был бы, по-видимому, титан, который очень мало корродирует даже в жестких окислительных средах [124, с. 114]. Однако запорную арматуру и регулирующую аппаратуру для реакторов таких процессов приходится изготовлять из нержавсталей.
До недавнего времени немногочисленные исследования коррозионной стойкости различных материалов ограничивались только водными растворами ГПК, не содержащими ванадия. В своей работе [125] мы постарались восполнить этот пробел за счет изучения коррозионной активности растворов Mo–V–Р-ГПК-х.
Материалами для коррозионных испытаний были титан (ВТ-0), нержавеющие стали 12Х18Н10Т и 10Х17Н13М2Т, железоникелевый сплав 06ХН28МДТ, а также никелевый сплав ХН65МВ (хастеллой С). Их составы приведены в табл. 7.
Таблица 7.
Составы исследованных сталей и сплавов*
| Марка стали (сплава) | Плотность, г/см | Содержание (%) | |||
|---|---|---|---|---|---|
| Fe | Cr | Ni | другие элементы | ||
| 12Х18Н10T | ~7.9 | ~70 | 17–19 | 9–11 | Mn (≤2.0), Ti (0.6–0.8), Si (≤0.8), Cu (≤0.3) |
| 10Х17Н13M2T | ~8.0 | ~65 | 16–18 | 12–14 | Mo (2–3), Mn (≤2), Si (≤0.8), Ti (≤0.7), Cu (≤0.3) |
| 06ХН28MДT | 7.95 | ~42 | 22–25 | 26–29 | Cu (2.5–3.5), Mo (2.5–3), Ti (0.5–0.9), Si (≤0.8) |
| Ni-сплав ХН65MВ (хастеллой C) |
9.03 | 4–5 | 14.5–16.5 | ~63 | Mo (15–17), W (3–5), Mn (≤2), Si (≤0.8), Ti (≤0.7) |
| Титан ВT-0 | 4.5 | 0.3 | – | – | Si (0.1), другие (0.3) |
* Составы сталей и никелевого сплава взяты из ГОСТа [126]. Прочерки означают, что соответствующие элементы в составе справа отсутствуют.
Скорость коррозии указанных образцов определяли гравиметрически в присутствии 0.25 М раствора ГПК-7 [125]. Оказалось, что наименьшая скорость коррозии наблюдалась для титана. Этот показатель для нержавеющих сталей и никелевых сплавов возрастала в ряду: 06ХН28МДТ < < 10Х17Н13М2Т < 12Х18Н10Т $ \ll $ ХН65МВ. Согласно классификации, приведенной в [124], образцы нержавсталей 06ХН28МДТ и 10Х17Н13М2Т можно отнести к 1-му классу, а образец 12Х18Н10Т – ко 2-му классу коррозионной стойкости. Интересно, что довольно дорогой никелевый сплав хастеллой С (ХН65МВ), считающийся наиболее коррозионноустойчивым в сильнокислых средах, оказался наименее стойким в растворах ГПК-7 и был отнесен к 4-му классу. Большая скорость коррозии сплава ХН65МВ в растворе ГПК-7 подтверждается и результатами определения Fe, Cr и Ni в растворе после коррозионного испытания; найдено, что [Fe] ≈ 0.50 г/л, [Cr] ≈ 0.09 г/л, [Ni] ≈ ≈ 0.44 г/л. В то же время после испытаний нержавстали 12Х18Н10Т содержание этих металлов в растворе ГПК-7 не превышало 0.01 г/л.
Как известно, титан обладает коррозионной стойкостью в окислительных средах, к которым относятся, например, растворы K2Cr2O7, CuCl2 или FeCl3 [124, с. 186, 205], а также растворы HNO3 в широких пределах концентраций. В этих средах на поверхности металлического титана образуется прочная окисная пленка TiO2, состоящая из его модификаций – анатаза и рутила. При этом происходит самопассивация поверхности металла. Титан коррозионно нестоек лишь при очень низких редокс-потенциалах раствора (E < < –0.3 В), которые в каталитических процессах с участием ГПК-х не встречаются.
Полученные в работе [125] результаты позволили признать, что растворы гомогенных катализаторов на основе Mo–V–P-ГПК имеют не только уникальные физико-химические, но и своеобразные коррозионные свойства.
При реализации каталитических окислительных процессов по схеме (I) + (II) в присутствии растворов ГПК-х необходимо использовать коррозионностойкую аппаратуру. Наиболее стойким материалом в таких средах является титан. Поскольку этот металл не усиливает коррозию контактирующих с ним нержавеющих сталей, то стали 06ХН28МДТ и 10Х17Н13М2Т вполне пригодны для изготовления отдельных узлов установок для процессов (III).
5. КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ В ПРИСУТСТВИИ РАСТВОРОВ ГПК-х
Растворы Мо–V–Р-ГПК зарекомендовали себя в качестве высокоэффективных катализаторов различных окислительных процессов. Их можно использовать как в составе многокомпонентных катализаторов, так и в однокомпонентном варианте.
5.1. Низкотемпературное окисление СО в СО2 в присутствии гомогенного двухкомпонентного катализатора Pd(II) + ГПК-х. Гетерогенный вариант системы
Процесс окисления СО в СО2 был детально изучен в 1980-х гг. [127–132]. Удалось создать эффективную двухкомпонентную каталитическую систему на основе комплекса Pd(II) и растворов кеггиновских ГПК-х (х = 3–4), в присутствии которой оксид углерода количественно окислялся в СО2 при низких (комнатных) температурах. Новый гомогенный катализатор выгодно отличался от существующих гетерогенных катализаторов (гопкалитов), используемых для низкотемпературной очистки воздуха от СО (в частности, в противогазах), поскольку гопкалиты не могут работать во влажной атмосфере, где их активность очень быстро падает.
Детальное исследование кинетики гомогенного окисления СО в СО2 в присутствии каталитической системы Pd(II) + ГПК-х позволило установить зависимость механизма реакции от значения Е раствора ГПК-х. Реакция в такой системе протекает через дипалладиевые комплексы Pd(I), образование которых обеспечивает возможность реализации быстрого (слитного) маршрута реакции, в котором перенос двух электронов с СО на дипалладиевый комплекс происходит синхронно с переносом электрона с Pd на ГПК. Была показана возможность регулирования активности каталитической системы [129, 132].
Итогом этих работ стало создание активного при комнатных температурах гомогенного катализатора окисления СО в СО2, представляющего собой водный раствор Рd(II) + ГПК-х [133]. Однако у него был недостаток, заключавшийся в невысокой окислительной емкости раствора, напрямую зависящей от концентрации в нем V(V). Так, в 0.2 М растворе ГПК-4 концентрация V(V) равна только 0.8 М. В этой связи следует заметить, что высокованадиевые модифицированные растворы ГПК-х, в которых удалось создать концентрации V(V) от 1.75 до 2.5 М, в то время еще разработаны не были.
Кроме того, в ходе исследования выявился и еще один недостаток двухкомпонентной каталитической системы [133] – это недостаточная стабильность палладия в растворе. Дело в том, что окисление СО происходит в комплексе палладия, который при этом восстанавливается и тут же окисляется ГП-кислотой. Таким образом, в ходе реакции (I) происходит постепенное восстановление раствора ГПК-х, значение Е снижается и с некоторого момента становится настолько низким, что восстановленный палладий уже не может быть окислен раствором ГПК-х. В итоге это приводит к постепенному выпадению Pd из раствора катализатора в виде металла (зеркала). Даже последующая регенерация катализатора кислородом по реакции (II), при которой значение Е сильно возрастает, не позволяет полностью вернуть палладий в раствор. Поэтому активность гомогенного катализатора при эксплуатации постепенно снижается.
Для решения этой проблемы были проведены исследования по созданию гетерогенного катализатора (Pd + ГПК-х) [134, 135]. Опробован ряд носителей, лучшие результаты были достигнуты на катализаторе (Pd + ГПК-х)/SiO2. Доказано, что в случае гетерогенного катализатора механизм окисления СО аналогичен таковому для гомогенного катализатора. Реакция протекает только в присутствии влажного газа, т.е. когда на поверхности гетерогенного катализатора образуется пленка воды, в которой растворяется ГПК-х и происходит окисление СО. Новый нанесенный катализатор (Pd + ГПК-х)/SiO2 для низкотемпературного окисления СО кислородом, работающий в условиях высокой влажности очищаемых газов, был запатентован [136]. Гетерогенный катализатор, как и ожидалось, обладает более высокой стабильностью, однако скорость реакции в его присутствии заметно ниже, чем в растворе.
5.2. Окисление низших олефинов (С2−С4) в карбонильные соединения в присутствии гомогенной двухкомпонентной каталитической системы Pd(II) + ГПК-х. МЭК-процесс
Реакция окисления этилена в ацетальдегид в присутствии гомогенного катализатора Pd(II) + + ГПК-х была первой реакцией, в которой в начале 1970-х гг. растворы ГПК-х были использованы в качестве обратимо действующих окислителей (см. Введение) [16]. Этот высокоселективный (>98%) процесс был весьма перспективен, он даже вышел на опытно-промышленные испытания на Омском заводе СК в середине 1970-х гг. Однако несовершенство применяемых в то время растворов ГПК-х (низкие концентрации ванадия, высокое содержание сульфатов и неэффективная регенерация) не позволило реализовать процесс окисления этилена в промышленности. Тем не менее, детальное изучение механизма этой реакции в растворах ГПК-х [62, 88, 137] положило начало фундаментальному подходу к исследованию широкого круга реакций окисления в их присутствии.
Следует отметить, что гомогенная каталитическая система Pd(II) + ГПК-х оказалась очень эффективной и при окислении пропилена в ацетон, которое протекает быстро с селективностью выше 99%. Данные по кинетике реакции представлены в работе [138].
Наиболее продвинутой разработкой по использованию растворов ГПК-х в окислительном катализе стал процесс окисления н-бутиленов в метилэтилкетон, так называемый МЭК-процесс. Он отчасти повторил судьбу ацетальдегидного процесса, когда в конце 1987 года вышел на опытно-промышленные испытания на Омском заводе СК [139]. Активность катализатора оказалась очень высокой, однако его стабильность была недостаточной. Сульфатсодержащие растворы низкованадиевых ГПК-х (в основном ГПК-4) и их кислых солей в ходе эксплуатации выделяли осадки, содержащие ванадий и приводящие к падению производительности гомогенного катализатора Pd(II) + ГПК-х. Осадки постепенно забивали узлы опытной установки синтеза МЭК, и стало понятно, что катализатор такого состава не может быть рекомендован для создания промышленной технологии синтеза МЭК. Требовалось кардинально изменить состав раствора ГПК-х для обеспечения высокой стабильности и производительности катализатора.
Вернуться к МЭК-процессу нам удалось только в начале 2000-х гг., когда был разработан способ синтеза модифицированных (некеггиновских) высокованадиевых растворов ГПК-х [70, 115]. Концентрация V(V) в новых растворах достигала значений 2.5 М, в 2.5−3 раза превышая ее значения в тех растворах, которые раньше были использованы в пилотных испытаниях МЭК-процесса. Термическая же стабильность ряда модифицированных растворов оказалась очень высокой [115], по нашему мнению, вполне достаточной для обеспечения эффективности гомогенного катализатора Pd(II) + ГПК-х. Наличие новых, перспективных растворов ГПК-х позволило нам возобновить на их основе работы по совершенствованию двухстадийной технологии синтеза МЭК.
Взрывобезопасность МЭК-процесса обеспечивалась тем, что реакции (XXI) и (II), в сумме составляющие каталитическую реакцию (III), проводили в двух разных реакторах при разных условиях.
(XXI)
$\begin{gathered} {m \mathord{\left/ {\vphantom {m 2}} \right. \kern-0em} 2}\,\,н{\text{ - }}{{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{8}} + {\text{ГПК - }}x + {m \mathord{\left/ {\vphantom {m 2}} \right. \kern-0em} 2}{{{\text{Н}}}_{{\text{2}}}}{\text{О}}~\xrightarrow{{{\text{P}}{{{\text{d}}}^{{{\text{II}}}}}}} \\ \to {m \mathord{\left/ {\vphantom {m 2}} \right. \kern-0em} 2}{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CO}}{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{5}} + {{{\text{Н}}}_{m}}{\text{ГПК - }}x, \\ \end{gathered} $(II)
${{{\text{Н}}}_{m}}{\text{ГПК - }}x + {m \mathord{\left/ {\vphantom {m 4}} \right. \kern-0em} 4}{{{\text{O}}}_{2}} \to {\text{ГПК - }}x + {m \mathord{\left/ {\vphantom {m 2}} \right. \kern-0em} 2}{{{\text{H}}}_{{\text{2}}}}{\text{O}}.$В реакции (XXI) газообразные н-бутилены реагировали с раствором катализатора (Pd + ГПК-х) в бутиленовом реакторе при 50−60°С. Восстановленный на m электронов катализатор вместе с МЭК поступал в отпарную колонну, где из него отпаривали водный азеотроп МЭК-сырца. Далее восстановленный катализатор в воздушном реакторе окисляли по реакции (II) кислородом воздуха с регенерацией ГПК-х при 170°С и ${{Р}_{{{{{\text{О}}}_{2}}}}}$ = 0.4 МПа. Таким образом, в нестационарном двухстадийном каталитическом процессе синтеза МЭК путем окисления н-бутиленов кислородом ГПК-x, являясь обратимо действующим окислителем, фактически играла роль катализатора.
Реакции (XXI) и (II) являются сложными, состоящими из ряда элементарных стадий. Детальное их изучение позволило установить, что кинетика и механизм реакции (XXI) в присутствии катализаторов Pd(II) + ГПК-x с кеггиновскими и модифицированными растворами одинаковы [140, 141]. Те же выводы относятся и к протеканию реакции (II) на обоих катализаторах. Сделанное сопоставление дало возможность использовать многочисленные данные по кинетике реакций (XXI) и (II), полученные ранее на кеггиновских катализаторах, для технологических расчетов, относящихся к новым, модифицированным катализаторам.
Селективность нового катализатора в реакции (XXI) высока и составляет, как и ранее, 97−98%. А такие параметры МЭК-процесса, как производительность, срок службы и физико-химические свойства катализатора Pd(II) + ГПК-x, целиком и полностью определяются составом модифицированного раствора ГПК-х. Детали оптимизации МЭК-процесса отражены в патентах [142–144]. Производительность новых катализаторов возросла почти в 6 раз (до 180 гМЭК${\text{л}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1) по сравнению с катализаторами на основе кеггиновских ГПК-х [139]. Изменение состава ГПК-х позволило добиться существенного повышения термостойкости катализатора Pd + ГПК-x при всех рабочих температурах: от 40−60°С в бутиленовой реакции (XXI) и до 160−180°С на стадии регенерации катализатора по реакции (II).
Итогом нашего исследования стала разработка новой малоотходной двухстадийной технологии синтеза МЭК [143, 144], которая призвана обеспечить высокую экономичность и экологичность промышленного производства МЭК.
Для отработки малоотходной технологии МЭК-процесса на новых катализаторах на базе ООО “Синтез-Инжиниринг” (Нижегородская область, г. Дзержинск) была создана опытно-промышленная установка производительностью до 110 т МЭК в год. В установке реализована усовершенствованная технологическая схема, использованы измененные (по сравнению с опытными работами 1987 г.) конструкции аппаратов, позволяющие обеспечить оптимальные режимы на всех стадиях процесса.
Малоотходным является также новый метод получения модифицированных растворов ГПК-х, реализованный на катализаторной фабрике при опытно-промышленной установке синтеза МЭК для получения катализатора Pd(II) + ГПК-x [73]. Для полного заполнения пилотной установки было наработано 1350 л раствора катализатора. Схема установки представлена в работе [72], там же обсуждаются отдельные узлы установки и основные результаты испытаний.
В итоге, гомогенный катализатор на основе модифицированных ГПК-х продемонстрировал в МЭК-процессе высокую производительность и термостойкость. Тем не менее, остались вопросы к устойчивости палладия в растворе катализатора, поскольку его стабилизаторы [143, 144] в ходе длительной эксплуатации постепенно окислялись на этапе регенерации ГПК-х кислородом при 170°С. Однако, на наш взгляд, проблема устойчивости палладия в растворе катализатора является разрешимой при обеспечении четкого контроля степени восстановления ГПК-х в бутиленовой реакции (XXI). Нельзя допускать перевосстановления раствора ГПК-х. Завершение реакции (XXI) при m ≤ 6, когда значение Е раствора катализатора еще довольно высокое (~0.8 В), обеспечит удержание палладия в растворе и без его стабилизатора.
5.3. Окисление алкилфенолов бензольного и нафталинового рядов в соответствующие пара-хиноны в присутствии растворов ГПК-х (однокомпонентные каталитические системы)
5.3.1. Окисление 2-метилнафтола-1 в 2-метил-1,4-нафтохинон (витамин К3). Разработка промышленной технологии “Викасиб” синтеза витаминов группы К. Менадион (МД, 2-метил-1,4-нафтохинон, витамин К3) является родоначальником всех витаминов группы К: из него получают викасол – водорастворимую форму витамина К3, витамины К4 и К1. МД в виде викасола широко применяют в медицинской практике для лечения многих заболеваний, а также в животноводстве для повышения продуктивности всех видов животных [145].
Основной способ промышленного получения МД – некаталитическое окисление 2-метилнафталина (МН) хромовой смесью с использованием 5-кратного избытка CrO3 [146]. Производство витамина К3 по этой технологии неэкологично вследствие обилия сточных вод, содержащих токсичные соединения хрома, смолы и кислоты. Производство также и неэкономично, поскольку МН – это труднодоступное сырье, а селективность целевой реакции не превышает 50%. Предложен ряд альтернативных способов синтеза МД, однако все они не более эффективны, чем существующий промышленный метод. К их недостаткам относятся многостадийность синтеза и неэкологичность.
В начале 1990-х гг. в ИК СО РАН была начата работа по использованию растворов ГПК-х в качестве катализаторов окисления 2-метилнафтола-1 в МД кислородом с целью создания новой малоотходной технологии синтеза витамина К3. На первом этапе работа велась совместно с коллегами из НИОХ СО РАН (г. Новосибирск).
В наших исследованиях показано, что наиболее селективными из всех существующих способов синтеза МД являются те, которые основаны на каталитическом окислении кислородом не 2-метилнафталина, как в промышленности, а другого субстрата − 2-метилнафтола-1 (2МН1). В качестве катализаторов были использованы кеггиновские растворы ГПК-х общей формулы H3 +хPVхMo12– хO40 (х ≤ 4) [89, 147–150]. Следует отметить, что, в отличие от МН, 2МН1 – вполне доступное сырьем, его можно получить метилированием нафтола-1 [151]. Этот способ, также разработанный в ИК СО РАН, был применен нами для наработки сырья.
Суммарный каталитический процесс окисления 2МН1 кислородом в МД в присутствии растворов ГПК-х описывается уравнением (XXII). Процесс осуществляли в двухстадийном варианте, при котором стадии окисления субстрата и регенерация ГПК-х кислородом протекали в разных условиях и разных реакторах.
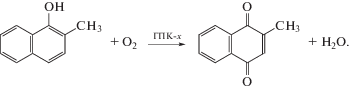
Окисление 2МН1 проводили в двухфазной системе: водный раствор ГПК-х и раствор Su и продуктов реакции в не смешивающемся с водой органическом растворителе. Реакцию вели в отсутствие О2 (в инертной атмосфере). Продукты реакции отделяли от катализатора путем разделения фаз, после чего катализатор регенерировали кислородом по реакции (II) [123] и использовали на следующем каталитическом цикле.
Результаты, полученные при изучении процесса окисления 2МН1 в МД в растворах ГПК-х [89, 147–150], позволили нам создать новую малоотходную технологию “Викасиб” (Викасол сибирский) синтеза витаминов группы К. Основные этапы этой технологии были запатентованы в 1994 году [147], после чего в течение ряда лет мы продолжали совершенствовать все ее стадии. Тем не менее, в присутствии кеггиновских ГПК-х так и не удалось добиться селективности катализаторов в целевой реакции свыше 83%. Только после того, как были предложены способы синтеза модифицированных растворов ГПК-х [70, 115], мы получили гораздо более эффективные катализаторы. Именно на основе растворов модифицированных ГПК-х был разработан новый способ окисления 2МН1 в менадион – ключевой стадии технологии “Викасиб” [22].
В ходе оптимизации процесса синтеза МД было установлено, что лучшие параметры реакции обеспечивают ГПК-х, содержащие не менее 6 атомов ванадия в своем составе (т.е. при [V] ≥ ≥ 1.2 M), при суммарном мольном отношении ГПК-х : 2МН1 ≥ 2.5. Для снижения вероятности побочных радикальных процессов целевую реакцию следует проводить в инертной атмосфере, так как на воздухе селективность (S) уменьшается на 8–10%.
Увеличение концентрации уксусной кислоты в системе до 15 об. % способствует некоторому росту селективности реакции окисления 2МН1 в МД, которая в оптимальных условиях достигает 90% [22].
Новый каталитический двухстадийный способ получения МД по реакции (XXII) в растворах ГПК-х перспективен для создания высокоэкологичной технологии “Викасиб” производства витаминов группы К [22, 150]. Основные элементы этой технологии [147] были отработаны нами совместно с коллегами из НИОХ СО РАН в пилотном варианте в 1994 г. на базе опытной установки НИОХ. Во время этих испытаний окисление 2МН1 проводили в присутствии кеггиновских ГПК-х. Было синтезировано более 1 кг МД высокой чистоты (> 98%), из которого в лабораторном масштабе в ИК были наработаны опытные партии викасола (водорастворимой формы витамина К3) по методике [152] и диацетата менадиола (витамина К4) [153]. Полученные по этой технологии викасол и витамин К4 прошли биологические испытания в животноводческих отраслях сельского хозяйства Новосибирской области и Алтайского края (1993−1999 гг.). На основании результатов испытаний был выдан сертификат, разрешающий использовать викасол (технология “Викасиб”, ИК СО РАН) в кормлении сельскохозяйственных животных.
5.3.2. Окисление 2,3,6-триметилфенола в 2,3,5-триметил-1,4-бензохинон – ключевой полупродукт синтеза витамина Е. В многостадийном синтезе витамина Е ключевым полупродуктом является 2,3,5-триметил-1,4-бензохинон (ТМХ). Как правило, его получают окислением 2,3,6-триметилфенола (ТМФ). Известно много способов каталитического окисления ТМФ кислородом в жидкой фазе по реакции (XXIII) [154]:
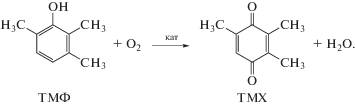
В промышленных способах, предложенных в 1970–1980-х гг., реакцию (XXIII) осуществляли гомогенно. Катализатором служила хлорная медь (СuCl2), а растворителем была водно-органическая смесь из диметилформамида (ДМФ), моноглима или ацетона [155]. При этом ДМФ выполнял функции как растворителя ТМФ и ТМХ, так и стабилизатора восстановленной формы катализатора (СuCl) в растворе. Во многих патентах применялся тот же катализатор в комбинации с водно-органическими смесями различных растворителей и комплексообразователей, обеспечивавших гомогенность системы [156–158]. Следует заметить, что селективность указанных методов не превышает 80%, а катализаторы имеют ряд недостатков, главный из которых − их способность хлорировать часть ТМХ в экологически опасные хлорхиноны.
Основным усовершенствованием в современных промышленных способах реализации реакции (XXIII) явилось использование двухфазных систем, впервые осуществленное фирмой “Mitsubishi” в 1987 г. [159]. Этот процесс предусматривал использование того же катализатора (CuCl2) в водном растворе с добавками МCl и NH4Cl. Его применяли в сочетании с органическим растворителем (алифатическим спиртом С4–С10), в котором находились ТМФ и ТМХ. В этом случае продукты реакции отделяли от катализатора простым разделением фаз. Способ обеспечивал селективность до 96%.
В последующих патентах различались составы органических растворителей, но составы катализаторов принципиальных изменений не претерпели [160, 161]. Следовательно, в новых процессах упростился только метод отделения продуктов реакции от катализатора, а хлорирующая способность и невысокая активность катализатора в целевой реакции остались прежними. Казалось, что исчерпаны все возможности оптимизации катализатора, поскольку для устранения его хлорирующей способности требовалось значительно снизить концентрацию ионов хлора в растворе, но это привело бы к полной потере его активности, так как в отсутствие большого избытка ионов хлора Cu2+-ионы теряют окислительные свойства [162] и не могут катализировать реакцию (XXIII).
Однако в начале 1990-х гг. наметился альтернативный путь реализации реакции (XXIII), когда были предложены способы окисления ТМФ в ТМХ, в которых в качестве катализаторов использовались растворы кеггиновских ГПК-х (1 ≤ ≤ x ≤ 6) или их кислых солей [163–165]. В частности, в работах сотрудников ИК СО РАН [164, 165] гомогенную реакцию проводили в 75–90%-ной водной уксусной кислоте при 20–70°С в атмосфере кислорода. Концентрацию ГПК-х варьировали в интервале 0.005−0.05 моль/л, а мольное отношение ТМФ/ГПК-х – в интервале 4–40. Лучшие результаты получены в присутствии ГПК-4 (S ≈ 86%) [164]. Продукты реакции отделяли экстракцией CCl4, а катализатор после регенерации применяли повторно (4 цикла). К ограничениям этого способа, кроме невысокой селективности, можно отнести сложность отделения катализатора и воспроизведения его состава от цикла к циклу.
Важно отметить, что, несмотря на умеренную селективность, катализаторы на основе ГПК-х не давали продуктов хлорирования и имели высокую начальную активность. Именно этим предложенный в патенте [164] уксуснокислый метод окисления ТМФ по уравнению (XXIII) заинтересовал нас с точки зрения оценки реальной возможности его оптимизации.
В итоге нашего исследования был разработан эффективный двухстадийный способ проведения реакции (XXIII) [166], в котором недостатки процесса с использованием уксусной кислоты удалось преодолеть применением двухфазной системы (водный раствор ГПК-х + раствор Su в органическом растворителе) в атмосфере О2 (при ${{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}$ = = 0.1 МПа, а также под давлением до ${{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}$ = 1.1 МПа). В первом варианте для окисления ТМФ в таких системах были использованы растворы ГПК-х структуры Кеггина.
Проведение реакции (XXIII) в среде несмешивающихся растворителей с последующей регенерацией катализатора на отдельной стадии при температурах до 150°С и давлении кислорода 0.4 МПа позволило решить проблему сильного недоокисления соли ГПК-4 в способе [164]. Более глубокая регенерация катализатора обеспечила рост S до 96%, однако вопрос о невысокой производительности в присутствии кеггиновских ГПК-х остался. Кроме того, эти растворы были недостаточно стабильны при высоких температурах (на этапе регенерации), поэтому при многоцикловой эксплуатации в них постепенно накапливались ванадийсодержащие осадки.
Гидролитически устойчивые и технологичные катализаторы окисления ТМФ в ТМХ [21] были созданы после разработки методов синтеза растворов модифицированных ГПК-х некеггиновских составов [70, 115]. Высокие концентрации ванадия в растворах модифицированных ГПК-х обеспечили бόльшие значения Е [115], что позволило существенно улучшить производительность реакции (XXIII). Новые катализаторы успешно прошли испытания на устойчивость в широком интервале концентраций (0.1−0.35 моль/л), они сохраняли свою гомогенность на всех этапах двухстадийного процесса окисления ТМФ по реакции (XXIII) [167–169].
Установлено, что природа органического растворителя сильно влияет на селективность катализатора. При вариации различных растворителей S убывала в ряду: н-спирты > н-карбоновые кислоты > их сложные эфиры > кетоны > простые эфиры. Лучшие значения S (>98%) получены в присутствии первичных неразветвленных спиртов C5−C12 [166].
Максимальных значений S (98.5−99.5%) и производительности катализатора (600−800 гТМХ${\text{л}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1) удалось достигнуть с модифицированными 0.2 М растворами ГПК-х, концентрация ванадия в которых находилась в интервале 1.2−2.4 М (рис. 7, по данным работы [166]).
Рис. 7.
Зависимость селективности (S) и производительности (П) катализатора окисления ТМФ в ТМХ от числа атомов ванадия в составе модифицированной ГПК-х' [166]. Условия: 60°С, атмосфера О2; [ГПК-х']= 0.2 М; органический растворитель – н-C6H13OH. Рисунок 7 адаптирован из работы [166] с разрешения журнала “Катализ в промышленности”.
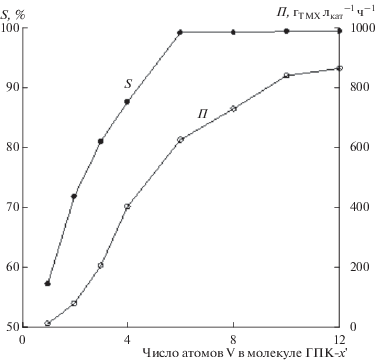
Возможность окисления восстановленных модифицированных растворов ГПК-х при температурах до 170°С обеспечивает их быструю глубокую регенерацию, что при высокой концентрации V(V) в растворе гарантирует сохранение селективности реакции до 99.5% на последующих циклах. Образцы модифицированных катализаторов были испытаны в 45−50 циклах, их селективность и производительность не менялись [166, 169].
В работе [170] были детально проанализированы различные технологические варианты использования растворов ГПК-х в реакции (XXIII): одностадийный гомогенный уксуснокислый способ окисления ТМФ [164] и двухфазный двухстадийный способ [21]. Доказана эффективность двухфазного варианта, обеспечивающего высокие показатели S (до 99.5%) и производительности катализатора (>600 гТМХ${\text{л}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1).
Селективность катализатора в реакции (XXIII) определяется относительными скоростями основного и побочных превращений (рекомбинация радикалов) на отдельных этапах реакции. Для быстрого переноса 4-х электронов с молекулы ТМФ на окислитель необходимо, чтобы редокс-потенциал раствора ГПК-х оставался высоким в ходе всей целевой реакции. Это обеспечивается повышенной концентрацией ГПК-х (≥0.2 М) и ее избытком по отношению к Su. Установлено, что при этих условиях Е раствора ГПК-х в ходе реакции всегда остается заведомо больше 0.85 В (относительно НВЭ) [169].
Для реакции (XXIII) каталитические системы на основе растворов модифицированных ГПК-х имеют самые лучшие показатели селективности и чистоты продукта по сравнению со всеми известными способами. Важно, что высокая чистота получаемого ТМХ может значительно увеличить эффективность последующих стадий синтеза витамина Е и облегчит его выделение. Поэтому разработанный нами метод [21] перспективен для использования в новой экологичной технологии синтеза витамина Е.
Избыток ГПК-х по отношению к ТМФ не является недостатком предлагаемого процесса [21], поскольку растворы на основе модифицированных ГПК-х обладают высокой гидролитической и термической стабильностью и способны долгое время (по нашим данным – несколько месяцев) работать без потери активности и производительности. Необходимо только очень аккуратно обращаться с этими растворами, не допуская их концентрирования и перегревания, тщательно собирать даже малые порции раствора ГПК-х при разделении фаз после целевой реакции. Правильная эксплуатация модифицированных растворов ГПК-х откроет широкие перспективы их длительного, многоциклового применения в качестве катализаторов не только для окисления ТМФ в ТМХ, но и в других окислительных процессах.
5.3.3. Окисление диалкилфенолов в соответствующие пара-бензохиноны. 1,4-Бензохиноны, обладающие несколькими реакционными центрами (С=С и С=О), могут участвовать в различных реакциях, таких как аминирование, ацилирование, алкилирование, арилирование, а также циклоприсоединение, образуя молекулы сложной структуры [171]. Интересующие нас диалкилбензохиноны (ДАБХ) также способны вступать в эти реакции, поэтому они широко используются в качестве предшественников для получения различных природных и синтетических соединений – лекарств и физиологически активных веществ. Например, 2,6-диметил-1,4-бензохинон (ДМХ) является предшественником для синтеза кемпанов (тетрациклических дитерпенов) и их производных [172–174], имеющих применение в качестве инсектицидов [175]. Диполярные соединения, такие как мезитонитрил N-оксид и азометин-илид, вступают в реакции циклоприсоединения по связи С=С хинонов с образованием производных изоксазолина [176] и изоиндола [177]. Подобным реакциям 1,3- и 1,4-диполярного циклоприсоединения уделяется особое внимание в органическом синтезе, поскольку они позволяют получать пяти- и шестичленные гетероциклы в одну стадию. Авторы [178] показали, что диметил-1,4-бензохиноны взаимодействуют с аллилтриметилсиланом в присутствии Bi(OSO2CF3)3, образуя аллилзамещенные гидрохиноны с селективностью 85–90%. Производные бензофурана, кислородсодержащего аналога индола, синтезированы авторами [179] при взаимодействии ДАБХ с 1,3-дикарбонильными соединениями в присутствии Cu(OSO2CF3)2 в качестве катализатора.
В ряду важных ДАБХ следует упомянуть ди-трет-бутил-1,4-бензохинон (ДТХ), который является полупродуктом синтеза разнообразных парфюмерных и фармацевтических препаратов [180], а получаемый из него 2,6-ди-трет-бутил-1,4-гидрохинон – высокоэффективный антистаритель и антиоксидант [181].
Легкость восстановления хинонов до гидрохинонов открывает возможность их применения в качестве окислителей и дегидрирующих агентов. Так, авторы работ [182, 183] описали метод редокс-конденсации в присутствии метилбензохинона, позволяющий превращать третичные спирты в соответствующие эфиры с почти полной инверсией конфигурации. Кроме того, ДАБХ имеют много потенциальных применений в составе амперометрических сенсоров и фоторефрактивных систем [184, 185]. Синтетический потенциал ДАБХ обширен, поэтому они и привлекли наше внимание.
В начале 2000-х гг. мы начали исследовать возможность создания эффективных способов окисления 2,6-диалкилфенолов (ДАФ) в соответствующие 2,6-ДАБХ на примере 2,6-диметил- и 2,6-ди-трет-бутилфенолов. Продукты этих реакций важны, но труднодоступны, их рыночная стоимость довольно высока. Поэтому поиск недорогих методов синтеза ДМХ и ДТХ был весьма актуален.
Сегодня наиболее распространены те методы, в которых синтез ДАБХ осуществляют путем каталитического окисления соответствующего ДАФ кислородом. Известно немало катализаторов таких реакций [186–191], однако у всех есть недостатки. Главными из них являются неустойчивость катализаторов к окислению, неполная регенерируемость, неполнота превращения субстрата, трудности выделения целевого продукта и неэкологичность (наличие отходов тяжелых металлов).
Нашей целью было найти каталитическую систему, подходящую для разработки промышленных способов двухстадийного жидкофазного окисления ДАФ в ДАБХ кислородом по реакциям (XXIV) + + (II). Критериями выбора оптимального катализатора реакции (XXIV) были высокая селективность, достаточная активность, технологичность использования и экологическая безопасность. Целевую реакцию осуществляли в двухфазной системе (водный раствор ГПК-х + раствор Su и продуктов реакции в органическом растворителе).
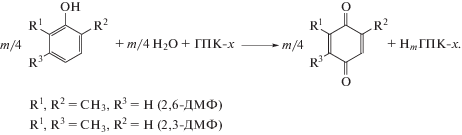
В наших первых работах [192, 193] (с использованием кеггиновских ГПК-х) показано, что в двухфазных системах в присутствии растворов ГПК-х можно с довольно высокой селективностью (до 85%) окислять ДАФ в соответствующие ДАБХ. Регенерацию катализатора кислородом проводили отдельно при температурах до 145°С [194].
Было установлено, что природа органического растворителя (ОР), состав ГПК-х, мольное отношение ГПК-х к субстрату, температура и концентрация уксусной кислоты в системе влияют на селективность целевой реакции [192, 193]. Особенно сильно S окисления ДАФ в ДАБХ зависит от природы ОР, являющегося компонентом двухфазной каталитической системы. Это подтверждают данные табл. 8 и 9 для ДМХ и ДТХ соответственно. Побочными продуктами таких реакций являются дифенохиноны и смолы различных составов.
Таблица 8.
Влияние природы органического растворителя на селективность катализатора окисления ДМФ в ДМХ (по данным работы [192], получено разрешение на использование данных)
| Номер цикла* | Органический растворитель | S, % |
|---|---|---|
| 1 | Трихлорэтилен | 85.3 |
| 2 | Хлороформ | 55.6 |
| 3 | Октанол-1 | 57.7 |
| 4 | н-Амилацетат | 62.5 |
| 5 | Октанон-2 | 59.8 |
| 6 | Каприловая кислота | 72.0 |
| 7 | м-Ксилол | 46.6 |
| 8 | Толуол | 49.3 |
| 9 | Бензиловый спирт | 41.9 |
| 10 | 3-Фенилпропанол | 48.7 |
| 11 | Ундекановая кислота | 37.6 |
Примечание. Условия реакции: 60°С, атмосфера СО2; [Co0.5Н6PMo8V4O40] = 0.3 М; [HOАс] = 15 об. %; [Co0.5ГПК-4]/ [ДМФ] = 7.5; время реакции 1 ч; конверсия 100%. * Все эксперименты проведены на одном и том же образце катализатора, регенерированном в каждом цикле по реакции (II) [194].
Таблица 9.
Влияние природы органического растворителя на активность и селективность катализатора окисления ДТФ в ДТХ (по данным работы [192], получено разрешение на использование данных)
| Номер цикла* | Органический растворитель | Время реакции, ч | S, % |
|---|---|---|---|
| 1 | Четыреххлористый углерод | >5 | 69.0 |
| 2 | Трихлорэтилен | >4 | 71.4 |
| 3 | Толуол | 3.5 | 77.0 |
| 4 | Мезитилен | 4 | 70.2 |
| 5 | м-Ксилол | 3 | 71.9 |
| 6 | п-Ксилол | 2.5 | 72.2 |
| 7 | Октанол-1 | <1 | 20.8 |
| 8 | Бензиловый спирт | <1 | 22.7 |
| 9 | Каприловая кислота | <1 | 69.9 |
Примечание. Условия реакции: 80°С, атмосфера СО2; [Н9PMo6V6O40] = 0.3 М; [HOАс] = 30 об. %; [ГПК-6]/[ДТФ] = = 7.5; конверсия 100%. * Все опыты проведены на одном и том же образце катализатора, регенерированном в каждом цикле по реакции (II) [194].
В окислении ДМФ наибольшее значение S (85%) достигается при использовании трихлорэтилена (табл. 8), тогда как для окисления ДТФ лучшим растворителем оказался толуол (табл. 9) [192]. В его присутствии S составляет 77%. Из данных табл. 9 видно, что при замене неполярных или слабо полярных растворителей на полярные (спирты) селективность катализатора окисления ДТФ падает до 20%.
Природа ОР сильно влияет не только на S катализатора, но и на его активность. Об этом можно судить по времени полного превращения субстрата, например, ДТФ (табл. 9). Подобрав оптимальный растворитель, можно увеличивать активность и селективность каталитической системы (ГПК-х + + ОР). При этом молекулы подходящих растворителей избирательно сольватируют “лишние” реакционные центры молекулы Su, подавляя побочные реакции с участием свободных радикалов, образующихся на промежуточных стадиях процесса (XXIV). Одновременно с этим растворитель, вероятно, наилучшим образом ориентирует молекулы субстрата относительно водной фазы с катализатором, обеспечивая ускорение целевой реакции (перенос электронов от Su на ГПК-х), протекающей на границе раздела фаз. Поэтому правильный подбор ОР – эффективный метод регулирования активности и селективности двухфазной каталитической системы (ГПК-х + ОР).
Реакции окисления ДМФ и ДТФ, как и реакции окисления других фенолов, протекают по радикальному механизму [195], первым этапом которого является образование феноксильного радикала, обладающего высокой реакционной способностью. Поэтому при окислении ДАФ помимо основной реакции могут идти побочные реакции димеризации этого радикала и дальнейшей полимеризации. Чтобы увеличить выход целевого хинона, потребовалось подобрать такие условия, которые обеспечивали бы скорость основной реакции, достаточную, чтобы снизить вклад побочных превращений. Мы показали [192, 193], что это достигается оптимизацией состава двухфазной каталитической системы (ГПК-х + ОР) и условий проведения реакции.
Так, было установлено, что в присутствии кислорода селективность катализатора падает на 30–40%, поскольку О2 инициирует побочные радикальные процессы. Поэтому суммарный каталитический процесс проводили нестационарно, в два этапа (XXIV) + (II), исключая присутствие О2 на стадии окисления фенолов раствором ГПК-х.
Введение катионов в раствор ГПК-х уменьшает его кислотность, но повышает скорость регенерации раствора по реакции (II) [101]. По этой причине в окислении ДАФ были исследованы не только свободные ГПК-х, но их кислые соли. Оказалось, что замещение одного протона в молекуле ГПК-х катионом Мz+ снижает селективность катализатора. Это показано, например, для кислых солей ГПК-4 в реакции окисления ДМФ [193]. Из полученных данных следует, что селективность солей ГПК-4, содержащих катионы Сo2+, Mg2+, Al3+, Mn2+, Na+, ниже таковой для ГПК-4. Наибольшее значение S, сравнимое с аналогичным значением для свободной ГПК-4 (73–76%), обеспечивают соли Mg0.5H6PMo8V4O40 и Со0.5H6PMo8V4O40. Снижение S окисления ДАФ при введении катионов в состав раствора ГПК-х объясняется уменьшением его редокс-потенциала [25, 90].
На примере окисления ДТФ показано, что концентрация уксусной кислоты в интервале 10−30 об. % слабо влияет на S, однако при ее дальнейшем увеличении значительно возрастает скорость целевой реакции [192, 193]. Так, при [НОAc] = 15 об. % за 4.5 ч конверсия ДТФ была 60%, а при [НОAc] = 40 об. %. ДТФ полностью окислялся за 1 ч. Эти результаты демонстрируют сильный сольватирующий эффект уксусной кислоты.
Зависимость селективности катализатора от температуры, найденная для окисления ДМФ в присутствии ГПК-4 [192], представлена на рис. 8. С ростом Т от 40 до 70°С значение S постепенно увеличивалось с 40 до 80%. Меньшая активность ДТФ по сравнению с ДМФ требовала для его окисления более высоких температур (≥80°С).
Рис. 8.
Зависимость селективности катализатора окисления ДМФ в ДМХ от температуры (по данным [192]). Условия: 60°С, атмосфера СО2; ОР – трихлорэтилен; [H7PMo8V4O40] = 0.3 М, [ГПК-4]/[ДМФ] = 7.5; [НОAc] = 20 об. Эксперименты проведены на одном и том же образце катализатора (с регенерацией). Рисунок 8 адаптирован из работы [192] с разрешения журнала Journal of Molecular Catalysis A: Chemical.
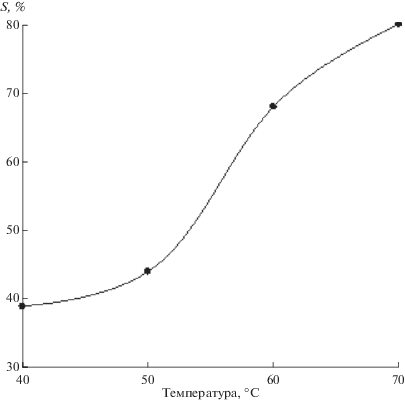
Представленные выше результаты по окислению ДМФ и ДТФ в соответствующие ДАБХ получены с использованием кеггиновских ГПК-х (х = = 2–6) [192, 193]. Однако, как было установлено при изучении окисления 2МН1 и ТМФ (разделы 5.3.1. и 5.3.2), кеггиновские растворы ГПК-х недостаточно эффективны в таких реакциях. Их недостаток заключается в том, что при х > 4 и [ГПК-х] > 0.2 М эти растворы при эксплуатации выделяют ванадийсодержащие осадки. Окислительная емкость катализаторов постепенно снижается, поскольку в растворах ГПК-х именно атомы ванадия претерпевают редокс-превращения (VV ⇄ VIV). Поэтому на следующем этапе исследований окисление ДМФ и ДТФ проводили уже в присутствии модифицированных ГПК-х некеггиновских составов [70, 115], обеспечивающих высокую стабильность ванадия при его большей концентрации в растворе.
Нам удалось выяснить, что в окислении ДМФ в ДМХ активность и селективность кеггиновских и модифицированных растворов ГПК-х сопоставимы для одного и того же растворителя, но вот в окислении ДТФ модифицированные ГПК-х были существенно более активны (табл. 10).
Таблица 10.
Влияние природы органического растворителя на активность и селективность катализатора окисления ДТФ в ДТХ в присутствии модифицированной ГПК-10
| Номер цикла* | Органический растворитель | Время реакции, мин | S, % |
|---|---|---|---|
| 1 | Изопропилбензол (кумол) | 50 | 83.0 |
| 2 | Кумол | 45 | 82.5 |
| 3 | Хлорбензол | 35 | 80.5 |
| 4 | Гептан | 50 | 82.0 |
| 5 | трет-бутилбензол | 60 | 80.5 |
| 6 | Толуол | 90 | 77.0 |
Примечание. Условия реакции: 80°С, воздух; [H17P3Mo16V10O89] = 0.2 М, [НОАс] = 30 об. %, [ГПК-10]/[ДТФ] = 5; конверсия 100%. * Все эксперименты проведены на одном и том же образце катализатора, регенерированном в каждом цикле по реакции (II) [194].
При сравнении данных табл. 9 и 10 видно, что окисление ДТФ в растворе модифицированной ГПК-10 протекает значительно быстрее. Так, если растворителем выступает толуол, то в присутствии кеггиновской ГПК-6 полная конверсия ДТФ происходит за 3.5 ч, а для модифицированной ГПК-10 – за 1.5 ч. При этом в обоих случаях S = 77%. Важно также отметить, что реакцию окисления ДТФ в ДТХ с модифицированными ГПК-х можно проводить не только в инертной атмосфере, но и на воздухе. Лучшим растворителем в окислении ДТФ в растворах модифицированных ГПК-х оказался кумол (S ≈ 83%).
В наших последующих работах [14, 196–200] были изучены особенности кинетики и механизма окисления ДАХ в ДАБХ в присутствии растворов модифицированных ГПК-х и их кислых солей. Кроме того, в качестве еще одного субстрата реакции (XXV) был опробован 2,3-диметилфенол [198].
Путем дальнейшего варьирования каталитической системы (модифицированные ГПК-х + + ОР) [198–200] удалось найти оптимальные условия для окисления 2,6-ДМФ в 2,6-ДМХ с S = = 95% при полной конверсии субстрата. При окислении 2,3-ДМФ в 2,3-ДМХ в оптимальных условиях селективность реакции достигает 97%. В этих работах впервые были рассмотрены принципы влияния природы органического растворителя на селективность реакций окисления ДМФ в ДМХ на основе строения субстратов и физических свойств исследованных ОР. Предложен ряд изменения эффективности растворителей, хорошо согласующийся с экспериментальными данными.
В итоге наше исследование показало, что каталитические системы (модифицированные ГПК-х + + ОР) являются высокоселективными (до 95–97%) катализаторами реакций окисления ДАФ в соответствующие ДАБХ. С модифицированными растворами ГПК-х процессы синтеза ДАБХ становятся экологически безопасными, поскольку катализаторы обладают высокой стабильностью [198–200]. Доказано, что активность и селективность остаются постоянными не менее 30 циклов. Подтверждено, что вариация природы ОР и состава раствора ГПК-х – эффективный метод увеличения активности и селективности таких каталитических систем.
По нашему убеждению, на основе растворов модифицированных ГПК-х могут быть созданы промышленные двухстадийные процессы окисления различных ДАФ в ДАБХ кислородом [14].
5.4. Каталитическое окисление 5-гидроксиметилфурфурола в 2,5-диформилфуран в присутствии ГПК-х
Важной задачей настоящего времени является разработка экологически чистых каталитических методов получения малотоннажных соединений и интермедиатов из продуктов переработки биомассы вместо ископаемых ресурсов. 2,5-Диформилфуран (ДФФ) – ценное соединение с симметричной и ненасыщенной структурой. Он применяется в синтезе различных полимеров с важными оптическими и электронными свойствами, гетероциклических лигандов, фунгицидов, лекарственных препаратов [201–203]. Наиболее перспективные методы получения ДФФ основаны на прямом окислении 5-гидроксиметилфурфурола (5-ГМФ), которые опробованы в присутствии различных катализаторов (VOx/TiO2 [204], V2O5/ цеолит [205], Ru/γ-Al2O3 [206] и др.). Однако нестабильность катализаторов, сложность их эффективного отделения от продуктов и повторного использования, жесткие реакционные условия ограничивают применение большинства предложенных систем.
В ряду катализаторов для окисления 5-ГМФ растворы Мо–V–P-ГПК-х выглядят весьма перспективно. Они эффективно катализируют реакции окисления, синтез их растворов осуществляют из простых оксидов с использованием безотходного “пероксидного” метода [68, 70], окислительную активность растворов ГПК-х можно регулировать путем изменения в них содержания V(V), они способны быстро регенерироваться по реакции (II) [70]. Поэтому одно из наших исследований последнего времени [207, 208] было направлено на разработку нового процесса окисления 5-ГМФ в ДФФ в присутствии растворов ГПК-х различных составов, включающих как кеггиновские, так и модифицированные высокованадиевые растворы. Процесс окисления 5-ГМФ в ДФФ представлен на схеме 1 .
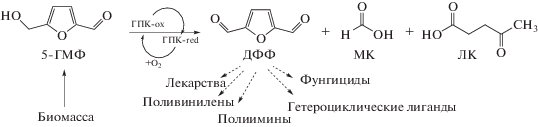
Схема 1 . Окисление 5-ГМФ в ДФФ в присутствии ГПК-х.
Реакция, протекающая по схеме 1 , была изучена в присутствии растворов ГПК-х пяти различных составов: ГПК-4 (H7PMo8V4O40), Na2ГПК-4 (Na2H5PMo8V4O40), ГПК-7 (H10P3Mo18V7O84), Co2ГПК-7 (Co2H6P3Mo18V7O84) и ГПК-10 (H17P3Mo16V10O89). Катализаторы обладают высоким значением Е и высокой бренстедовской кислотностью, причем оба этих свойства влияют на распределение продуктов [207, 208].
Однофазное окисление 5-ГМФ в водном 0.2 М растворе кеггиновской ГПК-4 в присутствии смешивающихся с водой растворителей, широко применяемых при получении 5-ГМФ из углеводов, привело к низкому выходу ДФФ (32%) из-за конкурентного процесса образования левулиновой (ЛК) и муравьиной (МК) кислот.
Проведение же реакции в двухфазной системе в присутствии трудно растворимых в воде растворителей позволило значительно увеличить выход целевого продукта (67% при использовании метилизобутилкетона, МИБК), а также решить проблемы, связанные с отделением катализатора. Было показано, что при повышении Т реакции и содержания V(V) в составе катализатора возрастает выход целевого продукта. В то же время рост кислотности раствора ГПК-х с увеличением количества ванадия оказывает противоположное влияние, способствуя образованию продуктов гидролиза – МК и ЛК.
Кислые соли ГПК-х имеют более низкую кислотность по сравнению с растворами свободных ГПК-х, что, как было установлено, повышает их каталитическую эффективность. В оптимизированных условиях выход ДФФ достигал 92% в присутствии ${\text{C}}{{{\text{o}}}_{2}}{\text{ГПК - }}7.$ Реакция проходила в среде Н2О/МИБК при Т = 110°С за 90 мин. Повторное использование раствора катализатора (после регенерации) доказало сохранение стабильности и активности в 4 циклах.
Таким образом, проведенное исследование [208] показало перспективу создания нового метода получения ДФФ в присутствии растворов ГПК-х.
5.5. Диеновый синтез. One-pot процессы
Растворы ГПК-х могут быть использованы в качестве эффективных бифункциональных катализаторов в процессах диенового синтеза (кислотно-каталитическая реакция), совмещенного с окислением образующихся аддуктов.
Ранее в работе [82] было продемонстрировано, что P–W-ГПК (H3PW12O40) катализирует реакцию диенового синтеза между хинонами (1,4-нафто-, толу- и бензохинонами) и 2,3-диметил-1,3-бутадиеном. Если же применять в качестве катализаторов Мо–V–P-ГПК-х (довольно сильные окислители), то образующиеся в ходе реакции аддукты могут быть сразу же окислены в соответствующие хиноны непосредственно в том же реакционном сосуде, т.е. в one-pot процессе возможно совмещение стадий диенового синтеза и окисления аддуктов. Мы разработали ряд таких one-pot процессов для получения 1,4-нафтохинонов (НХ) и 9,10-антрахинонов (АХ).
5.5.1. Получение 9,10-антрахинонов. В наших работах [209–215] были исследованы one-pot процессы синтеза АХ и его замещенных аналогов в присутствии растворов ГПК-x, протекающие в соответствии со схемой 2 :
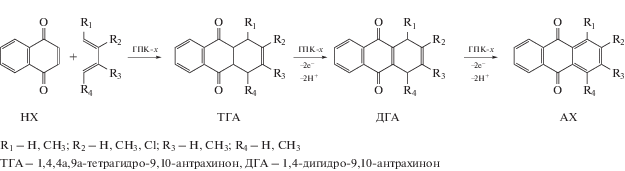
Схема 2 . Получение АХ из НХ и замещенных 1,3-бутадиенов [209–215].
В работах [209, 210] изучали реакцию НХ с 1,3-бутадиеном с использованием в качестве катализаторов водных растворов кеггиновской ГПК-4 и модифицированной ГПК-7. Было показано, что при осуществлении one-pot процесса синтеза АХ при температурах 80−135°С в отсутствие ОР или в присутствии несмешивающегося с водой растворителя (бензол, хлороформ, октанол-1) образуются только смеси продуктов (ТГА + ДГА + АХ), содержащие не более 50−55% АХ. В этих условиях исходная реакционная смесь является гетерогенной ввиду неполной растворимости НХ и гидрированных производных АХ в водном растворе ГПК-х или несмешиваемости водного раствора ГПК-х и раствора субстрата в органическом растворителе. Мы предположили, что одним из вариантов оптимизации условий one-pot процесса для получения чистого АХ может быть применение органических растворителей, растворяющих НХ, ТГА, ДГА и смешивающихся с водой. Это позволило бы сделать исходную реакционную смесь гомогенной и тем самым увеличить скорость процесса.
Действительно, использование 1,4-диоксана в качестве органического растворителя и раствора высокованадиевой модифицированной ГПК-10 как катализатора при температуре 90°С позволило за 7 ч получить продукт, содержащий 97% АХ и только 3% ДГА [211, 212]. Конверсия НХ в этом случае была 99.7%, выход АХ достигал 70%.
По аналогичной методике в работах [213–215] нами осуществлен также синтез ряда замещенных антрахинонов в присутствии раствора ГПК-10 из НХ и соответствующих замещенных 1,3-диенов. Условия синтеза и полученные результаты приведены в табл. 11.
Таблица 11.
Результаты экспериментов по диеновому синтезу замещенных антрахинонов в присутствии раствора ГПК-10 (по данным работ [213, 214], получено разрешение на использование данных)*, растворитель – 1,4-диоксан
| Заместитель и его положение | Объем диоксана, мл | Объем
0.2 М раствора ГПК-10, мл |
Мольное отношение ГПК-10/НХ |
Выход продукта, % |
Содержание основного вещества, % |
|---|---|---|---|---|---|
| R1 = R2 = R3 = R4 = H | 8 | 15.6 | 2 | 67 | 97 |
| R1 = R3= R4 = H R2 = СH3 |
4 | 3.9 | 0.5 | 72 | 96 |
| R2 = R3 = R4 = H R1 = СH3** |
8 | 7.8 | 1 | 91 | 99 |
| R1 = R4 = H R2 = R3 = CH3 |
8 | 15.6 | 2 | 78 | 98 |
| R1 = R3 = R4 = H R2 = Сl |
8 | 15.6 | 2 | 30 | 95 |
Выход 2-хлор-АХ, как видно из данных табл. 11, составляет всего около 30%. Мы пытались добиться его увеличения путем изменения мольных соотношений ГПК-10 : НХ и ГПК : диоксан, однако улучшить результат не удалось. Поскольку побочного образования смол в ходе процесса не наблюдалось, то для окончательного вывода о причинах низкого выхода 2-хлор-АХ [213, 214] необходимы дополнительные исследования.
Важно отметить, что разработанный нами способ синтеза антрахинонов применим только для диенов, устойчивых в сильнокислой среде. Так, 1,3-циклогексадиен при температуре 90°С в присутствии ГПК-х полимеризуется, поэтому в реакции с НХ наблюдалось только образование смолы. По аналогичной причине в реакции 2,4-гексадиена (90% транс–транс-изомера) с НХ получались смеси с содержанием соответствующего 1,4-диметил-АХ 29% (остальное – дигидро-АХ и НХ) при конверсии НХ, равной 85% [215].
Таким образом, в работах [209–215] были осуществлены one-pot синтезы АХ и его замещенных производных по реакциям 1,4-нафтохинона с 1,3-диенами с использованием водных растворов ГПК-x и смешивающихся с водой органических растворителей и показана принципиальная возможность синтеза таким путем антрахинонов с выходами до 90% и чистотой продукта 95–99%. Тот факт, что выход замещенных метил-АХ в наших экспериментах все же не превышал 70–90%, объясняется протеканием в присутствии ГПК-х побочных процессов, природа которых требует дополнительного исследования.
Катализатор (раствор ГПК-х) после отделения твердого продукта реакции и отгонки из реакционной смеси 1,4-диоксана в виде водного азеотропа (Ткип = 88°С) можно регенерировать кислородом [194] и многократно применять на последующих циклах one-pot процесса [216].
В качестве исходного субстрата для получения АХ диеновым синтезом возможно применение не только НХ, но и 1,4-бензохинона (БХ) [217]. В этом случае для реакции требуется не 1, а 2 моля 1,3-диена на 1 моль БХ. По нашим данным БХ количественно образуется при окислении гидрохинона (ГХ) в растворах ГПК-х, поэтому вполне логично ожидать получения производных АХ при проведении реакции между ГХ и замещенными диенами в растворах ГПК-х.
В работах [210, 218] была изучена реакция ГХ или БХ с 1,3-бутадиеном с использованием водного раствора ГПК-6 (H9PMo6V6O40), протекающая без органического растворителя либо в присутствии гидрофобных органических растворителей (хлороформ, трихлорэтилен). Установлено, что реакция ГХ с 2 молями 1,3-бутадиена может идти при комнатной температуре, давая смесь АХ, ТГА и ДГА при полной конверсии ГХ. Применение гидрофобного растворителя в этой реакции не улучшает, а ухудшает показатели процесса [210], поскольку катализатор и реагенты находятся в разных фазах.
Отметим, что реакция БХ с 1,3-бутадиеном в этих условиях проходит так же легко, как и в случае ГХ, который быстро и количественно окисляется в БХ раствором ГПК-х.
Для повышения выхода АХ и поиска оптимальных условий процесса в работе [219] была изучена возможность осуществления синтеза алкилзамещенного производного АХ – 2,3,6,7-тетраметилантрахинона (ТМАХ) – из ГХ и 2,3-диметилбутадиена (ДМБ) в присутствии раствора ГПК-10 и 1,4-диоксана в качестве гидрофильного органического растворителя. Установлено, что наибольшее влияние на показатели one-pot процесса оказывают следующие параметры: время реакции, мольное отношение ДМБ : ГХ и регенерация катализатора (ГПК-10) in situ в ходе процесса.
Однако во всех экспериментах, проведенных в выбранных условиях, в смеси продуктов, кроме ТМАХ, оставалось около 21% недоокисленного дигидропроизводного ТМАХ. Поэтому для достижения полного превращения всех промежуточных продуктов в ТМАХ намечены дополнительные исследования по оптимизации процесса. Тем не менее, полученный в новом one-pot процессе продукт, представляющий собой смесь 79% ТМАХ и 21% его дигидропроизводного, может и сейчас с успехом использоваться в качестве эффективного катализатора делигнификации древесины [217].
5.5.2. Получение 1,4-нафтохинонов. В упомянутых выше работах [210, 218] было показано, что в присутствии растворов ГПК-х реакция между ГХ и избытком бутадиена в отсутствие ОР в зависимости от температуры приводит к образованию смесей продуктов, содержащих НХ, АХ, а также гидрированные производные АХ, в различных соотношениях. Если при проведении процесса при комнатной температуре в смеси продуктов преобладает НХ (до 70%), то при повышенных температурах – АХ и его гидрированные производные. Поэтому вполне логично ожидать образование именно НХ при взаимодействии ГХ и 1,3-диенов в мольном соотношении, близком к 1 : 1, в присутствии растворов ГПК-х при комнатной температуре.
В работе [220] изучены реакции между ГХ и различными 1,3-диенами, протекающие согласно схеме 3 :
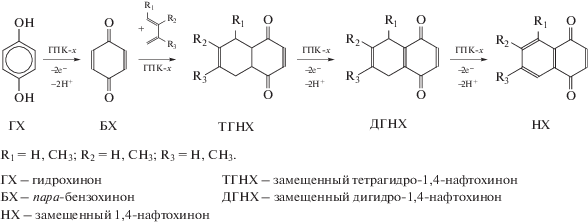
Схема 3 . Получение НХ из ГХ и 1,3-бутадиенов [220].
Кроме того, была исследована реакция ГХ с 1,3-циклогексадиеном, предположительно протекающая по схеме 4 :
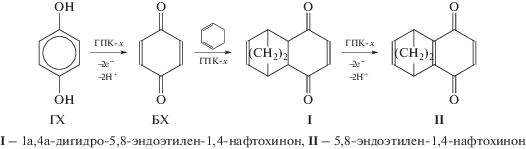
Схема 4 . Реакция диенового синтеза между ГХ и 1,3-циклогексадиеном в присутствии раствора ГПК-х [220].
Полученные результаты представлены в табл. 12.
Таблица 12.
Реакция ГХ с различными диенами в присутствии 0.25 М ГПК-x (по данным работы [220], получено разрешение на использование данных)*
| Диен | ГПК-x | Содержание НХ, % |
Выход НХ, % |
|---|---|---|---|
| Изопрен | ГПК-4 | 98 | 62.5 |
| ДМБ | ГПК-7 | 92** | 77 |
| Пиперилен | ГПК-4 | 97 | 62 |
| 1,3-Бутадиен | ГПК-4 | 99 | 51 |
| 1,3-Циклогексадиен | ГПК-4 | 87*** | 84*** |
* Условия реакции: 10 мл 0.25 М водного раствора ГПК-x; объемное отношение ГПК-x : диоксан = 1 : 1; мольное отношение диен : ГХ = 1 : 1; время реакции 30 ч, температура 25°С, полная конверсия ГХ и БХ. ** Также примесь двух производных АХ. В присутствии ГПК-4 содержание примесей увеличивается. *** Аддукт диенового синтеза I.
Как подтверждают данные табл. 12, нам удалось подобрать условия, при которых 4 из 5 перечисленных НХ образуются с хорошими выходами и высокой чистотой. При реакции ГХ с 2,3-диметилбутадиеном (ДМБ) продукт содержит, кроме соответствующего НХ, примесь гидрированных производных антрахинона (8%).
Интересно было оценить возможность реализации one-pot процесса при взаимодействии ГХ с 1,3-циклогексадиеном. Оказалось, что в этом случае основными продуктами реакции (данные хромато-масс-спектрометрии) являются моноаддукт диенового синтеза I (М = 188) диена с БХ и неидентифицированный продукт с М = 238. Отметим, что М продукта II окисления аддукта диенового синтеза равна 186. Аддукт же с двумя молями диена (М = 268) обнаружен лишь в следовых количествах (менее 1%). Повышение Т реакции выше комнатной ведет не к увеличению содержания аддукта с двумя молями 1,3-циклогексадиена, а к полимеризации последнего и осмолению реакционной смеси. Продукт II при любых температурах не образуется в ходе процесса.
Одним из важных представителей 1,4-нафтохинонов является уже рассмотренный выше (раздел 5.3.1) 2-метил-1,4-нафтохинон (витамин К3, менадион, МД). Однако синтезировать его аналогично описанным выше НХ не рационально ввиду труднодоступности исходного 2-метилгидрохинона. Вместе с тем известно, что 2-метил-1,4-бензохинон может быть получен окислением либо о-крезола (2-метилфенола), либо о-толуидина (2-метиланилина). Поэтому для поиска альтернативных путей синтеза МД мы использовали именно эти субстраты в реакции с 1,3-бутадиеном в присутствии ГПК-x, протекающей согласно схеме 5 [221, 222]:
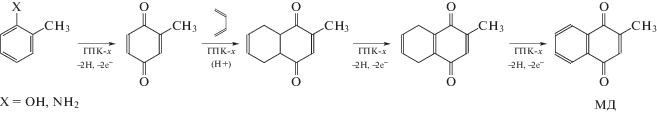
Схема 5 . One-pot процессы получения МД из 2-метилфенола и 2-метиланилина в растворах ГПК-х в атмосфере 1,3-бутадиена [221].
В ходе исследования были проварьированы условия процесса синтеза МД из указанных субстратов: температура в пределах 40–80°С, мольное отношение ГПК-х : субстрат в пределах 1.3–3, состав ГПК-х (x = 4, 7, 10). Показано, что в случае о-крезола образуется большое количество смол, а выход МД не превышает 20%. В случае о-толуидина удалось добиться лучших результатов – выход МД составил 32%. Варьирование условий процесса не позволило увеличить выход продукта, так как в случае использования этого субстрата также побочно образуется полимерный продукт окисления – политолуидин.
Подводя итог, можно сказать, что в работах [210, 218, 220–222] показана возможность синтеза 1,4-нафтохинонов из ГХ и 1,3-бутадиенов в присутствии модифицированных растворов ГПК-x в одну стадию с выходами 32–77% и чистотой 92–99%. До наших исследований одностадийные способы получения нафтохинонов из ГХ в литературе описаны не были. Полученные на данный момент результаты по диеновому синтезу, совмещенному с окислением аддуктов в растворах ГПК-х, были детально описаны в опубликованном недавно обзоре [223].
6. РЕГЕНЕРАЦИЯ РАСТВОРОВ ГПК-х КИСЛОРОДОМ
Во всех рассмотренных выше каталитических процессах растворы Мо–V–Р-ГПК-х используются в качестве обратимо действующих окислителей, стадия регенерации которых кислородом – общая для всех каталитических процессов в присутствии растворов ГПК-х. Как правило, именно стадия (II) определяет технологичность каталитического процесса окисления Su кислородом в целом. Необходимо, чтобы регенерация раствора ГПК-х происходила быстро и достаточно глубоко: остаточная концентрация V(IV) на этом этапе должна стать минимальной (низкие значения m). Окисленный раствор ГПК-х (уже с высоким значением Е) готов к работе на следующем цикле исследуемого каталитического процесса.
Принимая во внимание особую важность реакции (II), ее исследованию за эти годы было уделено много внимания. Изучить такую реакцию оказалось непростой задачей, поскольку растворы ГПК-х, как было показано выше (раздел 2), являются сложными равновесными смесями многих частиц, обладающих разной активностью по отношению к кислороду. В ходе регенерации растворов ГПК-х происходит смещение всех равновесий в системе [25, 51, 61, 62].
Отметим, что в наших работах впервые исследована кинетика и механизм окисления концентрированных (0.1−0.4 М) восстановленных растворов ГПК-х кислородом по реакции (II) [101]. По мере регенерации раствора ГПК-х повышается его кислотность, что способствует снижению скорости реакции (II), w2. Кроме того, снижается концентрация V(IV), от значения которой w2 зависит особенно сильно, резко падая при низких m. Получено кинетическое уравнение реакции, подтверждающее высказанную ранее гипотезу о роли числа ионов V(IV) в составе ГПА в процессах многоэлектронного восстановления молекулы О2 [101].
Нам удалось получить основные кинетические зависимости w2, необходимые для технологических расчетов и использованные, в частности, в процессах [21, 22, 143, 144]. Кинетику реакции (II) исследовали как при атмосферном давлении и температурах до 100°С [101, 224], так и под давлением О2 (до 0.809 МПа) при температурах до 190°С [194, 225]. Установлено, что восстановленные растворы ГПК-х можно глубоко окислить (т.е. максимально полно регенерировать катализатор) только при высоких температурах под давлением кислорода или воздуха. Кеггиновские же растворы ГПК-х не выдерживали температур >145°С, постепенно выделяя ванадийсодержащие осадки и снижая окислительную емкость катализаторов. Именно поэтому долгое время не удавалось на их основе получить действительно эффективные промышленные катализаторы жидкофазных двухстадийных процессов окисления органических субстратов кислородом, хотя высокая селективность и активность кеггиновских ГПК-х в окислении органических соединений была несомненной.
Разработка способов синтеза модифицированных некеггиновских высокованадиевых растворов ГПК-х, обладающих повышенной термостойкостью (до 170°С) (раздел 4.3.3), открыла перспективу для решения этой проблемы, обеспечив возможность быстрой регенерации модифицированных растворов ГПК-х кислородом. В работе [225] представлено кинетическое уравнение реакции (II) для модифицированных растворов, учитывающее сложную зависимость w2 от степени восстановления ГПК-х. Именно уравнение (3) было использовано при проектировании кислородного реактора идеального смешения для стадии регенерации катализатора в опытно-промышленной установке синтеза МЭК (раздел 5.2) [72, 143, 144]:
(3)
${{w}_{2}} = {{k}_{0}}{{P}_{{{{{\text{O}}}_{2}}}}}{{\left[ {{\text{V}}\left( {{\text{IV}}} \right)} \right]}^{{1 - 3}}}{\kern 1pt} {\text{exp}}{\kern 1pt} \left( {{{37.5} \mathord{\left/ {\vphantom {{37.5} {RT}}} \right. \kern-0em} {RT}}} \right).$Испытания модифицированных растворов ГПК-х в МЭК-процессе подтвердили их высокую активность и стабильность в качестве гомогенных катализаторов двухстадийных окислительных процессов. На данный момент стадию регенерации этих растворов кислородом (II) можно считать отработанной: она протекает полностью за 15–20 мин при Т = 150–160°С и ${{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}$ = 0.3–0.4 МПа. Следовательно, теперь основные усилия ученых на пути создания новых жидкофазных окислительных или one-pot процессов в присутствии модифицированных ГПК-х могут быть сосредоточены в основном на целевых реакциях окисления субстратов растворами ГПК-х, регенерация же таких катализаторов проблем не составит.
ЗАКЛЮЧЕНИЕ
В обзоре рассмотрены уникальные свойства растворов Мо–V–P-ГПК-х и каталитические жидкофазные процессы окисления, которые можно реализовать в их присутствии. Проанализированы проблемы, возникающие при использовании ГПК-х кеггиновских составов, и показаны пути их устранения. Доказано, что с технологической точки зрения наиболее перспективны модифицированные (некеггиновские) высокованадиевые растворы ГПК-х. Они обладают высокой окислительной емкостью и достаточной термостойкостью, чтобы выдерживать регенерацию при бόльших температурах, чем это возможно в присутствии кеггиновских ГПК-х. Поэтому модифицированные растворы ГПК-х становятся высокоэффективными катализаторами двухстадийных процессов окисления органических соединений кислородом. Ключевая стадия таких процессов (регенерации ГПК-х) в их присутствии протекает достаточно быстро, обеспечивая технологичность катализаторов и процессов на основе модифицированных растворов ГПК-х. Не вызывает сомнения, что круг жидкофазных каталитических реакций окисления в присутствии этих растворов будет существенно расширен.
Список литературы
Кожевников И.В., Матвеев К.И. // Успехи химии. 1982. Т. 51. № 11. С. 1875.
Kozhevnikov I.V., Matveev K.I. // Appl. Catal. 1983. V. 5. № 2. P. 135.
Pope M.T., Muller A. // Angew. Chem. Int. Ed. Eng. 1991. V. 30. P. 34.
Misono M., Nojiri N. // Appl. Catal. 1990. V. 64. P. 1.
Mizuno N., Misono M. // J. Mol. Catal. 1994. V. 86. № 1–3. P. 319.
Hill C.L., Prosser-McCartha C.M. // Coord. Chem. Rev. 1995. V. 143. № 1–2. P. 407.
Okuhara T., Mizuno N., Misono M. // Adv. Catal. 1996. V. 41. № 1–3. P. 113.
Kozhevnikov I.V. // Chem. Rev. 1998. V. 98. № 1–2. P. 171.
Okuhara T., Mizuno N., Misono M. // Appl. Catal. A: General. 2001. V. 222. № 1. P. 63.
Kozhevnikov I.V. Catalysis by Polyoxometalates. Chichester: John Wiley, 2002. 202 p.
López X., Carbó J.J., Bo C., Poblet J.M. // Chem. Soc. Rev. 2012. V. 41. P. 7537.
Zhou Yu, Guo Z., Hou W., Wang Q., Wang J. // Catal. Sci. Technol. 2015. V. 5. P. 4324.
Wang S.S., Yang G.Yu. // Chem. Rev. 2015. V. 115. P. 4893.
Rodikova Yu.A., Zhizhina E.G., Pai Z.P. // Chem. Select. 2016. V. 1. P. 2113.
Kale S.S., Armbruster U., Eckelt R., Bentrup U., Umbarkar S.B., Dongare M.K., Martin A. // Appl. Catal. A: General. 2016. V. 527. P. 9.
A.c. 421226 CCCP. Б.И. 1992. № 16. C. 34.
Pat. 1154798 DBR, 1963.
Cavani F. // Catal. Today. 1998. V. 41. № 1. P. 73.
Sano K., Uchida H., Wakabayashi S. // Catal. Surv. Japan. 1999. V. 3. P. 55.
Kozhevnikov I.V. // Chem. Rev. 1993. V. 62. № 1–2. P. 510.
Пaт. 2165406 PФ. Б.И. 2001. № 11.
Пaт. 2162837 PФ. Б.И. 2001. № 4.
Кожевников И.В. // Успехи химии. 1987. Т. 56. № 9. С. 1417.
Pope M.T. Heteropoly and Isopoly Oxometalates. Berlin-Heidelberg–New York–Tokyo: Spring–Verlag, 1983. 238 p.
Одяков В.Ф., Жижина Е.Г., Матвеев К.И. // Журн. неорг. химии. 2000. Т. 45. № 8. С. 1379.
Souchay P. Ions Mineraux condencer. Paris: Masson, 1969. 287 p.
Weakly T.J.R. // Struct. Bond. 1974. V. 18. P. 131.
Казанский Л.П., Торченкова Е.А., Спицын В.И. // Успехи химии. 1974. Т. 43. С. 1137.
Courtin P., Chauveau F., Souchay P.S // C. R. Acad. Sci. 1964. V. 258. P. 1247.
Courtin P. // Rev. Chim. Miner. 1971. V. 8. P. 75; 221.
Порай-Кошиц М.А., Атовмян Л.О. // Коорд. химия. 1975. Т. 1. № 6. С. 1271.
Grate J.H. // J. Molec. Catal. A: Chemical. 1996. V. 114. № 1–3. P. 93.
Souchay P., Courtin P. // C. R. Acad. Sci. 1970. V. 27OC. P. 1714.
Федотов М.А., Максимовская Р.И. // Докл. АН СССР. 1978. Т. 240. № 1. С. 128.
Максимовская Р.И., Федотов М.А., Мастихин В.М., Кузнецова Л.И., Матвеев К.И. / В сб. Исследование свойств и применение гетерополикислот в катализе. Новосибирск, 1978. С. 140.
Максимовская Р.И., Федотов М.А., Мастихин В.М., Кузнецова Л.И., Матвеев К.И.// Докл. АН СССР. 1978. Т. 240. № 1. С. 117.
Холдеева О.А., Максимовская Р.И. // Журн. неорг. химии. 1992. Т. 37. № 6. С. 1349.
Максимовская Р.И., Федотов М.А., Кузнецова Л.И., Мастихин Н.М., Матвеев К.И. // Докл. АН СССР. 1975. Т. 223. № 2. С. 385.
Сергиенко В.С., Порай-Кошиц М.А., Федотов М.А., Юрченко Э.Н., Кузнецова Л.И. // Журн. структ. химии. 1977. Т. 18. № 5. С. 976.
Сергиенко В.С., Порай-Кошиц М.А., Юрченко Э.Н. // Журн. структ. химии. 1980. Т. 21. № 1. С. 111.
Клевцова Р.Ф., Юрченко Э.Н., Глинская Л.А., Детушева Л.Г., Кузнецова Л.И. // Журн. структ. химии. 1981. Т. 22. № 1. С. 49.
Юрченко Э.Н., Соловьева Л.П., Клевцова Р.Ф., Детушева Л.Г., Глинская Л.А. // Журн. структ. химии. 1985. Т. 26. № 1. С. 118.
Fedotov M.A., Maksimovskaya R.A., Kazanskii L.P. // React. Kinet. Catal. Lett. 1981. V. 16. № 1. P. 185.
Юрченко Э.Н. Методы молекулярной спектроскопии в химии координационных соединений и катализаторов. Новосибирск: Наука, 1976. С. 56.
Максимовская Р.И., Кузнецова Л.И., Матвеев К.И. // Коорд. химия. 1977. Т. 3. № 3. С. 685.
Федотов М.А. Ядерный магнитный резонанс в растворах неорганических соединений. Новосибирск: Наука, 1986. 198 с.
Bruckman K., Haber J., Serwicka E.M., Yurchenko E.N., Lazarenko T.P. // Catal. Lett. 1990. V. 4. P. 181.
Kato R., Kobajashi A., Sasaky Y. // J. Amer. Chem. Soc. 1980. V. 102. P. 6571.
Юрченко Э.Н. Методы молекулярной спектроскопии в химии координационных соединений и катализаторов. Новосибирск: Наука, 1976. С. 100.
Pettersson L. // The International Workshop Polyoxometalates, Bielefield, July 15–17, 1992. Dordrecht: Kluwer Academic Publishers, 1993. P. 27.
Кузнецова Л.И., Юрченко Э.Н., Максимовская Р.И., Матвеев К.И. // Коорд. химия. 1976. Т. 2. № 1. С. 67.
Кузнецова Л.И., Юрченко Э.Н., Максимовская Р. И., Кирик Н.П., Матвеев К.И. // Коорд. химия. 1977. Т. 3. № 1. С. 51.
Максимовская Р.И. / В сб. Исследование свойств и применение гетерополикислот в катализе. Новосибирск, 1978. С. 177.
Детушева Л.Г., Юрченко Э.Н., Ткачев С.В. // Коорд. химия. 1983. Т. 9. № 9. С. 1220.
Pope M.T., Scully T.F. // Inorg. Chem. 1975. V. 14. № 5. P. 953.
Максимовская Р.И., Федотов М.А., Кузнецова Л.И., Мастихин Н.М., Матвеев К.И. // Докл. АН СССР. 1975. Т. 223. № 2. С. 385.
Pope M.T., O’Donnell S.E., Prados R.A. // J. Chem. Soc., Chem. Commun. 1975. № 1. P. 22.
Souchay P., Chauveau F., Courtin P. // Bull. Soc. Chim. France. 1968. № 6. P. 2384.
Детушева Л.Г., Юрченко Э.Н. // Коорд. химия. 1982. Т. 8. № 7. С. 948.
Детушева Л.Г., Юрченко Э.Н. // Коорд. химия. 1990. Т. 16. № 7. С. 930.
Буров Ю.В., Кожевников И.В., Матвеев К.И., Беляев В.Д. // Изв. АН СССР, Сер. хим. 1980. № 7. С. 1469.
Жижина Е.Г., Кузнецова Л.И., Юрченко Э.Н., Матвеев К.И. // Коорд. химия. 1980. Т. 6. № 12. С. 1846.
Maksimovskaya R.I., Chumachenko N.N. // Polyhedron. 1987. V. 6. № 10. P. 1813.
Максимов Г.М. // Успехи химии. 1995. Т. 64. № 5. С. 480.
Tsigdinos G.A., Hallada G.J. // Inorg. Chem. 1968. V. 7. № 2. P. 437.
Полотебнова Н.А., Нгуен Ван Чеу, Кальниболоцкая В.В. // Журн. неорг. химии. 1973. Т. 18. № 2. С. 413.
Charreton B., Bertho G. // Comptes Rendus Hebdomadaires des Seances de l’Academie des Sciences. 1965. V. 261. № 15. P. 2903.
Одяков В.Ф., Жижина Е.Г., Максимовская Р.И., Матвеев К.И. // Кинетика и катализ. 1995. Т. 36. № 5. С. 795. (Odyakov V.F., Zhizhina E.G., Maximovskaya R.I., Matveev K.I. // Kinet. Catal. 1995. V. 36. № 5. P. 733.)
Максимов Г.М., Максимовская Р.И., Кожевников И.В. // Журн. неорг. химии. 1994. Т. 39. № 4. С. 623.
Odyakov V.F., Zhizhina E.G., Rodikova Yu.A., Gogin L.L. // Eur. J. Inorg. Chem. 2015. V. 2015. № 22. P. 3618.
Пaт. 2230612 PФ. Б.И. 2004. № 17.
Matveev K.I., Zhizhina E.G., Odyakov V.F., Parmon V.N. // Catal. Indust. 2014. V. 6. № 3. P. 202.
Odyakov V.F., Zhizhina E.G., Matveev K.I., Parmon V.N. // Catal. Indust. 2015. V. 7. № 2. P. 111.
Rodikova Y.A., Zhizhina E.G. // Catal. Indust. 2019. V. 11. № 3. P. 179.
Timofeeva M.N. // Appl. Catal. A: General. 2003. V. 256. № 1–2. P. 19.
Федотов М.А., Максимов Г.М., Игнашин С.В. // Журн. неорг. химии. 2002. Т. 47. № 12. С. 2031.
Максимов Г.М. Головин А.В. // Журн. неорг. химии. 2004. Т. 49. № 8. С. 1362.
Okuhara T., Kimura M., Kawai T., Xu Z., Nakato T. // Catal. Today. 1998. V. 45. № 1–4. P. 73.
Izumi Y. // Catal. Today. 1997. V. 33. № 4. P. 371.
Ханхасаева С.Ц., Куликов С.М., Кожевников И.В. // Кинетика и катализ. 1990. Т. 31. № 1. С. 216.
Kozhevnilkov I.V., Kulikov S.M., Chukaeva N.G., Kirsanov A.T., Letunova A.B., Blinova V.I. // React. Kinet. Catal. Lett. 1992. V. 47. № 1. P. 59.
Meuzelaar, G.J., Maat, L., Sheldon, R.A., Kozhevnikov, I.V. // Catal. Lett. 1997. V. 45. P. 249.
Bardin B.B., Davis R.J. // Appl. Catal. A: General. 2000. V. 200. № 1–2. P. 219.
Hayashi H., Moffat J.B. // J. Catal. 1982. V. 77. № 2. P. 473.
Gayraud P.Y., Stewart I.H., Derouane-Abd Hamid S.B., Essayem N.E., Derouane E.G., Vedrine J.C. // Catal. Today. 2000. V. 63. № 2. P. 223.
Vaughan J.S., Oconnor C.T., Fletcher J.C.Q. // J. Catal. 1994. V. 147. № 2. P. 441.
Okuhara T., Nishimura T., Wanatabe H., Misono M. // J. Mol. Catal. 1992. V. 74. № 1–3. P. 247.
Матвеев К.И., Жижина Е.Г., Шитова Н.Б., Кузнецова Л.И. // Кинетика и катализ. 1977. Т. 18. № 2. С. 380.
Матвеев К.И., Жижина Е.Г., Одяков В.Ф., Пармон В.Н. // Изв. РАН, Сер. хим. 1994. № 7. С. 1208. (Matveev K.I., Zhizhina E.G., Odyakov V.F., Parmon V.N. // Russ. Chem. Bull. 1994. V. 43. Is. 7. P. 1142.)
Matveev K.I., Odyakov V.F., Zhizhina E.G. // J. Mol. Catal. A: Chem. 1996. V. 114. № 1–3. P. 151.
Одяков В.Ф., Кузнецова Л.И., Матвеев К.И. // Журн. неорг. химии. 1978. Т. 23. № 2. С. 457.
Алимарин И.П., Дорохова Е.Н., Казанский Л.П., Прохорова Г.В. // Журн. аналит. химии. 1980. Т. 35. № 10. С. 2000.
Хярсинг И.В., Филиппов А.П. // Журн. неорг. химии. 1988. Т. 33. № 5. С. 900.
Kaszonyi A., Hronec M., Harustiak M. // J. Mol. Catal. 1993. V. 80. № 3. P. L13.
Bard A.J., Parsons R., Jordan J. Standard Potentials in Aqueous Solution (IUPAC). New York–Basel: Marcer Dekker, 1985. P. 4.
Матвеев К.И. / В сб. Исследование свойств и применение гетерополикислот в катализе. Новосибирск. 1978, С. 3.
Жижина Е.Г., Кузнецова Л.И., Матвеев К.И. // Кинетика и катализ. 1988. Т. 29. № 1. С. 130.
Bard A.J., Parsons R., Jordan J. Standard Potentials in Aqueous Solution (IUPAC). New York–Basel: Marcer Dekker, 1985. P. 507.
Жижина Е.Г., Одяков В.Ф., Матвеев К.И. // Журн. прикл. химии. 2000. Т. 73. № 5. С. 777.
Jira R., Blau W., Grimm D. // Hydrocarbon Process. 1976. V. 55. № 3. P. 97.
Жижина Е.Г., Одяков В.Ф., Симонова М.В., Матвеев К.И. // Кинетика и катализ. 2005. Т. 46. № 3. С. 380. (Zhizhina E.G., Odyakov V.F., Simonova M.V., Matveev K.I. // Kinet. Catal. 2005. V. 46. № 3. P. 354.)
Новый справочник химика и технолога. Общие сведения и др. Санкт-Петербург: НПО “Профессионал”, 2006. Т. 12. С. 309, 316.
Yurchenko E.N., Matvienko L.G., Kuznetsova L.I., Pankratiev Yu.D., Matveev K.I. // React. Kinet. Catal. Lett. 1976. V. 4. № 2. P. 405.
Филиппов А.П., Хярсинг И.В. // Журн. неорган. химии. 1978. Т. 23. № 8. С. 1523.
Жижина Е.Г., Панкратьев Ю.Д., Одяков В.Ф., Матвеев К.И. // Журн. неорг. химии. 1999. Т. 44. № 4. С. 561.
Zhizhina E.G., Odyakov V.F., Matveev K.I. // Eur. J. Inorg. Chem. 1999. № 6. P. 1009.
Кальве Э., Прат А. Микрокалориметрия. Москва: ИЛ, 1963. С. 315.
Blanc P., Madic C., Launay J.P. // Inorg. Chem. 1982. V. 21. № 8. P. 2923.
Плетнев Р.Н., Ивакин А.А., Горшков В.В., Чирков А.К. // Докл. АН СССР. 1975. Т. 224. № 1. С. 106.
Sasaki Yu. // Nippon kesshogaku kaishi (J. Crystallogr. Soc. Japan). 1975. V. 17. № 2. P. 127.
Kirkegaard P., Longo J.M. // Acta Chem. Scand. 1965. V. 19. № 8. P. 1906.
Одяков В.Ф., Жижина Е.Г., Максимовская Р.И., Матвеев К.И. // Журн. неорг. химии. 1998. Т. 43. № 9. С. 1451.
Zhizhina E.G., Odyakov V.F. // Appl. Catal. A: General. 2009. V. 358. P. 254.
Zhizhina E.G., Odyakov V.F. // React. Kinet. Catal. Lett. 2008. V. 95. № 2. P. 301.
Odyakov V.F., Zhizhina E.G., Maksimovskaya R.I. // Appl. Catal. A: General. 2008. V. 342. P. 126.
Baes C.A., Mesmer R.E. // The Hydrolysis of Cations. New York: Wiley Intersci., 1976. P. 210.
Sadakane M., Steckhan E. // Chem. Revs. 1998. V. 98. Is. 1. P. 219.
Filippov A.P., Härsing I., Kirjanen E., Kirjanen I. // Eesti NSV Tead. Akad. Toim. Keemia. 1981. V. 30. P. 235 (in Russ.).
Blanc P., Madic C., Launay J.P. // Inorg. Chem. 1982. V. 21. P. 2923.
Jenkins H.D.B., Marcus Y. // Chem. Revs. 1995. V. 95. P. 2695.
Zhizhina E.G., Simonova M.V., Odyakov V.F., Matveev K.I. // Appl. Catal. A: General. 2007. V. 319. P. 91.
Родикова Ю.А., Жижина Е.Г. // Кинетика и катализ. 2019. Т. 60. №6. С.770. (Rodikova Yu.A., Zhizhina E.G. // Kinet. Catal. 2019. V. 60. № 6. P. 790).
Жижина Е.Г., Симонова М.В., Одяков В.Ф., Матвеев К.И. // Катализ в пром-сти. 2005. Т. 2. С. 17. (Zhizhina E.G., Simonova M.V., Odyakov V.F., Matveev K.I. // Kataliz v Prom. 2005. V. 2. P. 17).
Новый справочник химика и технолога. Электродные процессы и др. Санкт-Петербург: НПО “Профессионал”, 2004. С. 114 (A New Handbook of Chemistry and Chemical Engineering. Electrolytic Processes. St.-Petersburg: NPO “Professional”, 2004. P. 114.).
Zhizhina E.G., Odyakov V.F. // React. Kinet. Catal. Lett. 2009. V. 98. № 1. P. 51.
ГOCT 5632‑72 [№№ 6‑31, 6–22, 7–6, 8–2].
Жижина E.Г., Кузнецова Л.И., Матвеев К.И. // Кинетика и катализ. 1984. Т. 25. № 5. С. 1095.
Кузнецова Л.И., Матвеев К.И., Жижина Е.Г. // Кинетика и катализ. 1985. Т. 26. № 5. С. 1029.
Жижина Е.Г., Матвеев К.И., Кузнецова Л.И. // Кинетика и катализ. 1985. Т. 26. № 2. С. 461.
Жижина Е.Г., Кузнецова Л.И., Матвеев К.И. // Кинетика и катализ. 1985. Т. 26. № 7. С. 1349.
Zhizhina E.G., Kuznetsova L.I., Matveev K.I. // React. Kinet. Catal. Lett. 1986. V. 31. № 1. P. 113.
Жижина Е.Г., Кузнецова Л.И., Матвеев К.И. // Кинетика и катализ. 1988. Т. 29. № 1. С. 130.
A.c. 1027880 CCCP. Б.И. 1995. № 32.
Zhizhina E.G., Kuznetsova L.I., Maksimovskaya R.I., Pavlova S.N., Matveev K.I. // J. Molec. Catal. 1986. V. 38. № 1–2. P. 345.
Павлова С. Н., Кузнецова Л.И., Матвеев К.И., Сазонов В.А., Поповский В.В., Жижина Е.Г., Фенелонов В.Б., Гаврилов В.Ю. // Кинетика и катализ. 1987. Т. 28. № 2. С. 373.
A.c. 1135054 CCCP. Б.И. 1994. № 22.
Матвеев К.И., Шитова Н.Б., Жижина Е.Г. // Кинетика и катализ. 1976. Т. 17. № 4. С. 893.
Жижина Е.Г., Шитова Н.Б., Матвеев К.И. // Кинетика и катализ. 1981. Т. 22. № 6. С. 1451.
A.c. 1669109 CCCP. Б.И. 1989. № 3.
Zhizhina E.G., Simonova M.V., Odyakov V.F., Matveev K.I. // React. Kinet. Catal. Lett. 2003. V. 80. № 1. P. 171.
Жижина Е.Г., Одяков В.Ф., Балашов А.Л., Матвеев К.И. // Катализ в пром-ти. 2005. № 5. С. 28.
Пaт. 2230612 PФ. Б.И. 2004. № 17.
Пaт. 2243818 PФ. Б.И. 2005. № 1.
Пaт. 2275960 PФ. Б.И. 2006. № 13.
Одяков В.Ф., Титова Т.Ф., Матвеев К.И., Крысин А.П. // Хим.-фарм. журн. 1992. Т. 26. № 7–8. С. 69.
Шнайдман Л.О. Производство витаминов. 2-е изд. Москва: Пищепромиздат, 1973. С. 330.
A.c. 2022958 CCCP. Б.И. 1994. № 21.
Пaт. 2061669 PФ. Б.И. 1996. № 16.
Пaт. 2142935 PФ. Б.И. 1999. № 35.
Матвеев К.И., Жижина Е.Г., Одяков В.Ф. // Хим. пром-сть. 1996. № 3. С. 173.
Пaт. 2050345 PФ. Б.И. 1995. № 35.
Пaт. 2126792 PФ. Б.И. 1999. № 6.
Матвеев К.И., Жижина Е.Г., Одяков В.Ф., Коцаренко Н.С., Шмачкова В.П. // Кинетика и катализ. 1997. Т. 38. № 1. С. 76. (Matveev K.I., Zhizhina E.G., Odyakov V.F., Kotsarenko N.S., Shmachkova V.P. // Kinet. Catal. 1997. V. 38. № 1. P. 64.)
Mercier C., Chabardes P. // Pure Appl. Chem. 1994. V. 66. № 7. P. 1509.
Pat. 2221624 DE, 1972.
Pat. 0127888 EP, 1984.
Pat. 56140946 JP, 1981.
Pat. 60255745 JP, 1985.
Pat. 27888 EP, 1987.
Pat. 0475272 EP, 1992.
Pat. 181827 IN, 1998.
Bard A.J., Parsons R., Jordan J. Standard Potentials in Aqueous Solution (IUPAC). New York–Basel: Marcer Dekker, 1985. P. 292.
Lissel M., Jansen W., Neumann R. // Tetrahedron Lett. 1992. V. 33. № 13. P. 1795.
Пат. 1719392 СССР, 1992.
Kholdeeva O.A., Golovin A.V., Kozhevnikov I.V. // React. Kinet. Catal. Lett. 1992. V. 6. P. 107.
Жижина Е.Г., Одяков В.Ф., Матвеев К.И. // Катализ в пром-ти. 2005. № 6. С. 19.
Родикова Ю.А., Жижина Е.Г. // Изв. Вузов. Физика. 2011. № 12/2. С. 86.
Родикова Ю.А., Жижина Е.Г. // Химия в интересах устойчивого развития. 2012. Т. 20. № 6. С. 657. (Rodikova Yu.A., Zhizhina E.G. // Chemistry for Sustanable Development. 2012. V. 20. № 6. P. 605.)
Rodikova Yu.A., Zhizhina E.G. // J. Chem. Chem. Eng. 2013. V. 7. № 9. P. 808.
Родикова Ю.А., Жижина Е.Г. // Катализ в пром-ти. 2019. Т. 19. № 1. С. 19. (Rodikova Yu.A., Zhizhina E.G. // Kataliz v promyshlennosti. 2019. V.6. № 1. P. 243.)
Finley K.T. The chemistry of the quinonoid compounds. V. II. Part 1. / Eds.: Patai S., Rappoport Z. John Wiley & Sons Ltd., 1988. P. 537.
Dauben W.G., Farkas I., Bridon D.P., Chuang C.P., Henegar K.E. // J. Am. Chem. Soc. 1991. V. 113. № 15. P. 5883.
Liu C., Bao G., Burnell D.J. // J. Chem. Soc., Perkin Trans. 2001. V. 9. P. 2644.
Schuber M., Metz P. // Angew. Chem. 2011. V. 123. № 13. P. 3011.
Zhao L., Burnell D.J. // Org. Lett. 2006. V. 8. № 1. P. 155.
Shiraishi S., Ikeuchi S., Senō M., Asahara T. // Bull. Chem. Soc. Jpn. 1978. V. 51. № 3. P. 921.
Parker K.A., Cohen I.D., Padwa A., Dent W. // Tetrahedron Lett. 1984. V. 25. № 43. P. 4917.
Yadav J.S., Reddy B.V.S., Swamy T. // Tetrahedron Lett. 2003. V. 44. № 26. P. 4861.
Mothe S.R., Susanti D., Chan P.W.H. // Tetrahedron Lett. 2010. V. 51. № 16. P. 2136.
Pat. 5654163 USA, 1997.
Pat. 4263426 USA, 1981.
Mukaiyama T., Shintou T., Fukumoto K. // J. Am. Chem. Soc. 2003. V. 125. № 35. P. 10 538.
Shintou T., Fukumoto K., Mukaiyama T. // Bull. Chem. Soc. Jpn. 2004. V. 77. № 8. P. 1569.
Pat. 1436407 EP, 2006.
Tsutsumi N., Ito Y., Sakai W. // Chem. Phys. 2008. V. 344. № 1–2. P. 189.
Hirao T., Moriuchi T., Ishikawa T., Nishimura K., Mikami S., Ohshiro Y., Ikeda I. // J. Molec. Catal. A: Chem. 1996. V. 113. № 1–2. P. 117.
Pat. 3935247 USA, 1976.
Pat. 75-93. 931 JP, 1975.
Pat. 2221624 DE, 1972.
Pat. 3987068 USA, 1976.
Mostaghim R., Ahmadibeni Y. // Acta Chim. Slov. 2003. V. 50. № 3. P. 569.
Kolesnik I.G., Zhizhina E.G., Matveev K.I. // J. Mol. Catal. A: Chem. 2000. V. 153. № 1–2. P. 147.
Kolesnik I.G., Zhizhina E.G., Matveev K.I. // React. Kinet. Catal. Lett. 1999. V. 68. № 2. P. 339.
Жижина Е.Г., Симонова М.В., Одяков В.Ф., Матвеев К.И. // Химия в интересах устойчивого развития. 2004. Т. 12. № 6. С. 683. (Zhizhina E.G., Simonova M.V., Odyakov V.F., Matveev K.I. // Chemistry for Sustanable Development. 2004. V. 12. № 6. P. 663.)
Oxidation and reduction of phenols. The Chemistry of the Hydroxyl Group. Part 1. Chap. 10 / Ed. Patai S. The Hebrew University, Jerusalem. London: Wiley, 1971.
Родикова Ю.А., Жижина Е.Г., Пай З.П. // Международный научно-исследовательский журнал. 2017. Т. 65. № 11(4). С. 126. (Rodikova Yu.A., Zhizhina E.G., Pai Z.P. // Int. Research J. 2017. V. 65. № 11(4). P. 126.)
Родикова Ю.А., Жижина Е.Г., Пай З.П. // Международный научно-исследовательский журнал. 2017. Т. 66. № 12(4). С. 155. (Rodikova Yu.A., Zhizhina E.G., Pai Z.P. // Int. Research J. 2017. V. 66. № 12(4). P. 155.)
Rodikova Yu.A., Zhizhina E.G., Pai Z.P. // Appl. Catal. A: General. 2018. V. 549. P. 216.
Rodikova Yu.A., Zhizhina E.G., Pai Z.P. // React. Kinet. Mech. Catal. 2018. V. 124. P. 469.
Rodikova Yu.A., Zhizhina E.G., Pai Z.P. // Chem. Select. 2018. V. 3. № 16. P. 4200.
Ma J., Wang M., Du Z., Chen C., Gao J., Xu J. // Polym. Chem. 2012. V. 3. P. 2346.
van Putten R.-J., van der Waal J.C., E. de Jong, Rasrendra C.B., Heeres H.J., de Vries J.G. // Chem. Rev. 2013. V. 113. P. 1499.
Delidovich I., Hausoul P.J.C., Deng L., Pfützenreuter R., Rose M., Palkovits R. // Chem. Rev. 2016. V. 116. P. 1540.
Nie J.F., Liu H.C. // Pure Appl. Chem. 2012. V. 84. P. 765.
Sádaba I., Gorbanev Y.Y., Kegnaes S., Putluru S.S.R., Berg R.W., Riisager A. // ChemCatChem. 2013. V. 5. P. 284.
Antonyraj C.A., Jeong J., Kim B., Shin S., Kim S., Lee K.-Y., Cho J.K. // J. Ind. Eng. Chem. 2013. V. 19. P. 1056.
Родикова Ю.А., Жижина Е.Г. // Химия под знаком СИГМА: исследования, инновации, технологии. VI Всероссийская научная молодежная школа-конференция (18–20 мая 2020, Омск) [Электронный ресурс]: сборник тезисов докладов. ИК СО РАН. 2020. С. 05–106. УД-1-36–ISBN9785906376282.
Rodikova Yu.A., Zhizhina E.G. // Reac. Kinet. Mech. Catal. 2020. V. 130. № 1. P. 403.
Жижина E.Г., Симонова M.В., Русских В.В., Матвеев К.И. // Катализ в пром-ти. 2005. № 1. С. 12.
Zhizhina E.G., Odyakov V.F. // ChemCatChem. 2012. V. 4. P. 1405.
Gogin L., Zhizhina E. // Modern Res. Catal. 2013. V. 2. P. 90.
Гогин Л.Л., Жижина Е.Г., Пай З.П., Пармон В.Н. // Изв. РАН. Сер. хим. 2015. Т. 64. № 9. P. 2069. (Gogin L.L., Zhizhina E.G., Pai Z.P., Parmon V.N. // Russ. Chem. Bull. 2015. V. 64. № 9. P. 2069.)
Gogin L., Zhizhina E.// Modern Res. Catal. 2014. V. 3. № 2. P. 57.
Гогин Л.Л., Жижина Е.Г. // Катализ в пром-ти. 2014. Т. 4. С. 33. (Gogin L.L., Zhizhina E.G. // Kataliz v promyshlennosti. 2014. V. 6. № 4. P. 273.)
Гогин Л.Л., Жижина Е.Г., Пай З.П. // Кинетика и катализ. 2019. Т. 60. № 1. С. 75. (Gogin L.L., Zhizhina E.G., Pai Z.P. // Kinet. Catal. 2019. V. 60. № 1. P. 75.)
Гогин Л.Л., Жижина Е.Г., Пай З.П. // Катализ в пром-ти. 2016. Т. 16. № 3. С. 24. (Gogin L.L., Zhizhina E.G., Pai Z.P. // Kataliz v promyshlennosti. 2016. V. 8. № 4. P. 310.)
Anthraquinone, in Kirk-Othmer Encyclopedia of chemical technology, 5th Ed. 2006. V. 2.
Жижина Е.Г., Матвеев К.И., Русских В.В. // Химия в интересах устойчивого развития. 2004. Т. 1. №1. С. 47. (Zhizhina E.G., Matveev K.I., Russkikh V.V. // Chemistry for Sustanable Development. 2004. V. 1. № 1. P. 47.)
Гогин Л.Л., Жижина Е.Г., Пай З.П. // Кинетика и катализ. 2018. Т. 59. № 5. С. 566. (Gogin L.L., Zhizhina E.G., Pai Z.P. // Kinet. Catal. 2018. V. 59. № 5. P. 578.)
Gogin L.L., Zhizhina E.G., Pai Z.P. // Modern Res. Catal. 2019. V. 8. P. 1.
Симонова М.В., Жижина Е.Г. // Химия в интересах устойчивого развития. 2005. Т. 13. № 3. С. 479. (Simonova M.V., Zhizhina E.G. // Chemistry for Sustanable Development. 2005. V. 13. № 3. P. 477.)
Гогин Л.Л., Жижина Е.Г. // Кинетика и катализ. 2020. Т. 61. № 2. С. 254. (Gogin L.L., Zhizhina E.G. // Kinet. Catal. 2020. V. 61. № 2. P. 276.)
Гогин Л.Л., Жижина Е.Г. // Кинетика и катализ. 2020. Т. 61. № 4. С. 469. (Gogin L.L., Zhizhina E.G. // Kinet. Catal. 2020. V. 61. № 4. P. 516.)
Жижина Е.Г., Одяков В.Ф., Симонова М.В. // Кинетика и катализ. 2008. Т. 49. № 6. С. 814. (Gogin L.L., Zhizhina E.G. // Kinet. Catal. 2008. V. 49. № 6. P. 773.)
Zhizhina E.G., Odyakov V.F. // Int. J. Chem. Kinet. 2014. V. 46. № 9. P. 567.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ