

Кинетика и катализ, 2021, T. 62, № 3, стр. 296-304
Активация пероксида водорода ацетонитрилом в окислении тиоэфиров. Кинетика и механизм реакций
А. К. Любимова a, Т. В. Безбожная a, В. Л. Лобачев a, *
a Государственное учреждение Институт физико-органической химии и углехимии
им. Л.М. Литвиненко
83114 Донецк, ул. Р. Люксембург, 70, Украина
* E-mail: lobachev.vl51@yandex.ru
Поступила в редакцию 03.07.2020
После доработки 14.11.2020
Принята к публикации 16.12.2020
Аннотация
Кинетическим распределительным методом изучена кинетика окисления диэтилсульфида (Et2S) пероксидом водорода (Н2О2) в водных растворах ацетонитрила (MeCN). Установлено, что порядок реакции по субстрату зависит от рН и изменяется от первого при рН 8.06 до близкого к нулевому при рН 11.02. Начальные скорости убыли Et2S растут с увеличением рН и линейно зависят от концентраций гидропероксид-аниона (НОО–) и MeCN. Предположено, что реакция включает неравновесное образование в медленной стадии пероксиимидата при взаимодейстии НОО– с MeCN, который далее быстро окисляет Et2S.
ВВЕДЕНИЕ
Окисление органических сульфидов (RSR′) широко применяется для разложения активных компонентов пестицидов и отравляющих веществ [1], обессеривания углеводородного сырья и топлив [2], синтеза сульфонов и сульфоксидов, являющихся важными интермедиатами в синтезе биологически активных соединений и лекарств [3]. Проблема создания систем для быстрого селективного окисления RSR′ включает решение двух основных задач: 1) поиск новых эффективных, отвечающих требованиям “зеленой” химии, окислителей и 2) выбор растворителей, позволяющих, с одной стороны, существенно увеличить растворимость зачастую практически нерастворимых в воде сульфидов и, с другой – сохранить высокую скорость их окисления.
Среди многочисленных окислителей [4], используемых для окисления RSR′, наиболее экологически чистым и дешевым является пероксид водорода (Н2О2). Однако сам по себе пероксид водорода имеет низкую активность в окислении сульфидов, что зачастую требует его специфической активации. Один из путей активации Н2О2 – его превращение в пероксокислоты с применением таких активаторов, как бикарбонаты [5, 6], бораты [7–10], молибдаты [11], силикаты [12], нитриты [13, 14] и других соединений [4].
Для повышения растворимости сульфидов, а, следовательно, и скорости их окисления используют водно-спиртовые смеси [5, 6, 10]. Альтернативным путем увеличения скорости реакций гидрофобных субстратов в воде является их солюбилизация поверхностно-активными веществами (ПАВ).
В этом плане особый интерес представляют растворы вода/ацетонитрил (МеСN). Использование смеси Н2О/МеСN должно приводить к повышению растворимости RSR′. Кроме того, известно, что в щелочных средах нитрилы (RCN) взаимодействуют с Н2О2 с образованием короткоживущих пероксиимидных кислот, RC(O2H)=NH2, или пероксиимидатов, RC(O2H)=NH– [15–17], которые эффективно окисляют алкены в эпоксиды [18], амины в N-оксиды [15] и сульфиды в сульфоксиды и сульфоны [16, 17].
Окисление арилметилсульфидов (ArSMe) пероксидом водорода в присутствии МеСN было изучено в растворах метанол–К2СО3 [16] и в водных растворах NaOH (0.001–0.002 М) при [H2O2] $ \gg $ $ \gg $ [NaOH] [17]. В последней системе скорость окисления тиоанизола не зависит от концентрации субстрата (нулевой порядок по ArSMe), но линейно зависит от [HОО–] и [МеCN]. Кинетические данные были интерпретированы в рамках механизма, включающего образование в медленной стадии пероксиимидата (или пероксиимидной кислоты) и в их быстрых конкурентных реакциях с ArSMe и Н2О2 с образованием сульфоксида и амида соответственно [17].
Целью настоящей работы было установление механизма реакции и природы активной частицы в процессе окисления диэтилсульфида системой Н2О2/МеСN. Для этого было проведено исследование зависимости начальных скоростей реакций Et2S с пероксидом водорода от кислотности среды в широком диапазоне изменения рН и от концентраций Н2О2 и МеСN.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные вещества и реагенты
Диэтилсульфид Et2S синтезировали по методике [19]. Для приготовления рабочих растворов использовали бидистиллированную воду, 30%-ный раствор Н2О2, H3PO4 (х. ч.) и NaOH (х. ч.). Ацетонитрил очищали по методике [20].
Кинетические измерения
Кинетику окисления Et2S в системе Н2О/MeCN изучали кинетическим распределительным методом [21] по убыли субстрата в газовой фазе при [MeCN] $ \gg $ [H2O2] $ \gg $ [Et2S]. Рабочие растворы готовили непосредственно перед кинетическими измерениями. В газовую (воздушную) фазу термостатируемого встряхиваемого реактора типа “каталитическая утка” (полный объем V = 62.3 см3) вводили смесь паров Et2S и толуола, стабильного в условиях реакции и используемого в качестве внутреннего стандарта. В определенные промежутки времени пробы газовой фазы (0.1 см3) отбирали стеклянным шприцем через отверстие в пробке реактора, закрытое резиновой и тефлоновой прокладками. За изменением концентрации субстрата в газовой фазе следили с помощью ГЖХ, хроматограф ЛХМ-80 (Россия), пламенно-ионизационный детектор, колонка 2 м, неподвижная фаза – 5% SE-30 на носителе Chromaton N-AW.
Большая частота встряхивания реактора (500 мин–1) позволяет исключить диффузионные осложнения, связанные с массопереносом Et2S из газовой фазы в раствор [21].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Наблюдаемая кинетика окисления диэтилсульфида
Согласно данным [6], реакция окисления диэтилсульфида пероксидом водорода в воде имеет первый порядок как по Et2S, так и по Н2О2 в области изменения рН от 9 до 12. В растворах Н2О/MeСN порядок реакции по субстрату (n), определенный дифференциальным методом [22], зависит от рН и изменяется от первого при рН 8.06 до близкого к нулевому при рН 11.02 (табл. 1).
Таблица 1.
Зависимости начальных скоростей (WМеCN) окисления Et2S и порядка реакции по субстрату (п) от рН среды в растворах Н2О/MeСN; вклады маршрутов реакций с пероксидом водорода (WРН)а и пероксиимидатом (WPI)б при [H2O2] = 0.006 М
| pH | W МеCN × 109, M c–1 | n | WРН × 109, M c–1 | WPI × 109, M c–1 | [HOО–] × 104, Mв | WPI/ [HOО–][MeCN] × × 104, M–1 c–1 |
|---|---|---|---|---|---|---|
| 8.06 | 5.8 | 1.0 | 5.8 | 0 | 0.0434 | 0 |
| 8.63 | 5.9 | 0.95 | 5.8 | 0.1 | 0.161 | 0.32 |
| 9.08 | 6.7 | 0.54 | 5.8 | 0.90 | 0.451 | 1.1 |
| 9.49 | 8.4 | 0.30 | 5.8 | 2.6 | 1.147 | 1.2 |
| 9.97 | 13 | 0.25 | 5.7 | 7.3 | 3.34 | 1.2 |
| 10.0 | 14 | 0.21 | 5.6 | 8.4 | 3.57 | 1.2 |
| 10.5 | 29 | 0.15 | 5.3 | 24 | 9.98 | 1.3 |
| 11.02 | 60 | 0.10 | 4.4 | 56 | 23.9 | 1.2 |
Примечание. Условия реакции: [МеCN] = 0.19 М (1 об. %), NS = 8.4 × 10–7 моль, объем реактора V = 62.2 мл, λ = 2.12; α = 0.06, Т = 25°С. аРасчет по уравнению (9) при kНООН = 2.7 × 10–2 М–1 с–1, ${{k}^{{{\text{HO}}{{{\text{O}}}^{ - }}}}}$ = 0.41 × 10–2 М–1 с–1; рКа = 11.5 [6]. бWPI = WМеCN – WРН. вРасчет по уравнению (10).
Скорость убыли Et2S из газовой фазы в системе Н2О/MeСN при постоянном значении рН линейно растет с увеличением концентрации Н2О2 (табл. 2), при этом порядок реакции по субстрату в пределах точности измерений практически не изменяется. Скорость реакции также линейно возрастает с повышением концентрации MeСN, а порядок реакции по Et2S не зависит от [MeСN] (табл. 3).
Таблица 2.
Зависимости начальных скоростей (WМеCN) окисления Et2S и порядка реакции по субстрату (п) от концентрации Н2О2 в растворах Н2О/MeСN при рН 10; вклады маршрутов WРН и WPI*
| [H2O2] × 103, М | WМеCN × 109, M c–1 | n | WРH × 109, M c–1 |
WPI × 109, M c–1 | [HOО–] × 104, M | WPI/[HOО–] × [СH3CN] × 104, M–1 c–1 |
|---|---|---|---|---|---|---|
| 3 | 6.9 | 0.28 | 2.8 | 4.1 | 1.79 | 1.2 |
| 5 | 12 | 0.33 | 4.7 | 7.3 | 2.98 | 1.3 |
| 6 | 14 | 0.21 | 5.6 | 8.4 | 3.57 | 1.2 |
| 7 | 17 | 0.21 | 6.6 | 10 | 4.17 | 1,3 |
| 9 | 21 | 0.19 | 8.5 | 13 | 5.36 | 1.2 |
* Условия см. в табл. 1.
Таблица 3.
Зависимости начальных скоростей (WМеCN), порядка реакции по субстрату (n) и вклада маршрута реакций с пероксиимидатом (WPI) от концентрации MeСN для окисления Et2S пероксидом водорода при [Н2О2] = 0.006 М, рН 10
| [MeCN], об. % | [MeCN], M | WМеCN × 109, M c–1 а |
n | WPI × 109, M c–1 б | WPI/
[HOО–] × 105, c–1 в |
WPI/[HOО–] × [MeCN] × 104, M–1 c–1 |
|---|---|---|---|---|---|---|
| 0.5 | 0.095 | 10 | 0.26 | 4.4 | 1.2 | 1.3 |
| 1.0 | 0.19 | 14 | 0.21 | 8.4 | 2.4 | 1.2 |
| 1,5 | 0.29 | 19 | 0.22 | 13 | 3.8 | 1.3 |
| 2.0 | 0.38 | 21 | 0.25 | 15 | 4.3 | 1.1 |
| 2.5 | 0.48 | 26 | 0.24 | 20 | 5.7 | 1.2 |
| 5.0 | 0.95 | 47 | 0.22 | 41 | 12 | 1.2 |
* Условия см. в табл. 1.аРасчет при α = 0.06.бWPI = WМеCN – WРН при WPH = 5.6 × 10–9 M c–1. в[HOО–] = 3.57 × 10–4 М.
Поэтому для анализа кинетических данных в растворах Н2О/MeСN при различных условиях и сравнения их с данными по окислению Et2S пероксидом водорода в воде мы использовали значения начальных скоростей реакции.
Расчет жидкофазных скоростей реакций окисления Et2S в растворах Н2О/MeСN
Кинетический распределительный метод (КРМ) был предложен, широко изучен и применен в различных вариантах для исследования кинетики реакций летучих малорастворимых субстратов, таких как алканы, в водных или сернокислотных средах [21]. Большое значение в КРМ имеет интенсивное перемешивание фаз, что позволяет исключить осложнения, связанные с диффузией и межфазным массопереносом.
В “простой” модели КРМ [21] предполагается, что субстрат S равновесно распределяется между газовой (g) и жидкой фазами (l) закрытого реактора, но реакция с реагентом Х протекает только в растворе. При [S] $ \ll $ [X] кинетика убыли субстрата из газовой фазы следует уравнению:
(1)
${{ - {\text{d}}{{{[{\text{S}}]}}_{{\text{g}}}}} \mathord{\left/ {\vphantom {{ - {\text{d}}{{{[{\text{S}}]}}_{{\text{g}}}}} {{\text{d}}\tau }}} \right. \kern-0em} {{\text{d}}\tau }} = {{k}_{\lambda }}{{[{\text{S}}]}_{{\text{g}}}}[{\text{X}}] = {{{{k}_{2}}{{{[{\text{S}}]}}_{{\text{l}}}}[{\text{X}}]} \mathord{\left/ {\vphantom {{{{k}_{2}}{{{[{\text{S}}]}}_{{\text{l}}}}[{\text{X}}]} {(1 + \alpha \lambda )}}} \right. \kern-0em} {(1 + \alpha \lambda )}},$Рассмотрим модель КРМ для скорости убыли субстрата (W). Введем обозначения: $N_{{\text{g}}}^{{\text{S}}}$ и [S]g = = $N_{{\text{g}}}^{{\text{S}}}$/Vg – количество и концентрация субстрата S в газе соответственно; $N_{{\text{l}}}^{{\text{S}}}$ и [S]l = $N_{{\text{l}}}^{{\text{S}}}$/Vl – то же для раствора. Тогда полное количество вещества S (NS) в замкнутой системе равно:
(2)
$\begin{gathered} {{N}^{{\text{S}}}} = N_{{\text{g}}}^{{\text{S}}} + N_{{\text{l}}}^{{\text{S}}} = {{[{\text{S}}]}_{{\text{g}}}}{{V}_{{\text{g}}}} + \\ + \,\,{{[{\text{S}}]}_{{\text{l}}}}{{V}_{{\text{l}}}} = {{[{\text{S}}]}_{{\text{g}}}}{{V}_{{\text{g}}}}(1 + {1 \mathord{\left/ {\vphantom {1 {\alpha \lambda }}} \right. \kern-0em} {\alpha \lambda }}). \\ \end{gathered} $Если реакция протекает только в растворе, то скорость убыли субстрата в системе равна:
(3)
$ - ({{{\text{d}}{{N}^{{\text{S}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{N}^{{\text{S}}}}} {{\text{d}}\tau }}} \right. \kern-0em} {{\text{d}}\tau }}) = {{W}_{{\text{l}}}}{{V}_{{\text{l}}}} = {{{{W}_{{\text{l}}}}{{V}_{{\text{g}}}}} \mathord{\left/ {\vphantom {{{{W}_{{\text{l}}}}{{V}_{{\text{g}}}}} \lambda }} \right. \kern-0em} \lambda },$В то же время согласно уравнению (2)
(4)
$\begin{gathered} - ({{{\text{d}}{{N}^{{\text{S}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{N}^{{\text{S}}}}} {{\text{d}}\tau }}} \right. \kern-0em} {{\text{d}}\tau }}) = - {{V}_{{\text{g}}}}(1 + {1 \mathord{\left/ {\vphantom {1 {\alpha \lambda }}} \right. \kern-0em} {\alpha \lambda }})({{{\text{d}}{{{[{\text{S}}]}}_{{\text{g}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{{[{\text{S}}]}}_{{\text{g}}}}} {{\text{d}}\tau }}} \right. \kern-0em} {{\text{d}}\tau }}) = \\ = {{V}_{{\text{g}}}}(1 + {1 \mathord{\left/ {\vphantom {1 {\alpha \lambda }}} \right. \kern-0em} {\alpha \lambda }}){{W}_{{\text{g}}}}. \\ \end{gathered} $Из равенства
следует зависимость, связывающая измеряемую (наблюдаемую) скорость убыли субстрата в газовой фазе Wg со скоростью реакции в раствореWl:(6)
${{W}_{{\text{g}}}} = {{{{W}_{{\text{l}}}}\alpha } \mathord{\left/ {\vphantom {{{{W}_{{\text{l}}}}\alpha } {(1 + \alpha \lambda )}}} \right. \kern-0em} {(1 + \alpha \lambda )}}.$При вариации λ из линейной зависимости
(7)
${1 \mathord{\left/ {\vphantom {1 {{{W}_{{\text{g}}}}}}} \right. \kern-0em} {{{W}_{{\text{g}}}}}} = {\lambda \mathord{\left/ {\vphantom {\lambda {{{W}_{{\text{l}}}}}}} \right. \kern-0em} {{{W}_{{\text{l}}}}}} + {1 \mathord{\left/ {\vphantom {1 {\alpha {{W}_{{\text{l}}}}}}} \right. \kern-0em} {\alpha {{W}_{{\text{l}}}}}},$Рис. 1.
Выполнения зависимости (7) для реакции окисления Et2S пероксидом водорода в растворах Н2О/MeСN при [Н2О2] = 0.006 M, [MeCN] = 1 об. % (0.19 М), NS = 8.4 × 10–7 моль и pH 10.0.
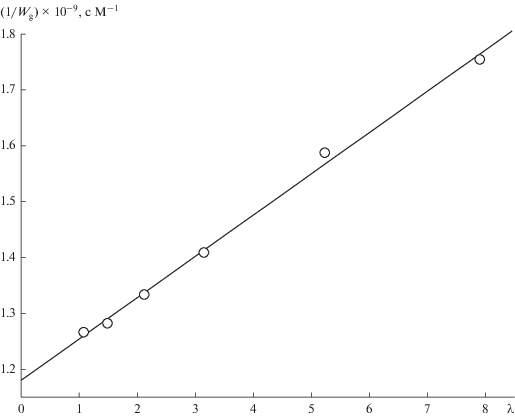
Наблюдаемые начальные скорости определяли из зависимости измеряемой концентрации субстрата в газовой фазе $({{N_{\tau }^{{\text{S}}}} \mathord{\left/ {\vphantom {{N_{\tau }^{{\text{S}}}} {{{V}_{{\text{g}}}}}}} \right. \kern-0em} {{{V}_{{\text{g}}}}}})$ от времени в области убыли NS < 20%. Текущую концентрацию субстрата в газовой фазе $({{N_{\tau }^{{\text{S}}}} \mathord{\left/ {\vphantom {{N_{\tau }^{{\text{S}}}} {{{V}_{{\text{g}}}}}}} \right. \kern-0em} {{{V}_{{\text{g}}}}}})$ рассчитывали по уравнению
(8)
$({{N_{\tau }^{{\text{S}}}} \mathord{\left/ {\vphantom {{N_{\tau }^{{\text{S}}}} {{{V}_{{\text{g}}}}}}} \right. \kern-0em} {{{V}_{{\text{g}}}}}}) = ({{{{\varphi }_{\tau }}} \mathord{\left/ {\vphantom {{{{\varphi }_{\tau }}} {{{\varphi }_{0}}}}} \right. \kern-0em} {{{\varphi }_{0}}}})({{N_{0}^{{\text{S}}}} \mathord{\left/ {\vphantom {{N_{0}^{{\text{S}}}} {{{V}_{{\text{g}}}}}}} \right. \kern-0em} {{{V}_{{\text{g}}}}}}),$Влияние кислотности среды на скорость окисления Et2S
По данным [6], в водных растворах в области рН 8–12 окисление Et2S пероксидом водорода (РН) протекает по двум параллельным маршрутам: с участием НООН и НОО–. В этом случае зависимость начальной скорости убыли диэтилсульфида (WРН) от кислотности имеет вид:
(9)
$\begin{gathered} W_{{}}^{{{\text{PH}}}} = {{W}^{{{\text{HOOH}}}}} + {{W}^{{{\text{HO}}{{{\text{O}}}^{ - }}}}} = \\ = \left( {\frac{{{{k}^{{{\text{HOOH}}}}}[{{{\text{H}}}^{ + }}] + {{k}^{{{\text{HO}}{{{\text{O}}}^{ - }}}}}{{K}_{{\text{a}}}}}}{{[{{{\text{H}}}^{ + }}] + {{K}_{{\text{a}}}}}}} \right){{[{\text{PH]}}}_{{\text{0}}}}{{{\text{[E}}{{{\text{t}}}_{{\text{2}}}}{\text{S]}}}_{{\text{0}}}}{\text{,}} \\ \end{gathered} $Зависимость начальных скоростей окисления Et2S в воде (${{W}^{{{{{\text{Н}}}_{{\text{2}}}}{\text{О}}}}}$), рассчитанных по данным [6], хорошо описывается уравнением (9) при значениях параметров kНООН = 2.7 × 10–2 М–1 с–1, ${{k}^{{{\text{HO}}{{{\text{O}}}^{ - }}}}}$ = 0.41 × 10–2 М–1 с–1 и рKа = 11.5 (рис. 2, кривая 1).
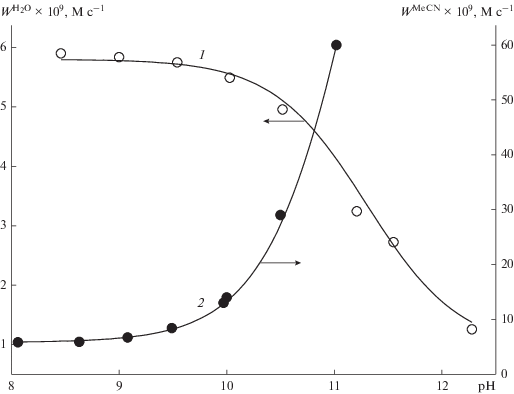
В отличие от водных растворов скорость реакции в смеси Н2О/MeСN (1 об. %) экспоненциально растет с уменьшением кислотности (рис. 2, кривая 2) и при рН 11 более чем в 10 раз превышает скорость окисления в воде (табл. 1). Эти результаты говорят о том, что в указанной системе с увеличением рН повышается концентрация активных частиц, участвующих в окислении Et2S. Отметим, что при рН 8–9 скорость окисления в растворах Н2О/MeСN слабо зависит от кислотности среды и близка к соответствующим значениям в воде.
Полученные данные о порядках реакции и зависимости скорости от кислотности среды позволяют предположить, что при рН < 9 основной вклад в скорость окисления Et2S в системе Н2О/MeСN (WМеCN) вносят маршруты реакций с пероксидом водорода, уравнение (9). Об этом свидетельствуют порядки реакции по Et2S (n > 0.5), постоянство скоростей в изученном интервале рН и их близость к соответствующим значениям для реакции в воде.
Рост скорости реакции при рН ≥ 10 указывает на то, что в этих условиях основную роль в окислении Et2S играет маршрут с активированной формой Н2О2 (PI) – пероксиимидной кислотой МеC(O2H)=NH2 или пероксиимидатом МеC(O2H)=NH–, которые образуются при взаимодействии НОО– с MeСN [15–17, 23], схема 1 :
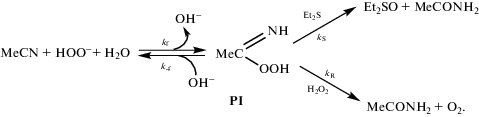
Схема 1 .
Здесь kf и k–f – константы скорости прямой и обратной стадий реакций образования PI; kS – константа скорости реакции Et2S с PI; kR – константа скорости маршрута PI с пероксидом водорода, приводящему к образованию ацетиламида [23].
Разделение маршрутов окисления Et2S в системе Н2О/MeСN
Вклад маршрута WРН в реакцию окисления диэтилсульфида пероксидом водорода в растворах Н2О/MeСN рассчитывали по уравнению (9) с использованием констант скорости kНООН = 2.7 × × 10–2 М–1 с–1, ${{k}^{{{\text{HO}}{{{\text{O}}}^{ - }}}}}$ = 0.41 × 10–2 М–1 с–1, рKа = = 11.5 [6]11, рассчитанной концентрацией диэтилсульфида в растворе при α = 0.06 и в предположении, что скорости окисления Et2S в данной системе и в воде близки. Об этом может свидетельствовать22 совпадение скоростей реакций в водных растворах и в системе Н2О/MeСN при рН < 9 (табл. 1). Вклад маршрута с пероксиимидатом (WPI) (схема 1) определяли как разность между измеряемой начальной скоростью окисления Et2S в растворах Н2О/MeСN (WМеCN) и скоростью окисления пероксидом водорода (WРН).
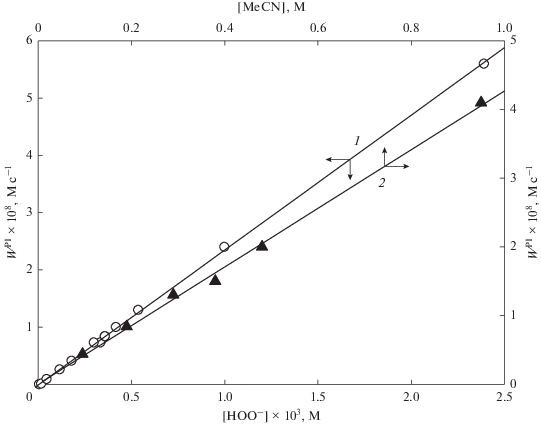
Скорость маршрута WPI растет с увеличением рН и линейно зависит от концентрации НОО– (рис. 3, кривая 1). Концентрации НОО– (табл. 1–3) рассчитывали по уравнению:
(10)
$[{\text{HO}}{{{\text{O}}}^{ - }}] = \frac{{{{K}_{{\text{a}}}}{{{[{\text{PH}}]}}_{0}}}}{{{{K}_{{\text{a}}}} + [{{{\text{H}}}^{ + }}]}}$Рис. 3.
Зависимости начальных скоростей убыли Et2S в маршруте реакции с пероксиимидатом в системе Н2О/MeСN от концентрации HOО– (1) и концентрации MeСN (2). Условия см. в табл. 1.
при рКа = 11.5.
В области рН 9.08–10.02, когда вклад маршрута с пероксиимидатом становится существенным или основным, значения WPI/[HOО–][MeCN] одинаковы и равны (1.2 ± 0.1) × 10–4 M–1 c–1 (табл. 3). Значения WPI при постоянном рН также линейно зависят от [HOО–] при варьировании начальной концентрации пероксида водорода (табл. 2, рис. 3) и концентрации MeCN (рис. 3, кривая 2).
Найденная величина WPI/[HOО–][MeCN] = = (1.2 ± 0.1) × 10–4 M–1 c–1 отличается от полученного в работе [17] значения WМеCN/[HOО–][MeCN] = = (7.8 ± 0.8) × 10–4 M–1 c–1 для окисления тиоанизола пероксидом водорода в водных растворах NaOH (0.01–0.02 М) в присутствии ацетонитрила. Возможно это связано с тем, что в работе [17] использовались измеряемые скорости WМеCN и не учитывался вклад маршрута с пероксидом водорода.
О механизме окисления Et2S пероксиимидатом
Согласно данным [16, 17] окисление тиоэфиров пероксиимидатами (или пероксиимидными кислотами) протекает по схеме 1 , включающей медленную стадию образования активного интермедиата PI в реакции MeCN с НОО– и его быструю реакцию с субстратом, о чем свидетельствуют первые порядки реакции по MeCN и НОО– и нулевой порядок по RSR'. Полученные нами данные (табл. 1–3) также согласуются с предложенным в [16, 17] механизмом.
В литературе нет однозначных сведений о характере первой стадии в схеме 1 и степени протонирования интермедиата PI [17]. В работах [16, 17], посвященных изучению ArSMe, полагают, что эта стадия является медленной (возможно обратимой), но неравновесной. В то же время, по данным [15] константы равновесия K = kf/k–f × 104, определенные спектофотометрическим методом в фосфатном буфере (рН 7), равны 2.0, 3.3 и 5.2 М–1 при 20, 30 и 40°C соответственно.
Для схемы 1 с быстрым равновесным образованием пероксиимидата скорость убыли Et2S должна описываться уравнением
(11)
$\begin{gathered} {{W}^{{{\text{PI}}}}} = K{{k}_{{\text{S}}}}[{\text{MeCN][HO}}{{{\text{O}}}^{ - }}]{{[{\text{E}}{{{\text{t}}}_{{\text{2}}}}{\text{S]}}}_{{\text{0}}}} = \\ = \frac{{K{{K}_{{\text{a}}}}{{k}_{{\text{S}}}}[{\text{MeCN][PH}}{{{\text{]}}}_{{\text{0}}}}{{{{\text{[E}}{{{\text{t}}}_{{\text{2}}}}{\text{S}}]}}_{0}}}}{{{{K}_{{\text{a}}}} + {\text{[}}{{{\text{H}}}^{ + }}]}}. \\ \end{gathered} $Согласно уравнению (11) в случае, когда стадия образования пероксиимидата является равновесной, скорость убыли диэтилсульфида, в отличии от экспериментальных данных, должна следовать первому порядку по субстрату во всей области рН.
Рассмотрим механизм (схема 1 ) с обратимым, но неравновесным образованием пероксиимидата. В предположении о стационарности концентрации интермедиата PI наблюдаемая начальная скорость убыли Et2S описывается уравнением
(12)
${{W}^{{{\text{PI}}}}} = \frac{{{{k}_{{\text{f}}}}{{k}_{{\text{S}}}}[{\text{MeCN][HO}}{{{\text{O}}}^{ - }}]{{{[{\text{E}}{{{\text{t}}}_{{\text{2}}}}{\text{S]}}}}_{{\text{0}}}}}}{{{{k}_{{{\text{ - f}}}}} + {{k}_{{\text{S}}}}{{{[{\text{E}}{{{\text{t}}}_{{\text{2}}}}{\text{S]}}}}_{{\text{0}}}} + {{k}_{{\text{R}}}}{{{[{\text{PH]}}}}_{0}}}}.$Если предположить, что k–f$ \ll $ kS[Et2S] $ \gg $ kR[PH]0, то выражение для начальной скорости окисления Et2S пероксиимидатом в изученной области рН имеет вид:
(13)
${{W}^{{{\text{PI}}}}} = {{k}_{{\text{f}}}}[{\text{MeCN}}][{\text{HO}}{{{\text{O}}}^{ - }}]{\text{.}}$То есть медленной стадией реакции является взаимодействие ацетонитрила с гидропероксид-анионом с образованием перокисимидата PI, а скорость убыли Et2S следует нулевому порядку по субстрату. Постоянство значений WPI/[MeCN][HOО–] в изученной области рН (табл. 1–3) согласуется с этим предположением и данными работы [16], в которой было найдено, что kS$ \gg $ kR$ \gg $ kf.
Реакционная способность пероксиимидатов
Реакции RSR′ с Н2О2, пероксимуравьиной, пероксиуксусной, пероксиазотистой кислотами и с пероксосульфат-анионом [14], пероксокарбонат-анионом, моно- и дипероксоборат-анионами [8] имеют первый порядок по сульфиду. Константы скорости реакции второго порядка Et2S с В(ОН)3(ООН)–, ${\text{В}}{{\left( {{\text{ОН}}} \right)}_{2}}\left( {{\text{ООН}}} \right)_{2}^{ - }$ и ${\text{HCO}}_{4}^{ - }$ в 2.5, 100 и 97 раз выше, чем с Н2О2 [8].
Пероксиимидаты быстро реагируют с сульфидами, и наблюдаемая кинетика нулевого порядка в окислении Et2S и PhSMe [17] может свидетельствовать о том, что RC(O2H)=NH2 (или RC(O2H)=NH–) значительно более реакционноспособны по сравнению с другими типичными пероксокислотами, а скорость убыли субстрата лимитируется скоростью реакции MeCN с НОО–. Иное объяснение нулевого порядка по RSR′ состоит в том, что реакция образования пероксиимидата является неравновесным процессом и протекает гораздо медленнее, чем в случае образования других пероксокислот.
ЗАКЛЮЧЕНИЕ
Установлено, что скорость окисления Et2S пероксидом водорода в водных растворах МеCN растет с увеличением рН среды. Порядок реакции по субстрату близкий к нулевому указывает на то, что лимитирующей стадией процесса является реакция НОО– с МеCN, приводящая к образованию активного пероксиимидата PI, который затем в быстрой стадии взаимодействует с Et2S, схема 1 . При значении рН 10 скорость убыли Et2S в системе МеCN/Н2О почти в 15 раз превышает скорость окисления диэтилсульфида пероксидом водорода в воде. Отсюда можно заключить, что ацетонитрил – один из наиболее эффективных активаторов Н2О2 в реакции окисления органических сульфидов.
Список литературы
Wagner G.W., Yang Y.C. // Ind. Eng. Chem. Res. 2002. V. 41. № 8. P. 1925.
Анисимов А.В., Тараканова А.В. // Рос. хим. журн. (Журн. Рос. хим. об-ва им. Д.И. Менделеева). 2008. Т. LII. № 4. С. 32.
Fernandez I., Khiar N. // Chem. Rev. 2003. V. 103. № 9. P. 3651.
Das R., Chakraborty D. // Tetrahedron Lett. 2010. V. 51. № 48. P. 6255.
Richardson D.E., Yao H., Frank K.M., Bennett D.A. // J. Amer. Chem. Soc. 2000. V. 122. № 8. P. 1729.
Лобачев В.Л., Савелова В.А., Прокопьева Т.М. // Теорет. и эксперим. химия. 2004. Т. 40. № 3. С. 157.
Davies D.M., Deary M.E., Quill K., Smith R.A. // Chem. Eur. J. 2005. V. 11. № 12. P. 3552.
Лобачев В.Л., Зимцева Г.П., Матвиенко Я.В., Рудаков Е.С. // Теорет. и эксперим. химия. 2007. Т. 43. № 1. С. 38.
Durrant M.C., Davies D.M., Deary M.E. // Org. Biomol. Chem. 2011. V. 9. № 20. P. 7249.
Лобачев В.Л., Дятленко Л.М., Зимцева Г.П. // Теорет. и эксперим. химия. 2012. Т. 48. № 3. С. 168.
Chiarini M., Gillitt N.D., Bunton C.A. // Langmuir. 2002. V. 18. № 10. P. 3836.
Лобачев В.Л., Дятленко Л.М., Зимцева Г.П. // Теорет. и эксперим. химия. 2012. Т. 48. № 5. С. 322.
Лобачев В.Л., Зимцева Г.П., Рудаков Е.С. // Теорет. и эксперим. химия. 2005. Т. 41. № 5. С. 290.
Amels P., Elias H., Wannowius K.-J. // J. Chem. Soc., Faraday Trans. 1997. V. 93. № 15. P. 2537.
Laus G. // J. Chem. Soc., Perkin Trans. 2. 2001. P. 864.
Bethell D., Graham A.E., Heer J.P., Markopoulou O., Page P.C.B., Park B.K. // J. Chem. Soc., Perkin Trans. 2. 1993. P. 2161.
Gillitt N.D., Domingos J., Bunton C.A. // J. Phys. Org. Chem. 2003. V. 16. P. 603.
Payne G.B., Deming P.H., Williams P.H. // J. Org. Chem. 1961. V. 26. № 3. P. 659.
Вейганд-Хильгентаг. Методы эксперимента в органической химии. Москва: Химия, 1969. 944 с.
Вайсбергер А., Проскауэр Э., РиддикДж., Тупс Э. Органические растворители. Москва: Инлит, 1958. 519 с.
Рудаков Е.С. Реакции алканов с окислителями, металлокомплексами и радикалами в растворах. Киев: Наук. думка, 1985. 247 с.
Семиохин И.А., Страхов Б.В., Осипов А.И. Кинетика химических реакций. Москва: Изд-во МГУ, 1995. 351 с.
McLsaac Jr.J.E., Ball R.E., Behrman E.J. // J. Org. Chem. 1971. V. 36. № 9. P. 3048.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ