

Кинетика и катализ, 2021, T. 62, № 3, стр. 316-324
Анализ кинетических параметров ферментативного катализа реакций этерификации в среде гексана
В. С. Гамаюрова a, Е. С. Воробьев a, Г. А. Давлетшина a, Л. Э. Ржечицкая a, *
a ФГБОУ ВО Казанский национальный исследовательский технологический университет
420065 Казань, ул. К. Маркса, 68, Россия
* E-mail: larisa.edvard@gmail.com
Поступила в редакцию 28.10.2020
После доработки 09.01.2021
Принята к публикации 03.02.2021
Аннотация
С помощью метода математического моделирования выполнен анализ некоторых кинетических параметров реакции этерификаций алифатических кислот и спиртов, проводимых с участием биокатализаторов в неводных средах. Для решения кинетических уравнений применен модифицированный метод Рунге–Кутта–Мерсона, обеспечивающий заданную точность. В качестве доноров ацильных групп использованы кислоты ряда С3–С8, в качестве акцепторов – спирты ряда С4–С11, биокатализаторов – неиммобилизованные ферменты Lipozyme CALB и панкреатическая липаза. Анализ расчетных данных позволил дать количественную оценку протекания в исследуемой системе трех процессов: этерификации, гидролиза и вторичной этерификации, аналогичной автокатализу. Вторичная этерификация возникает за счет того, что вода, образовавшаяся при первичной этерификации, входит в гидратную оболочку активного центра фермента, усиливая его каталитические свойства.
ВВЕДЕНИЕ
Ферментативный катализ реакций этерификации, приводящий к синтезу сложных эфиров различного строения и назначения, в последние годы интенсивно развивается и приобретает все большее значение. Эта тенденция определяется различными факторами, прежде всего, расширением сфер использования сложных эфиров, которые и ранее производились в промышленных масштабах и находили применение, особенно в химической промышленности. Теперь, в связи с исследованиями и ростом потребления биодизельного топлива, основу которого составляют сложные эфиры низших спиртов и кислот растительных масел, роль сложных эфиров приобретает другой масштаб и значение [1]. Кроме того, происходит расширение объема и спектра ароматических веществ класса сложных эфиров, используемых в пищевой промышленности, производстве синтетических моющих средств и в ароматерапии [2].
Процессы этерификации и переэтерификации с применением липолитических ферментов стали важным направлением в пищевой промышленности для модификации жиров и растительных масел с целью получения жировых продуктов с функциональными свойствами [3, 4].
Ведение ферментативного катализа в неводных средах сделало возможным реализацию синтеза сложных эфиров. В настоящее время для ферментативного катализа используется ряд органических растворителей с определенными гидрофильно-гидрофобными свойствами, в основном, с lg Р ≥ 4 (Р – коэффициент распределения растворителя в системе октанол–вода). Самым распространенным из органических растворителей является гексан как наиболее доступный и применяемый в промышленности. Нужно отметить, что практически во всех работах по ферментативному катализу реакций этерификации в неводных растворителях в реакционную среду добавляли воду в количестве 2–20%. Считалось, что она необходима для запуска процесса, т.е. фактически катализ протекает в водно-органической среде. При использовании гексана часто обходятся без добавления воды, при этом достигаются высокие выходы целевых продуктов. Это означает, что воды, содержащейся в исходных субстратах и растворителе, если они не были подвергнуты специально осушению, достаточно для старта процесса [5–10]. Обзор указанных работ приведен в статье [11].
Подробный кинетический анализ ферментативной этерификации в водно-органических средах проведен в работах [12, 13]. Была исследована реакция этерификации изопропанола пальмитиновой кислотой при использовании биокатализатора Novozym 435 из Candida аntarctica в интервале температур 65–75°С, рассчитаны основные кинетические параметры процесса. Рассмотрены три возможные модели механизмов процесса этерификации: рандом-процесс (random–беспорядочный), пинг-понг (ping-pong) и би-би (Bi-Bi) механизмы. Во всех этих моделях стартовой реакцией является образование связи фермента с карбоксильной группой кислоты (ацил-энзимный интермедиат). Важная роль карбоксильной группы в создании активной конформации фермента подтверждена и в более поздних работах [14]. Авторы [12] пришли к заключению, что ферментативный синтез изопропилпальмитата лучше всего согласуется с упорядоченной кинетической моделью би-би-механизма, которая включает пять этапов:
1) образование ацил-ферментного комплекса:
2) образование тройного комплекса:
3) превращение его в сложный комплекс фермент–эфир–вода:
4) отделение от тройного комплекса сложного эфира:
5) отделение воды от фермента:
где Ас – кислота, Al – спирт, Es – эфир, W – вода.Были определены кинетические параметры процесса этерификации глицерина олеиновой кислотой в водно-органических средах [4]. Максимальная скорость этерификации наблюдалась при использовании среды гексан–вода при активности воды (аW) в диапазоне 0.5–0.8. Снижение скорости реакции этерификации при увеличении активности воды свыше 0.8 авторы объясняют конкуренцией между реакциями алкоголиза и гидролиза при участии промежуточного продукта – ацил-ферментного комплекса.
В этих и аналогичных работах не рассматривается тот факт, что в активном центре фермента протекают и другие процессы. Прежде всего, липаза катализирует не только реакцию этерификации, но и конкурентную ей реакцию гидролиза сложного эфира. Кроме того, в литературе не всегда рассматривается включение воды, добавленной или образовавшейся в результате синтеза, в гидратную оболочку фермента. Именно вода обеспечивает равновесие гидрофильно-гидрофобных сил, водородных связей, электростатического взаимодействия в белковой глобуле, обуславливая способность к ферментативному катализу [15].
В настоящей работе для решения дифференциальных уравнений, описывающих кинетические кривые процесса этерификации, применен модифицированный метод Рунге–Кутта–Мерсоном, проведены расчеты некоторых кинетических параметров этерификации кислот и спиртов алифатического ряда и их гидролиза в среде гексана без добавления воды и обнаружено явление вторичной этерификации, аналогичной автокатализу.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
В работе использованы: коммерческий препарат панкреатической липазы (Lipase from porcine pancreas) Type II лиофильно высушенный с активность 25.0 ед./мг белка при применении оливкового масла в качестве субстрата (США); коммерческий препарат Lipozyme CALB – неспецифическая липаза, жидкая, активность 5000 ед./г, (“Novozymes”); масляная, пропионовая, валериановая, каприловая кислоты марки “ч.”; ледяная уксусная кислота марки “х. ч.”; бензиловый, амиловый, изоамиловый спирты марки “ч.”; октиловый, ундециловый спирты марки “х. ч.”; бутиловый, изобутиловый спирты марки “ч. д. а.”; гексан марки “ч.”.
Проведение синтеза
Процесс осуществляли в органическом растворителе – гексане. Ферментные препараты липаз без дополнительной иммобилизации смешивали с органическим растворителем. Концентрация кислоты в реакционной среде составляла 0.1 моль/л, мольное соотношение кислоты и спирта – 1 : 1 и 1 : 2. Ферментативный синтез осуществляли при температуре от 30 до 40°С в течение 4–48 ч. Конверсию кислоты в процессе синтеза определяли титриметрически по убыли кислоты в системе. Титрование проводили 0.1 Н спиртовым раствором NaOH в 80% этиловом спирте. Контрольный образец не содержал ферментного препарата. Конверсию кислоты (В, %) рассчитывали по формуле:
Результаты обрабатывали с помощью пакета статистических программ Statistica 6.0. Достоверность отличий контрольных и экспериментальных результатов оценивали при помощи t-критерия Стьюдента. Погрешность измерений не превышала 7%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Расчет кинетических параметров процесса этерификации сложных эфиров алифатического ряда с использованием биокатализа был выполнен методом моделирования кинетических характеристик реакций, позволяющим вычислять скорости, подбирая различные модели их протекания. При решении обратной задачи химической кинетики применяли модифицированный метод Рунге–Кутта–Мерсона, который позволяет решать системы дифференциальных уравнений, подбирая шаг с заданной точностью. Поиск неизвестных констант скорости реакций проводили средствами оптимизации в надстройке MS Excel “Поиск решения”. Подробное описание решения показано в [16, 17]. При решении поставленной задачи были рассмотрены две кинетические модели этерификации сложных эфиров, каждая из которых включает систему из нескольких химических реакций.
1) Первая модель описывает реакцию этерификации как обратимую, протекающую под воздействием фермента, и представляет собой систему из двух процессов: реакции этерификации (константа скорости kэтр) и обратной реакции гидролиза (константа скорости kгид):
Система кинетических уравнений для первой модели (Sys_1):
2) Вторая модель учитывает активирующее действие воды, выделяющейся при образовании сложного эфира, и включает соответственно три процесса: первичную этерификацию (константа скорости kэтр), вторичную этерификацию, условно названную “автокаталитической” (константа скорости kавк) и гидролиз образовавшегося эфира (константа скорости kгид).
Система кинетических уравнений для второй модели (Sys_2):
Достоверность аппроксимации выбранных моделей вычисляли через критерий Пирсона путем сравнения набора экспериментальных и расчетных значений концентраций компонентов. Для подбора неизвестных констант реакции использовали метод наименьших квадратов. В MS Excel он реализовывался через функцию СУММКВРАЗН, которая вычисляет сумму квадратов разностей между наборами экспериментальных и расчетных значений концентрации компонентов.
Влияние спиртовых субстратов при ферментативном катализе синтеза сложных эфиров алифатического ряда исследовано на примере синтеза эфиров масляной кислоты. Процесс вели в гексане, в качестве катализатора использовали панкреатическую липазу Type II. Концентрацию фермента варьировали в диапазоне 5–30 мг на 0.1 мМ кислоты, температура реакции 30°С. Донором ацильных групп выступала масляная кислота, а акцепторами следующие спирты: н-бутиловый, изобутиловый, н-амиловый, изоамиловый, н-октиловый, н-ундециловый. Соотношение кислота : спирт составляло 1 : 2. Для расчета кинетических параметров были использованы экспериментальные данные, приведенные в более ранней работе [18].
Сравнительный анализ достоверности моделей Sys_1 и Sys_2 для расчета констант скорости процесса синтеза бутилбутирата дан в табл. 1.
Таблица 1.
Расчетные кинетические параметры этерификации эфиров масляной кислоты и бутилового спирта (бутилбутират) с использованием панкреатической липазы Type II
| Вариант модели | Концентрация фермента, мг/0.1 мМ | Константы скорости, моль–1 ч–1 | Минимум | Критерий Пирсона |
||
|---|---|---|---|---|---|---|
| kэтр | kавт | kгид | ||||
| Sys_1 | 5 | 0.0966 | – | 0.0000 | 0.2001 | 0.9283 |
| Sys_2 | 0.0078 | 0.6166 | 0.0011 | 0.0084 | 0.9943 | |
| Sys_1 | 10 | 0.1334 | – | 0.0000 | 0.1961 | 0.9185 |
| Sys_2 | 0.0050 | 0.9903 | 0.0031 | 0.0171 | 0.9896 | |
| Sys_1 | 20 | 0.1596 | – | 0.0000 | 0.1462 | 0.9108 |
| Sys_2 | 0.0121 | 1.1010 | 0.0141 | 0.0152 | 0.9857 | |
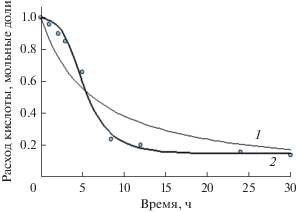
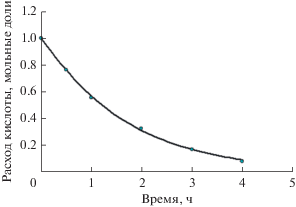
Как видно из представленных в табл. 1 расчетных данных, критерий Пирсона для модели Sys_2 на 7–11% выше, чем для модели Sys_1 при всех значениях концентрации фермента. Это подтверждается графически при сравнении экспериментальных и расчетных концентрационных кривых расхода кислоты при синтезе эфиров масляной кислоты, описанных с помощью моделей Sys_1 и Sys_2 (рис. 1). Все это говорит о том, что модель Sys_2 наиболее адекватно отражает процессы, протекающие в системе, по сравнению с моделью Sys_1.
Рис. 1.
Сравнение экспериментальных (точки) и расчетных (линии) данных по этерификации масляной кислоты бутиловым спиртом. Концентрация панкреатической липазы Type II – 10 мг/0.1 мМ кислоты. Критерий Пирсона 0.9185, критерий оптимизации 0.1961 для функции Sys_1 (1); критерий Пирсона 0.9896, критерий оптимизации 0.0171 для функции Sys_2 (2).
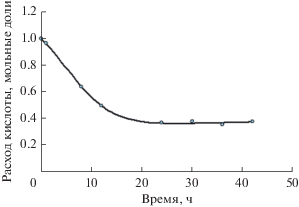
В подтверждение этого на рис. 2 в качестве примера приведен график сходимости расчетных и экспериментальных концентрационных кривых при синтезе ундецилового эфира масляной кислоты в системе кинетических уравнений второй модели (Sys_2).
Рис. 2.
Сравнение экспериментальных данных (точки) и расчетных значений, описанных моделью Sys_2 (линия), при синтезе ундецилбутирата. Концентрация панкреатической липазы Type II– 30 мг/0.1 мМ кислоты.
Таким образом, получены статистически значимые доказательства, что модель Sys_2 хорошо описывает экспериментальные данные, и именно она взята за основу при дальнейших расчетах. Таблица 2 содержит сводные результаты проведенных расчетов процесса этерификации масляной кислоты различными спиртами под действием панкреатической липазой Type II с использованием модели Sys_2.
Таблица 2.
Расчетные кинетические параметры этерификации различных спиртов масляной кислотой (модель Sys_2) с использованием панкреатической липазы Type II
| Наименование эфира |
Концентрация фермента, мг/0.1 мМ | Константы скорости, моль–1 ч–1 | Минимум | Критерий Пирсона |
||
|---|---|---|---|---|---|---|
| kэтр | kавт | kгид | ||||
| 5 | 0.0078 | 0.6166 | 0.0011 | 0.0084 | 0.9943 | |
| н-Бутилбутират | 10 | 0.0050 | 0.9903 | 0.0031 | 0.0171 | 0.9896 |
| 20 | 0.0121 | 1.1010 | 0.0141 | 0.0152 | 0.9857 | |
| 5 | 0.0093 | 0.3024 | 0.0000 | 0.0069 | 0.9775 | |
| н-Амилбутират | 10 | 0.0417 | 0.3434 | 0.0000 | 0.0014 | 0.9905 |
| 20 | 0.0076 | 1.0217 | 0.03120 | 0.0013 | 0.9284 | |
| 5 | 0.0047 | 0.0842 | 0.0000 | 0.0005 | 0.9989 | |
| изо-Амилбутират | 10 | 0.0064 | 0.1768 | 0.0107 | 0.0026 | 0.9974 |
| 20 | 0.0243 | 0.1290 | 0.0144 | 0.0031 | 0.9959 | |
| 5 | 0.0094 | 0.0555 | 0.0054 | 0.0011 | 0.9976 | |
| изо-Бутилбутират | 10 | 0.0132 | 0.0785 | 0.0199 | 0.0011 | 0.9976 |
| 20 | 0.0122 | 0.0902 | 0.0177 | 0.0000 | 1.0000 | |
| 10 | 0.0050 | 0.2609 | 0.0690 | 0.0001 | 0.9998 | |
| Октилбутират | 20 | 0.0150 | 0.1947 | 0.0509 | 0.0008 | 0.9983 |
| 30 | 0.524 | 0.0789 | 0.0571 | 0.0011 | 0.9955 | |
| 10 | 0.0052 | 0.0704 | 0.0283 | 0.0033 | 0.9912 | |
| Ундецилбутират | 20 | 0.0201 | 0.0647 | 0.0354 | 0.0011 | 0.9969 |
| 30 | 0.0188 | 0.1276 | 0.0604 | 0.0004 | 0.9988 | |
Анализ представленных результатов показывает хорошую сходимость набора расчетных и экспериментальных данных в том случае, когда учтены все три процесса: первичная этерификация, гидролиз и вторичная этерификация, названная нами автокатализ и протекающая при включении вновь образованной воды в ферментную систему. Полученные данные свидетельствуют о том, что в активном центре фермента кроме процессов этерификации и гидролиза протекает еще и процесс включения воды, образовавшейся в результате синтеза эфира, в гидратную оболочку фермента. Известно, что гидратная оболочка в активном центре необходима для формирования нативной конформации фермента, поскольку она обеспечивает сложный комплекс водородных связей, электростатического и гидрофобного взаимодействий. Утрата гидратной оболочки ведет к потере каталитических свойств фермента. Процесс включения воды условно назван нами “автокатализ”, поскольку он обеспечивает сохранение ферментативной активности и продолжение синтеза эфиров. При этом автокатализ, как показывают данные табл. 2, преобладает над прямой этерификацией во всех использованных вариантах эксперимента (kавт > kэтр), в то время как гидролиз вносит меньший вклад в суммарный процесс.
Наличие автокатализа подтверждено ранее экспериментально [19] при использовании панкреатической липазы Type II в реакциях этерификации в неводных средах. Было установлено, что при многократном применении фермента (рециклизации) наблюдается не снижение, а напротив, повышение ферментативной активности в течение 10 рециклов. Это объясняется включением воды, образующейся при этерификации, в ферментную систему, что ведет к сохранению и даже к возрастанию ее активности (автокатализ).
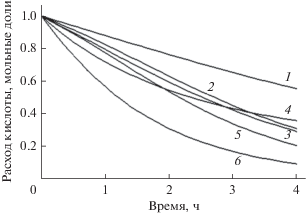
На рис. 3 приведена зависимость суммарного расхода кислоты от времени в процессе этерификации масляной кислоты различными спиртами при концентрации фермента 20 мг/0.1 мМ кислоты.
Рис. 3.
Расчетные концентрационные кривые (Sys_2) расхода масляной кислоты в процессе этерификации спиртами: 1 – изобутиловым, 2 – ундециловым, 3 – октиловым, 4 – гептиловым, 5 – изоамиловым, 6 – н-бутиловым. Концентрация панкреатической липазы Type II – 20 мг/0.1 мМ кислоты.
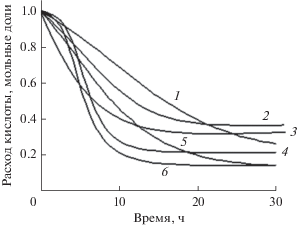
Как видно из рис. 3, н-бутиловый спирт наиболее быстро подвергается этерификации, а ундециловый спирт, имеющий самую длинную углеводородную цепь, этерифицируется медленнее всех. Сравнение скорости расхода масляной кислоты в процессе образования эфиров позволяет построить следующий ряд снижения скорости реакции этерификации в зависимости от природы спиртовых субстратов:
Таким образом, скорость реакции этерификации алифатических спиртов снижается с увеличением длины углеводородной цепи спиртов и их разветвленности независимо от концентрации фермента.
Кинетический анализ проведен также для реакций этерификации, в которых в качестве акцептора ацильных групп использовали бензиловый спирт. Донорами ацильных групп были кислоты алифатического ряда: уксусная, пропионовая, масляная, валериановая и каприловая. Все эксперименты вели в растворе гексана, фермент – липаза Lipozyme CALB.
Кинетические параметры анализировали аналогично тому, как было описано выше, путем сравнения расчетных величин критерия Пирсона для обеих моделей. Например, в табл. 3 приведено сравнение достоверности аппроксимации моделей Sys_1 и Sys_2 при расчете констант скорости синтеза бензилбутирата с использованием в качестве донора ацильной группы масляной кислоты.
Таблица 3.
Расчетные кинетические параметры этерификации эфира масляной кислоты и бензилового спирта (бензилбутирата) с использованием Lypozyme CALB*
| Модель | Константы скорости, моль–1 ч–1 | Минимум | Критерий Пирсона |
||
|---|---|---|---|---|---|
| kэтр | kавт | kгид | |||
| Sys_1 | 0.1485 | – | 0.0000 | 0.0175 | 0.9568 |
| Sys_2 | 0.0798 | 0.4299 | 0.0000 | 0.0042 | 0.9847 |
На рис. 4 показаны экспериментальные и расчетные концентрационные зависимости процесса этерификации бензилового спирта пропионовой кислотой, описанные с помощью моделей Sys_1 и Sys_2.
Рис. 4.
Сравнение расчетных (точки) и экспериментальных (линии) данных по этерификации бензилового спирта и пропионовой кислоты при концентрации биокатализатора Lypozyme CALB – 30 мкл/0.1 мМ кислоты. Критерий Пирсона 0.9901, критерий оптимизации 0.0213 для функции Sys_1 (1); критерий Пирсона 0.9985, критерий оптимизации 0.0005 для функции Sys_2 (2).
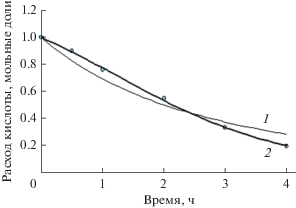
График сходимости расчетных и экспериментальных концентрационных кривых при синтезе бензилового эфира уксусной кислоты (бензилацетата) в системе кинетических уравнений второй модели (Sys_2) представлен на рис. 5.
Рис. 5.
Сравнение экспериментальных данных (точки) и расчетных значений, описанных моделью Sys_2 (линия), при синтезе бензилацетата. Концентрация Lypozyme CALB – 30 мкл/0.1 мМ кислоты.
Эти результаты также свидетельствуют, что модель Sys_2 более адекватно описывает экспериментальным данные. Таблица 4 содержит сводные расчетные значения кинетических параметров этерификации бензилового спирта алифатическими кислотами с использованием модели Sys_2.
Таблица 4.
Расчетные кинетические параметры этерификации бензилового спирта алифатическими кислотами с использованием модели Sys_2 при концентрации биокатализатора Lypozyme CALB*
| Наименование эфира | Константы скорости, моль–1 ч–1 | Минимум | Критерий Пирсона |
||
|---|---|---|---|---|---|
| kэтр | kавт | kгид | |||
| Бензилацетат | 0.2622 | 0.4643 | 0.0000 | 0.0005 | 0.9983 |
| Бензилпропионат | 0.1011 | 0.5332 | 0.0000 | 0.0005 | 0.9985 |
| Бензилбутират | 0.0798 | 0.4299 | 0.0000 | 0.0042 | 0.9847 |
| Бензилвалерат | 0.0984 | 0.3858 | 0.0000 | 0.0011 | 0.9960 |
| Бензилкаприлат | 0.0595 | 0.1817 | 0.0000 | 0.0002 | 0.9985 |
Приведенные в табл. 4 кинетические параметры говорят об отсутствии обратной реакции – гидролиза эфиров – для всех использованных кислот. При этом автокатализ значительно преобладает над первичной “прямой” реакцией этерификации. На рис. 6 показана зависимость суммарного расхода кислоты от времени в процессе этерификации бензилового спирта алифатическими кислотами.
Рис. 6.
Расчетные концентрационные кривые (Sys_2) расхода кислот в процессе этерификации бензилового спирта и алифатических кислот: 1 – каприловой, 2 – масляной, 3 – валериановой, 4 – капроновой, 5 – пропионовой, 6 – уксусной. Концентрация фермента Lypozyme CALB – 30 мкл/0.1 мМ кислоты.
Анализ концентрационных кривых (рис. 6) свидетельствует, что скорость образования эфира зависит от длины углеводородной цепи кислоты и снижается в ряду: уксусная > пропионовая> масляная > валериановая > каприловая, что соответствует расходу кислот в процессе этерификации бензилового спирта.
ЗАКЛЮЧЕНИЕ
Применение модифицированного метода Рунге–Кутта–Мерсона для решения дифференциальных уравнений при описании процесса биокатализа реакций этерификации алифатических кислот и спиртов позволило установить одновременное протекание трех процессов: первичной (прямой) этерификации, гидролиза образовавшегося эфира (обратной реакции) и вторичной этерификации (автокатализа). Процесс автокатализа обусловлен включением воды, образовавшейся в первичной этерификации, в гидратную оболочку фермента, тем самым усиливая его каталитические свойства.
Список литературы
Самойлова Ю.В., Сорокина К.Н., Пилигаев А.В., Пармон В.Н.// Катализ в промышленности. 2018. Т. 18. № 6. С. 61. https://doi.org/10/1842/1816-0387-2018-6-61-73
Смирнов Е.В. Пищевые ароматизаторы. Справочник. Санкт-Петербург: ЦОП “Профессия”, 2008. 736 с.
Шеламова С.А. Биотехнологические основы конверсии триглицеридов: монография. Воронеж: Научная книга, 2008. 145 с.
Тырсин Ю.А., Шеламова С.А. Механизм гидролиза, синтеза и переэтерификации в пищевой биотехнологии: монография. Воронеж: Научная книга, 2012. 124 с.
Bartling K., Thompson J.U.S., Pfromm P.H., Czermak P., Rezac M.E. // Biotechnol. Bioeng. 2011. V. 75. P. 676. https://doi.org/10.1002/bit.1193
Aissa I., Sellami M., Kamoun A., Gargouri Y., Miled N. // Curr. Chem. Biol. 2012. V. 6. P. 77. https://doi.org/10.2174/187231312799984376
Larios A., Garcia H.S., Oliart R.M., Valerio-Alfaro G. // Appl. Microbiol. Biotechnol. 2004. V. 65. P. 373. https://doi.org/10.1007/s00253-004-1602-x
Гамаюрова В.С., Зиновьева М.Е., Елизарова Е.В. // Катализ в промышленности. 2008. Т. 3. С. 54.
Matle C.R., Bordinhao C., Poppe J.K., Rodrigues R.C., HertzP.F. // J. Mol. Catal. B: Enzymatic. 2016. V. 127. P. 67.
Гамаюрова В.С., Шнайдер К.Л., Джамай М.Д. // Катализ в промышленности. 2016. Т. 16. № 3. С. 64. https://doi.org/10.18412/1816-0387-2016-3-64-68
Гамаюрова В.С., Зиновьева М.Е., Шнайдер К.Л., Давлетшина Г.А. // Катализ в промышленности. 2020. Т. 20. С. 216. https://doi.org/10.18412/1816-0387-2020-3-216-233
Garcia T., Sanchez N., Martinez M., Aracil J. // Enzyme Microb. Technol. 1999. V. 25. P. 584. https://doi.org/10.1016/s0141-0229(99)00082-4
Garcia T., Sanchez N., Martinez M.,Aracil, J. // Enzyme Microb. Technol. 1999. V. 25. P. 591. https://doi.org/10.1016/s0141-0229(99)00083-6
Шеламова С.А., Тырсин Ю.А. // Научные ведомости. 2014. Т. 3(174). Вып. 2. С. 103.
Аксенов С.И. Вода и ее роль в регулировании биологических процессов. Москва–Ижевск: Институт компьютерных исследований. 2004. 212 с.
Коробов В.И., Очков В.Ф. Химическая кинетика: введение с Mathcad/Мaple/MCS. Москва: Горячая линия – Телеком. 2009. 384 с.
Ерандаева Ю.В., Воробьев Е.С., Воробьева Ф.И. // Вестник Казанского технологического университета. 2011. Т. 11. С. 88.
Gamayurova V.S., Shnaider K.L., Zaripova S.K., Mataz J., Jamai M.D. // J. Thermodyn. Catal. 2016. V. 7. P. 161. https://doi.org/10.4172/2157-7544.1000161
Гамаюрова В.С., Давлетшина Г.А. // Известия ВУЗов. Прикладная химия и биотехнология. 2020. Т. 10. № 3. С. 515. https://doi.org/10.21285/2227-2925-2020-10-3-515-521
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ