

Кинетика и катализ, 2021, T. 62, № 3, стр. 283-295
ГЛИФОСАТ: СПОСОБЫ ПОЛУЧЕНИЯ
Д. Ю. Ющенко a, *, Т. Б. Хлебникова a, З. П. Пай a, В. И. Бухтияров a
a ФГБУН ФИЦ “Институт катализа им. Г.К. Борескова СО РАН”
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: dyy@catalysis.ru
Поступила в редакцию 29.12.2020
После доработки 15.01.2021
Принята к публикации 15.01.2021
Аннотация
В представленном обзоре описаны основные химические процессы получения глифосата и выделены их особенности. Рассмотрены способы синтеза глифосата как по направлениям, реализуемым в настоящее время в промышленности (из глицина и через окисление N-(фосфонометил)-иминодиуксусной кислоты), так и перспективные так называемые “атом-эффективные” методы (деалкилирование N-замещенных глифосатов).
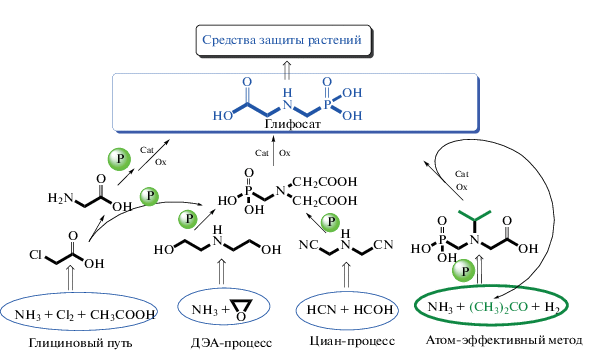
ВВЕДЕНИЕ
N-(фосфонометил)-глицин, широко известный как глифосат, является действующим веществом препаратов, используемых в качестве средств защиты культивируемых растений (Раундап®, Торнадо®, Глисол® и др. – растворы с концентрацией до 35% глифосата из расчета на его аммонийную соль). В настоящее время глифосатсодержащие препараты находятся в числе наиболее востребованной агрохимической продукции как на мировом, так и на внутреннем рынке РФ. Основная область применения глифосатсодержащих препаратов – обработка посевных площадей культур, имеющих устойчивость (естественную или добавленную методами селекции) к глифосату, после которой сорные растения погибают, а урожайность культивируемых увеличивается. Обработка посевных площадей стратегически важных культур (пшеница, кукуруза, соя) гербицидными препаратами, содержащими глифосат, обуславливает повышенное требование к его качеству. При этом зачастую именно выбор метода синтеза в значительной степени определяет чистоту получаемого глифосата, а также нормы образования отходов и расхода электроэнергии на единицу готовой продукции. В приведенном обзоре рассмотрены различные способы синтеза глифосата, в том числе с применением катализаторов и экологически благоприятных окислителей.
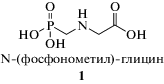
Впервые N-(фосфнометил)-глицин (1, глифосат) получил в 1950 г. Генри Мартин (Dr. Henry Martin), работавший в швейцарской фармацевтической фирме “Cilag”, однако синтезированная молекула оказалось не интересна для фармацевтической промышленности. Спустя десятилетия “Aldrich Chemical Co.” приобрела образцы биологически активных веществ кoмпании “Cilag”, которые поступили в продажу как химические реагенты. Так, образцы N-(фосфнометил)-глицина были приобретены компанией “Monsanto” в числе других реактивов для исследовательских работ. Позже Джон Е. Франц (Dr. John E. Franz), работавший в “Monsanto” в рамках направления разработки умягчителей воды, заметил, что ряд фосфонометильных кислот проявляют гербицидную активность в отношении многолетних сорняков [1]. Синтезировав их аналоги, он выяснил, что именно N-(фосфнометил)-глицин, позже получивший коммерческое название глифосат, обуславливает наблюдаемую активность. С целью подтверждения этого факта в 1970 г. в фирме “Monsanto” был разработан собственный способ синтеза глифосата и проведено систематическое исследование по выявлению его воздействия на растения [2].
В целом открытие гербицидной активности глифосата по значимости можно сравнить с открытием пенициллина [3], и, как показало время, именно глифосат оказался наиболее востребованным гербицидом в XX веке [4]. При этом N‑(фосфонометил)-глицин (глифосат) представляет собой относительно простую молекулу замещенной аминометилфосфоновой кислоты 1, существующую в твердой фазе в виде двух полиморфных модификаций (бесцветные кристаллы), отличающихся типами кристаллической решетки (моноклинная кристаллическая система, группы симметрии P21 и P21/С) [5], либо в форме конформеров в водном растворе [6]. Уникальная биохимическая активность 1 обусловлена специфическим ингибированием 5-еноилпирувил-шикимат-3-фосфат-синтетазы (EPSP, EC 2.5.1.19) – фермента [7], ответственного за синтез практически всех ароматических соединений, в том числе аминокислот, в водорослях и высших растениях [8, 9], а также бактериях, грибах и апикомплексе простейших [10–12]. При попадании глифосата в клетку происходит эквимолярное взаимодействие глифосата с шикимат-3-фосфатом [13], приводящее к образованию ингибитора, блокирующего действие фермента EPSP [14]. Инактивация фермента достигается за счет нековалентных взаимодействий между остатками аминокислот на сайтах белка EPSP при Lys-23, Asp-50, Gly-101, Thr-102, Arg-105, Arg-131, Asp-331, Glu-359, Arg-362, Arg-404, Lys-429 и комплекса ингибитора [15]. Замещение аминокислот на этих сайтах, например, Gly-101 на Ala у прокариот обуславливает устойчивость к действию глифосата [16]. Блокирование EPSP приводит к прекращению синтеза хоризмата ключевого предшественника в биохимическом синтезе ароматических соединений (L-триптофан, L-фенилаланин, L-тирозин), что заканчивается гибелью организма [17]. В целом, ввиду отсутствия шикиматного пути синтеза органических соединений у млекопитающих, блокирование EPSP может стать важным направлением в разработке противомикробных препаратов, подавляющих развитие бактериальных, паразитарных и грибковых патогенов [11, 18]. Однако наибольшую распространенность глифосат нашел в агропромышленном комплексе, где препараты на его основе (в части действующего вещества) являются самыми распространенными средствами защиты растений – гербицидами широкого спектра действия [19]. Начиная с 1970-х гг., транснациональные компании (“Monsanto”, “BASF”, “Syngenta” и др.) проводили серьезные исследования, направленные на разработку эффективных способов получения глифосата. В патентной литературе представлено множество методов его синтеза, однако лишь некоторые нашли применение в промышленности. Различные подходы к получению глифосата описаны в монографии Джона Е. Франца (Dr. John E. Franz), опубликованной в 1991 г. [20]. Фактически эта работа является единственной, в которой выполнен аналитический обзор различных способов получения глифосата. Целью настоящего обзора является обобщение методов синтеза, используемых в процессах крупнотоннажного производства глифосата, и новых, появившихся в последние десятилетия подходов.
МЕТОДОЛОГИЧЕСКИЕ ПОДХОДЫ К ПОЛУЧЕНИЮ ГЛИФОСАТА
В настоящее время известны различные пути получения N-(фосфонометил)-глицина [20–22], отличающиеся набором исходных веществ для синтеза. Однако общим для всех способов является выделение целевого продукта путем осаждения из воды (растворимость 12 г/л при 298 K) либо из водно-спиртовых растворов [23, 24].
Получение глифосата из глицина (аминоуксусной кислоты)
При использовании глицина (аминоуксусной кислоты, 2) в качестве исходной азотсодержащей молекулы для синтеза глифосата на всех стадиях применяют сравнительно простые превращения, в которых протекание побочных процессов легко прогнозируемо (схема 1 ).
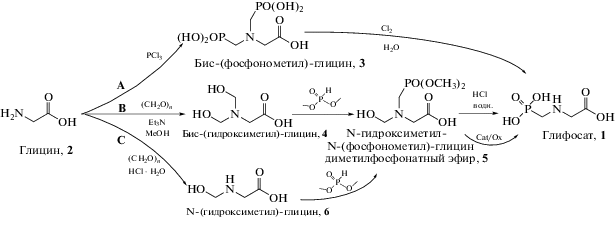
Схема 1 . Способы получения глифосата из глицина.
Одним из первых таких способов стал предложенный в СССР синтез глифосата, протекающий через стадию получения промежуточного N,N-бис-(фосфонометил)-глицина – глифосина 3 (схема 1 , путь А). Особенностью способа являлось расходование двух атомов фосфора на одну молекулу глицина, где промежуточно образующийся глифосин 3 окисляли молекулярным хлором до глифосата [25]. Необходимость использования больших количеств высокотоксичных хлорида фосфора(III) и хлора была серьезным недостатком предложенного решения. Поэтому при организации производства глифосата из глицина следует опираться на альтернативные методы, исторически наиболее распространенные в КНР. В одном из таких способов на первой стадии в результате взаимодействия глицина с 2-х кратным избытком формальдегида (в форме параформа) в неводной среде метанол/триэтиламин образуется бис-(гидроксиметил)-глицин 4 (схема 2 , путь В). Взаимодействие получающегося диола 4 с одним эквивалентом диметилфосфита приводит к диметилфосфонату 5, гидролиз которого концентрированной соляной кислотой завершает синтез глифосата с выходом около 85% [26]. Триэтиламин можно заменить на алкоголяты щелочных металлов (Li, Na, K), выход глифосата 1 при этом превышает 95% [27]. Добавление триэтиламина или щелочи в избыточных количествах существенно снижает Е-фактор синтеза и требует установки скрубберов для поглощения амина или систем очистки от щелочи. Уход от применения оснований позволяет реализовать эквимолярное взаимодействие формальдегида с глицином в кислой среде (схема 1 , путь С), где вместо диола 4 получается N-(гидроксиметил)-глицин 6. В результате фосфонилирования соединения 6 с последующим гидролизом диметилфосфоната 5 глифосат образуется с выходом около 80% [26]. Однако необходимость использования диметилфосфита остается общим недостатком этих способов.
Недавно, в 2017 г., был предложен вариант, в котором вместо диметилфосфита применяли фосфористую кислоту, реагирующую с бис-(гидроксиэтил)-глицином (~1 : 1). Особенностью способа является проведение реакции фосфонилирования при 263 K, а также удаление метилольной группы (превращение 5 в 1) окислением кислородом в присутствии активированного угля или водным пероксидом водорода в присутствии вольфраматов или молибдатов с выходом глифосата до 87% [28]. Описанные выше способы требуют осуществления синтеза при строгом контроле дозировки реагентов и температурного режима. При отклонении этих параметров от оптимальных значений основным продуктом становится глифосин 3 [29], а побочным – N-(метил)-глицин [30].
В целом, несмотря на простоту вышеуказанных способов, их существенными недостатками являются:
• применение большого количества кислот и/или оснований;
• тщательность контроля соотношения реагентов в реакционной смеси.
Поэтому в дальнейшем “глициновый” путь синтеза глифосата вытесняется более эффективным, основанным на использовании иминодиуксусной кислоты (ИДУК, 7, схема 2 ), при котором завершающей стадией является окисление N-(фосфонометил)-иминодиуксусной кислоты (ФИДУК, 8), и этот подход будет рассмотрен ниже.
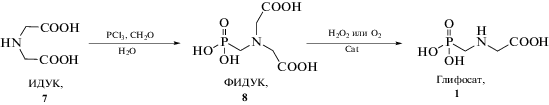
Схема 2 . Синтез глифосата из иминодиуксусной кислоты (ИДУК).
Получение глифосата из N-(фосфонометил)-иминодиуксусной кислоты
Часто качество крупнотоннажного продукта определяется именно завершающей стадией его синтеза. В ходе поисковых исследований многие научные группы пришли к выводу, что использование именно ФИДУК в качестве предшественника глифосата и проведение стадии ее каталитического окисления позволяют обеспечить производство глифосата оптимального качества с наилучшей эффективностью и выходом целевого продукта более 85%. Синтез исходной ФИДУК, применяемой в этих процессах, можно рассматривать как неотъемлемую часть полного цикла технологического процесса получения глифосата. Синтез ФИДУК проводят путем фосфонометилирования ИДУК формальдегидом и треххлористым фосфором [31] возможна замена PCl3 на фосфористую кислоту или фосфит натрия [22]. Образующаяся ФИДУК плохо растворима в воде и практически нерастворима в водно-спиртовых растворах [32, 33], поэтому возможно ее отделение декантированием и/или фильтрованием.
В промышленных процессах получения глифосата через ИДУК (схема 3 ) сырьем служат: монохлоруксусная кислота (схема 3 , способ А), диэтаноламин (синтезируемый из оксида этилена и аммиака) (схема 3 , способ В) или синильная кислота (схема 3 , способ С). Завершающими стадиями всех трех способов являются фосфонометилирование ИДУК и окисление ФИДУК до глифосата (схема 2 ).
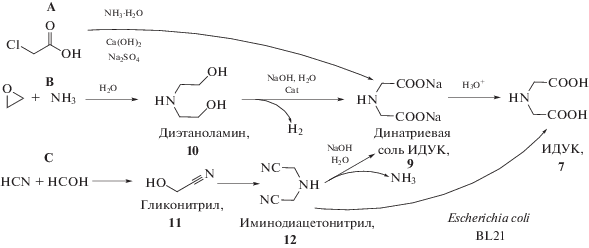
Схема 3 . Способы получения ИДУК в процессах производства глифосата.
Синтез динатриевой соли ИДУК осуществляется следующим образом:
• (А) аммонолизом хлоруксусной кислоты в присутствии гидроксида кальция [2, 34];
• (В) каталитическим дегидрированием ДЭА, 10 на меди и/или никеле Ренея [35–37];
• (С) исходя из синильной кислоты с образованием на промежуточных стадиях гликонитрила 11 [38] и иминодиацетонитрила 12 [39], с последующим щелочным гидролизом до динатриевой соли ИДУК 9 [40] или микробиологическим синтезом до ИДУК 7 [41].
При этом выбор стартового вещества (синильная кислота, хлоруксусная кислота или оксид этилена–аммиак) для синтеза глифосата зачастую определяется доступностью сырья. Однако сопоставление приведенных выше способов получения глифосата позволяет сделать вывод, что наиболее привлекательным из них является так называемый ДЭА-процесс, поскольку нормы образования отходов на единицу продукции в этом случае наименьшие, а использования высокотоксичных хлоруксусной и синильной кислот (а также ее производных) не требуется [42]. Дополнительное преимущество при организации процесса, включающего стадию синтеза ИДУК – отсутствие побочных продуктов бисфосфонилирования, неизбежно образующихся при проведении реакции Кабачника–Филдса с первичными аминами или гидроксиэтиламинами [43]. Таким образом, в настоящее время синтез глифосата из ДЭА (оксида этилена и аммиака) путем окисления промежуточного продукта – ФИДУК – представляет собой оптимальный способ как с точки зрения снижения техногенной нагрузки на окружающую среду, так и минимизации себестоимости продукта. Окислителями чаще всего служат кислород или водные растворы пероксида водорода, сочетающие в себе эффективность, технологичность и экологическую безопасность. Применение каждого из этих реагентов имеет как свои преимущества, так и недостатки, а использование катализаторов окисления ФИДУК до глифосата является общим и ключевым фактором в этих методах.
Каталитическое окисление ФИДУК кислородом
Выбор катализатора окисления ФИДУК в первую очередь зависит от применяемого окислителя. Например, окисление кислородом может протекать как в присутствии растворимых, так и твердых катализаторов.
Окисление кислородом в присутствии растворимых в реакционной смеси катализаторов – сульфатов кобальта, марганца или ванадила – предположительно идет по гомолитическому пути (схема 4 ) с образованием в качестве промежуточного вещества метилен-глифосата 13. Превращаясь по целевому пути, интермедиат 13 окисляется до N-формил глифосата 14, который гидролизуется, давая глифосат и основной побочный продукт – формальдегид (возвращаемый в процесс получения глифосата на стадию синтеза ФИДУК). По нецелевому пути метилен-глифосат 13 превращается в N-(метил)-глифосат 15, который окисляется в N,N-(диметил)-аминофосфоновую кислоту 16 [44].
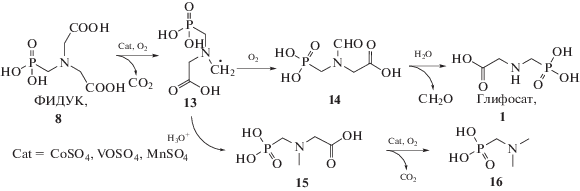
Схема 4 . Предполагаемый механизм образования глифосата при окислении ФИДУК кислородом в условиях гомогенного катализа.
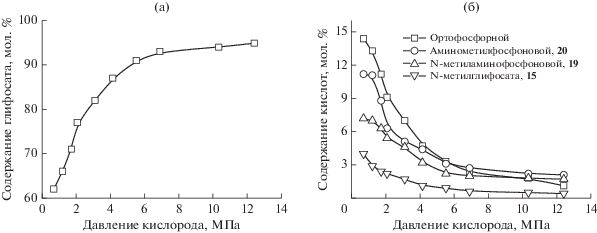
При использовании в качестве катализатора сульфатов кобальта и/или ванадила реакция имеет первые порядки по всем реагентам. При этом, как следует из графических зависимостей, построенных нами на основании данных, представленных в работе [44], с ростом давления кислорода селективность по глифосату увеличивается (рис. 1), а количество побочных продуктов, обнаруживаемых в смеси через 200 мин, уменьшается.
Рис. 1.
Зависимости содержания продуктов окисления ФИДУК в реакционной смеси от давления кислорода: а – глифосат; б – побочные продукты. Условия: [ФИДУК] = 0.10 М, [CoSO4] = 0.0067 М, Т = 358 K, τ = 200 мин.
Авторы работ [45, 46] связывают образование нежелательных продуктов с восстановлением кислорода солями Co(II) до Н2О2. В результате этого параллельно целевому окислению ФИДУК кислородом протекает окисление пероксидом водорода, приводящее к отличным от глифосата продуктам. Для решения этой проблемы в реакционную смесь вводят бромиды, разрушающие Н2О2 [45, 46].
Общим недостатком описанных выше методов, основанных на использовании в качестве катализаторов растворимых в водной среде соединений тяжелых металлов, является неизбежное загрязнение ими целевого продукта – глифосата.
В условиях гетерогенного катализа ФИДУК окисляют кислородом (схема 5 ), используя углеродные материалы либо благородные металлы на углеродных носителях. В этом случае наряду с целевым глифосатом побочно образуются N-замещенные аминофосфоновые кислоты 14, 15, 18 и 19 [47].
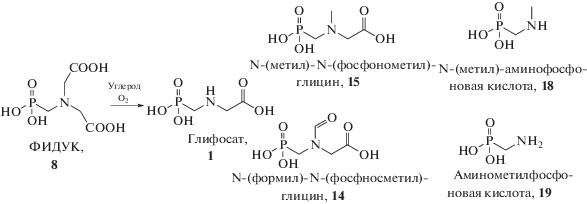
Схема 5 . Возможные продукты окисления ФИДУК кислородом в присутствии активированных углей.
Окисление ФИДУК кислородом в водном растворе до глифосата в присутствии активированного угля, выступающего в роли катализатора, протекает с полной конверсией субстрата 8 только при больших загрузках катализатора в количествах до 25% от массы субстрата [48, 49]. Кроме того, основным недостатком указанных катализаторов является дальнейшее окисление образующегося глифосата. При этом и реакция окисления ФИДУК 8 до глифосата 1, и реакция его дальнейшего окисления имеют первые порядки по субстрату, а значения энергий активации этих реакций равны 13.0 и 10.9 ккал/моль соответственно [50]. Отметим, что уменьшение количества загружаемого катализатора возможно в случае применения катализаторов на основе платины и/или палладия, в том числе в композиции с другими металлами, нанесенными на углеродные материалы [51]. Катализаторы Pd/C более активны по сравнению с Pt/C, а оптимальная загрузка катализатора (Pd/C) составляет 45 моль/м3 при массовом соотношении ФИДУК : H2O = 1 : 1 [52]. Селективность по глифосату достигает максимального значения 95% при давлении кислорода 300 кПа и времени реакции около 3 ч [47]. Недостатком описанных методов синтеза глифосата является необходимость использования избыточного давления для достижения необходимой концентрации О2 в реакционной смеси, поэтому при организации производства применение эмалированных или стеклянных реакторов невозможно. В связи с этим синтез необходимо вести в металлических реакторах из легированных марок стали или титана. В противном случае, учитывая великолепные комплексообразующие свойства фосфоновых кислот, например, за счет образования сильных ионных связей P–O–Fe [53], а также то, что реакция идет в кислой среде [50–52], загрязнение целевого продукта соединениями металлов неизбежно.
Каталитическое окисление ФИДУК пероксидом водорода
Жидкофазное окисление ФИДУК пероксидом водорода более технологично по сравнению с вышеуказанными способами с применением молекулярного кислорода, поскольку реакция протекает в относительно мягких условиях: при атмосферном давлении и температурах не выше 373 K [42], что делает возможным использование эмалированных или стеклянных реакторов. В литературе описаны некаталитические способы окисления ФИДУК до глифосата. Так, в одном из первых патентов Д. Е. Франца (Dr. John E. Franz) [54] в качестве окислителя предлагается применять пероксид водорода, неорганические окислители или пероксосоединения совместно с 1–2 эквивалентами сильной кислоты, например, серной, при температуре от 343 до 373 K. При этом расход H2O2 должен составлять два или более эквивалента H2O2 на один эквивалент ФИДУК. Выходы глифосата составляют около 76%, а основным недостатком данного подхода является необходимость использования больших количеств экологически неблагоприятных окислителей и сильных кислот. Синтез в этом случае протекает через стадии окисления ФИДУК до N-оксида 17 и его превращения в глифосат (схема 6 ), предположительно, путем перегруппировки по типу реакции Полоновского [55], где при внутримолекулярном взаимодействии кислорода оксидной группы с кислотным протоном происходит образование имина 20, изомеризующегося в спирт 21, разложение которого дает глифосат и глиоксалевую кислоту [56].
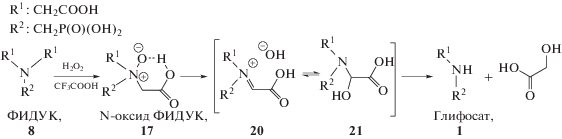
Схема 6 . Окисление ФИДУК пероксидом водорода в присутствии трифторуксусной кислоты.
При использовании в качестве катализатора активированного угля (марки PM–KS, PC, Taiko KW, NORIT, DARKO и др.) его загрузка составляет от 0.16 до 0.25 г на один грамм ФИДУК, а расход H2O2 – от 2 до 5 моль на один моль ФИДУК. При этом выход глифосата не превышает 77% [57]. Большие загрузки катализатора требуют проработки отдельной задачи по утилизации катализатора, что является существенным недостатком, поскольку может значительно снизить экологические показатели процесса.
Описаны способы окисления ФИДУК до глифосата, где в качестве катализаторов применяют соли Co(II) или V(IV) совместно с хинонами и/или гидрохинонами [58], или бипиридилами [59]. К недостатками этих методов относятся большие загрузки катализатора [ФИДУК] : [Cat] = 20–30, невозможность отделения катализатора от целевого продукта, а значит и его загрязнение тяжелыми металлами.
Наиболее простым среди описанных методов получения глифосата путем каталитического жидкофазного окисления ФИДУК пероксидом водорода является 2-х стадийный процесс (схема 7 ), запатентованный “Monsanto” [60].
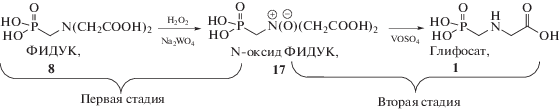
Схема 7 . Синтез глифосата через стадию получения N-оксида ФИДУК.
Первая стадия окисление ФИДУК 47%-ным водным раствором H2O2 осуществляется при 338–348 K в присутствии растворимых в воде соединений молибдена и/или вольфрама, например, вольфрамата натрия. Ключевой результат этого процесса – образование N-оксида 17. Вторую стадию, представляющую собой превращение интермедиата 17 в глифосат, проводят добавлением в полученную реакционную смесь 0.3 мол. % сульфата ванадила (VOSO4). Вероятно, в данном случае имеет место, как и при окислении ФИДУК кислородом, неклассический вариант реакции Полоновского [61], катализируемый не солями железа [62], а комплексом ванадия [45].
Дальнейшие попытки усовершенствования процесса синтеза глифосата путем каталитического жидкофазного окисления ФИДУК водным раствором H2O2 в рамках описанного выше метода отличались способом осуществления стадии превращения N-оксида 17 в глифосат, где вместо сульфата ванадила использовали:
• металлические Fe, Zn, Al, V, Cu или их соединения, растворимые в воде [63];
• метабисульфиты [64];
• тионилгалиды, дитионит аммония или щелочных металлов, диалкилсульфиты, дихлорид серы, диоксид серы или неорганические серосодержащие кислоты [65];
• тиоциановую кислоту или ее соли [66];
• хромовую кислоту [67].
Следующим шагом в улучшении вышеописанного метода получения глифосата стало использование стехиометрических и/или избыточных количеств сульфата железа(II) для превращения N-оксида 17 в глифосат [42], при этом для получения 17 так же, как и в [60, 63–67], предполагается проводить окисление ФИДУК водным раствором H2O2 в присутствии вольфрамата натрия. Сульфат железа(II) в этом случае применяется в форме водного 10%-ного раствора в мольном избытке к субстрату от 1.5 до 3.6, а целевой продукт не очищается от этого реагента. Последнее, несмотря на простоту предложенного способа, относится к недостаткам, поскольку ухудшает качество готового продукта [68, 69].
Хотя эффективность описанных выше подходов к окислению ФИДУК водным раствором H2O2 до глифосата достаточно высока, их общими минусами является использование:
• эквивалентных количеств минеральных (в том числе пероксокислот);
• специальных катализаторов для окисления ФИДУК до N-оксида ФИДУК;
• специальных добавок или нестехиометрических реагентов для превращения N-оксида ФИДУК в глифосат.
Дальнейшее развитие описанных выше способов было предложено в работе [70], где в качестве катализаторов окисления ФИДУК пероксидом водорода применяли бифункциональные пероксополиоксокомплексы вольфрама, а реакцию окисления проводили в двухфазной жидкой системе толуол–вода. Среди всех протестированных катализаторов на основе тетракисоксодипероксовольфрамофосфата(IV) в реакции жидкофазного окисления ФИДУК водным раствором Н2О2 наилучшие результаты были получены при использовании [(Octn)3NMe]3{PO4[WO(O2)2]4} в сочетании с порционной дозировкой Н2О2 [70].
Согласно этому способу окисление ФИДУК до глифосата идет через стадию образования N‑оксида ФИДУК [71, 72], а механизм превращения 17 в глифосат аналогичен рассмотренному выше на схеме 6 . Необходимость порционного дозирования окислителя (табл. 1), наиболее вероятно, связана с протеканием параллельных реакций глубокого окисления фосфоновых кислот пероксидом водорода, например, как при окислении глифосата пероксидом водорода [73], а также с частичным разложением H2O2 во времени при относительно высоких первоначальных концентрациях в реакционной смеси.
Таблица 1.
Выход глифосата в зависимости от количества пероксида водорода*
| № | [H2O2]/[ФИДУК] | Выход глифосата от теоретического, мас. % |
|---|---|---|
| 1 | 1.51 | 84.9 |
| 2 | 1.61 | 88.1 |
| 3 | 1.71 | 90.9 |
| 4 | 1.81 | 92.2 |
| 5 | 2.01 | 85.3 |
| 6 | 0.8 | 45.7 |
| 7 | 1.8 | 46.8 |
* Условия реакции: [H2O]/[ФИДУК] = 44, [ФИДУК]/[(Octn)3NMe]3{PO4[WO(O2)2]4}] = 6000, T = 371 K, τ = 3 ч. 1Дозирование раствора пероксида водорода отдельными порциями во времени.
Получение глифосата из N-(алкил)-N-(фосфонометил)-глицинов
Несмотря на то, что описанные выше способы получения N-(фосфонометил)-глицина путем окисления ФИДУК позволяют получать глифосат с высокими выходами и относительно просты в реализации, они обладают общим недостатком – потерей одного атома углерода в форме оксида углерода(IV) на одну образующуюся молекулу глифосата. Повышение эффективности этого процесса возможно при изменении способа синтеза глифосата на “атом-эффективный”, в котором побочные продукты реакций используются повторно [74] (схема 8 ).
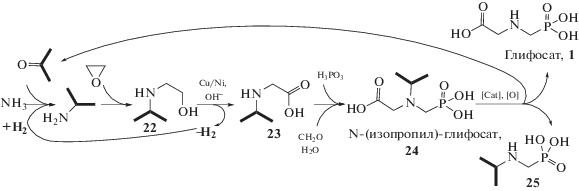
Схема 8 . “Атом-эффективный” процесс получения глифосата.
Ключевой реакцией в этом случае является удаление изопропильной группы N-(изопропил)-N-(фосфонометил)-глицина 24, защищающей аминогруппу от бисфосфонилирования на стадии взаимодействия аминокислоты 23 с формальдегидом и соединением фосфора(III) (фосфористой кислотой). В свою очередь N-(изопропил)-аминоуксуная кислота 23 образуется при дегидрировании аминоспирта 22, который получают по реакции оксиэтилирования изопропиламина, синтезируемого из ацетона, аммиака и водорода. Повышение эффективности процесса в этом методе достигается за счет повторного использования ацетона, образующегося на стадии удаления алкильной защиты [75]. В общем случае в зависимости от типа N-защитной группы можно применять разные методы для ее удаления, например, путем кислотного гидролиза в случае N‑бензильных [76] или N-третбутильных производных глифосата с образованием в качестве побочных веществ толуола или изобутилена [77]. Известно удаление N-бензильных групп гидрированием в присутствии палладия, при этом побочным продуктом является толуол [78]. Предложен способ, в котором деалкилирование выполняют путем щелочного гидролиза в 50%-ном водном растворе NaOH при температуре 573 K в автоклаве [79]. Общий недостаток методов (табл. 2) – невозможность их применения для решения задач “атом-эффективного” синтеза глифосата 1, в котором побочные продукты используются повторно (в рецикле).
Таблица 2.
Выход глифосата в зависимости от метода удаления N-алкильной защиты N-замещенных глифосатов и типа используемого окислителя Ox

Способ удаления N-аклильных групп, позволяющий получать оба основных продукта деблокирования (ацетона и ключевого фософнометилглицина) с высокими выходами, основан на окислении N-замещенных глифосатов кислородом в присутствии гетерогенных катализаторов [75]. При проведении исследований фирма “Monsanto” запатентовала широкий спектр катализаторов, в том числе мезопористые углеродные носители [80], позволяющие решать задачу селективного удаления изопропильной группы. Однако наилучшие результаты наблюдаются в случае использования платиновой черни в каталитических количествах, а оптимальным окислителем является кислород, барботируемый через реакционную смесь при температуре 423 K. Для достижения удовлетворительных величин конверсии субстрата (90–98%) и селективности по глифосату (80–90%) загрузка катализатора (платиновой черни) должна составлять 0.3 г на 1.0 г N‑(изопропил)-N-(фосфонометил)-глицина 23 или 1 г на 10 г N-(метил)-N-(фосфонометил)-глицина. Основная проблема этого метода – снижение селективности, обусловленное протеканием побочной реакции декарбоксиметилирования с образованием аминофосфоновой кислоты 25 или реакции глубокого окисления фосфоновых кислот до неорганических. Это происходит вследствие переокисления катализаторов кислородом при величинах конверсии N-аклилглифосата 50–80% и требует остановки реакции до полного превращения субстрата и организации его рецикла после выделения продукта из реакционной смеси [81].
Перспективным решением проблемы снижения селективности вследствие переокисления глифосата на платиновых катализаторах является метод, предложенный в работах [82, 83], в котором удаление N-защитной группы происходит в условиях окисления N-замещенных глифосатов пероксидом водорода при атмосферном давлении в присутствии наноструктурированных Au/C-катализаторов (при соотношении [24]/[Au] = 100–700). Конверсия субстрата 22 составляет 98%, а селективность по глифосату – 93% [82]. Недостаток этого подхода – особенность методики приготовления катализатора [84], где для воспроизводимости его характеристик требуется достижение в разных партиях количественной и качественной однотипности кислородсодержащих функциональных групп на поверхности углеродного носителя (УН), что представляет сложную задачу [85].
Воспроизводимость свойств синтезируемых наноструктурированных Au/C-катализаторов была достигнута в результате разработки нового метода. В нем для получения наноразмерных частиц Au в качестве предшественников использовали генерируемые in situ комплексы золота(III) с диаминомонокарбоновыми кислотами (лизин, аргинин, гуанидин) [86]. Приготовленные таким способом катализаторы показали активность в реакции каталитического окисления N-(изопропил)-N-(фосфонометил)-глицина 24 как воздухом, так и пероксидом водорода, но следует отметить, что при окислении водным раствором Н2О2 конверсия 24 выше на порядок. Селективность катализатора по глифосату обуславливается именно наличием наночастиц Au на УН, а в их отсутствиe основным продуктом является N-(изопропил)-аминофосфоновая кислота 25 (схема 8 ), образующаяся при декарбоксиметилировании изопропилглифосата 24 (табл. 3).
Таблица 3.
Сравнение активности и селективности наноструктурированных катализаторов в реакции каталитического деалкилирования N-замещенных глифосатов*
| № | Катализатор | Лиганд предшественника | $\bar {d}$; WXRD | Ox | Выход глифосата, % |
|---|---|---|---|---|---|
| 1 | Сибунит | – | – | H2O2 | –*** |
| 2** | 2% Au/Сибунит | Аргинин | 1.59; 0 | воздух | 10.8 |
| 3 | H2O2 | 79.3 | |||
| 4 | Лизин | 1.68; 0.30 | 71.5 | ||
| 5 | Гуанидин | 3.71; 0.32 | 44.5 |
Примечание. Ox – окислитель. $\bar {d}$ – средний размер частиц Au, нм. WXRD – масссовая фракция кристаллитов с размеров 2.5–7 нм, определяемая методом рентгеновской дифракции. * Условия реакции: [24]0 = 0.082 M, [H2O2]0 = 0.11 M, [24]/[Au] = 100, T = 343 K, n = 1000 об/мин, τ = 120 мин. ** Атмосферное давление. *** Реакция протекает по пути декарбоксиметилирования.
Также, основываясь на результатах исследования, описанных в работе [87], можно заключить, что выход глифосата, наблюдаемый в присутствии катализаторов, синтезированных из комплексов Au(III), увеличивается в ряду гуанидин → лизин → → аргинин (табл. 3), что мы связываем с морфологией наноструктурированных частиц, т.е. размерным эффектом.
Описанные выше методы позволяют с разной эффективностью решить задачу деалкилирования N-замещенных глифосатов и, по-видимому, будут востребованы при организации “атом-эффективного” процесса получения глифосата. При этом наиболее перспективным представляется метод, основанный на удалении N-защитных групп пероксидом водорода, поскольку в этом случае реакция протекает в мягких условиях при атмосферном давлении, что не требует использования специального оборудования.
ЗАКЛЮЧЕНИЕ
В настоящее время разработаны способы получения глифосата из различных исходных материалов. Это позволяет выбрать подходящий метод для промышленного внедрения в зависимости от доступного сырья.
Результаты многолетних исследований по усовершенствованию многостадийного синтеза глифосата позволили:
– значительно упростить технологический процесс за счет использования эффективных катализаторов;
– снизить техногенную нагрузку на окружающую среду (исключить токсичные реагенты, уменьшить количество отходов на единицу продукта).
Отметим, что оптимизация некоторых существующих процессов, например, получения глифосата путем окисления ФИДУК пероксидом водорода, возможна с сохранением принципиальных технологических схем, снижением издержек производства и повышением качества целевого глифосата. В целом же при организации отечественного производства глифосата следует ориентироваться, прежде всего, на наличие сырьевой базы и с ее учетом осуществлять выбор наиболее эффективного способа.
Список литературы
Szekacs A., Darvas B. // Herbic.-Prop. Synth. Control Weeds. 2012. P. 247.
Dill G.M., Sammons R.D., Feng P.C.C., Kohn F., Kretzmer K., Mehrsheikh A., Bleeke M., Honegger J.L., Farmer D., Wright D., Haupfear E.A. // Glyphosate Resist. Crop. Weeds. 2010. P. 1.
Powles S.B. // Proc. Natl. Acad. Sci. 2010. V. 107. № 3. P. 955.
Duke S.O., Powles S.B. // Pest Manag. Sci. 2008. V. 64. № 4. P. 319.
Krawczyk H., Bartczak T.J. // Phosphorus. Sulfur. Silicon Relat. Elem. 1993. V. 82. № 1–4. P. 117.
Peixoto M.M., Bauerfeldt G.F., Herbst M.H., Pereira M.S., da Silva C.O. // J. Phys. Chem. A. 2015. V. 119. № 21. P. 5241.
Duke S.O. // Pest Manag. Sci. 2018. V. 74. № 5. P. 1027.
Wang C., Lin X., Li L., Lin S. // PLoS One. 2016. V. 11. № 3. P. e0151633.
Maeda H., Dudareva N. // Annu. Rev. Plant Biol. 2012. V. 63. № 1. P. 73.
Morjan W.E., Pedigo L.P., Lewis L.C. // Environ. Entomol. 2002. V. 31. № 6. P. 1206.
Tanney J.B., Hutchison L.J. // Can. J. Microbiol. 2010. V. 56. № 2. P. 138.
Roberts C.W., Roberts F., Lyons R.E., Kirisits M.J., Mui E.J., Finnerty J., Johnson J.J., Ferguson D.J.P., Coggins J.R., Krell T., Coombs G.H., Milhous W.K., Kyle D.E., Tzipori S., Barnwell J., Dame J.B., Carlton J., McLeod R. // J. Infect. Dis. 2002. V. 185. № s1. P. S25.
Sammons R.D., Gruys K.J., Anderson K.S., Johnson K.A., Sikorski J.A. // Biochem. 1995. V. 34. № 19. P. 6433.
Marzabadi M.R., Gruys K.J., Pansegrau P.D., Walker M.C., Yuen H.K., Sikorski J.A. // Biochem. 1996. V. 35. № 13. P. 4199.
Li J., Peng Q., Han H., Nyporko A., Kulynych T., Yu Q., Powles S. // J. Agric. Food Chem. 2018. V. 66. № 30. P. 7880.
Funke T., Han H., Healy-Fried M.L., Fischer M., Schonbrunn E. // Proc. Natl. Acad. Sci. 2006. V. 103. № 35. P. 13010.
Schonbrunn E., Eschenburg S., Shuttleworth W.A., Schloss J. V., Amrhein N., Evans J.N.S., Kabsch W. // Proc. Natl. Acad. Sci. 2001. V. 98. № 4. P. 1376.
Samac D., Foster-Hartnett D. // Plant Dis. 2012. V. 56. № 2. P. 1104.
Duke S.O. // Pest Manag. Sci. 2018. V. 74. № 5. P. 1025.
Molin W.T. // Weed Technol. 1998. V. 12. № 03. P. 564.
Zhantasov K.T., Shalataev S.S., Kadirbayeva A.A., Zhantasova D.M., Dzhanmuldaeva Z.M., Ismailov B., Petropavlovsky I.A. // Orient. J. Chem. 2017. V. 33. № 3. P. 1381.
Montchamp J.-L. // Phosphorus. Sulfur. Silicon Relat. Elem. 2013. V. 188. № 1–3. P. 66.
Fu W., Guo Q., Wang J. // J. Chem. Eng. Data. 2010. V. 55. № 9. P. 3915.
Jia D., Wang L., Li C., Wang X. // Fluid Phase Equilib. 2012. V. 327. № 5. P. 1.
Pat. SU1112760, 1983.
Zhou J., Li J., An R., Yuan H., Yu F. // J. Agric. Food Chem. 2012. V. 60. № 25. P. 6279.
Pat. US4486359, 1984.
Pat. CN106543222, 2017.
Mao J.-G., Wang Z., Clearfield A. // New J. Chem. 2002. V. 26. № 8. P. 1010.
Pine S.H., Sanchez B.L. // J. Org. Chem. 1971. V. 36. № 6. P. 829.
Pat. CN102952159, 2013.
Chen N., Wang S., Yao C.H., Hui L., Qu Y. // J. Chem. Eng. Data. 2010. V. 55. № 7. P. 2613.
Jia D., Zhang X., Li J., Li C. // Fluid Phase Equilib. 2014. V. 382. P. 54.
Ogata Y., Kawasaki A., Goto T. // J. Org. Chem. 1968. V. 33. № 3. P. 1107.
Andreev D.V., Sergeev E.E., Gribovskii A.G., Makarshin L.L., Prikhod’ko S.A., Adonin N.Y., Pai Z.P., Parmon V.N. // Chem. Eng. J. 2017. V. 330. № 3. P. 899.
Ющенко Д.Ю., Жижина Е.Г., Пай З.П. // Катализ в промышленности. 2018. Т. 6. С. 33.
Андреев Д.В., Сергеев Е.Е., Грибовский А.Г., Макаршин Л.Л., Приходько С.А., Адонин Н.Ю., Пай З.П., Пармон В.Н. // Катализ в промышленности. 2012. № 5. С. 23.
Pollak P., Romeder G., Hagedorn F., Gelbke H.-P. // Ullmann’s Encycl. Ind. Chem. 2000. V. 12. № 4. P. 365.
Polstorff K., Meyer H. // Berichte der Dtsch. Chem. Gesellschaft. 1912. V. 45. № 2. P. 1905.
Duan P.-G., Wang X., Dai L.-Y. // Chem. Eng. Technol. 2007. V. 30. № 2. P. 265.
Liu Z.-Q., Zhou M., Zhang X.-H., Xu J.-M., Xue Y.-P., Zheng Y.-G. // J. Mol. Microbiol. Biotechnol. 2012. V. 22. № 1. P. 35.
ian J., Shi H., Li X., Yin Y., Chen L. // Green Chem. 2012. V. 14. № 7. P. 1990
Черкасов Р.А., Галкин В.И. // Успехи Химии. 1998. Т. 67. № 10. С. 940.
Riley D.P., Fields D.L. // J. Am. Chem. Soc. 1992. V. 114. № 5. P. 1881.
Riley D.P., Fields D.L., Rivers W. // Inorg. Chem. 1991. V. 30. № 22. P. 4191.
Riley D.P., Fields D.L., Rivers W. // J. Am. Chem. Soc. 1991. V. 113. № 9. P. 3371.
Kuil M., E. W. Beers A., Alsters P.L. // Liq. Phase Aerob. Oxid. Catal. Ind. Appl. Acad. Perspect. 2016. V. 91. P. 267.
Pinel C., Landrivon E., Lini H., Gallezot P. // J. Catal. 1999. V. 182. № 2. P. 515.
Besson M., Gallezot P., Perrard A., Pinel C. // Catal. Today. 2005. V. 102–103. P. 160.
Bao Y., Wu J., Hu X.P. // Adv. Mater. Res. 2012. V. 455–456. P. 872.
Pat. US7067693, 2006.
Liu Y.B., Liang E.X., Yi J.M., Wu X., Zhang P.L. // Appl. Mech. Mater. 2014. V. 556–562. P. 213.
Demin A.M., Mekhaev A. V., Esin A.A., Kuznetsov D.K., Zelenovskiy P.S., Shur V.Y., Krasnov V.P. // Appl. Surf. Sci. 2018. V. 440. P. 1196.
Pat. US3954848, 1976.
Franz J.E. // Adv. Pestic. Sci. 1979. P. 139.
Губарев Ю.А., Лебедева Н.С., Андреев В.П., Нижник Ю.П., Вьюгин А.И. // ЖОХ. 2007. Т. 77. № 6. С. 1006.
Pat. US5948938, 1999.
Pat. US5072033, 1999.
Pat. US5061820, 1991.
Pat. US5023369, 1991.
Bur S.K. // Compr. Org. Synth. II. 2014. V. 6. P. 755.
Thavaneswaran S., Scammells P.J. // Bioorg. Med. Chem. Lett. 2006. V. 16. № 11. P. 2868.
Pat. US5043475, 1991.
Pat. EP0439445, 1991.
Pat. US6528680, 2003.
Pat. US6696602, 2004.
Pat. WO2004014925, 2004.
Pat. CN1482130, 2004.
Pat. CN1557820, 2004.
Pat. RU2618629, 2016.
Pai Z.P., Yushchenko D.Y., Khlebnikova T.B., Parmon V.N. // Catal. Commun. 2015. V. 71. P. 102.
Pai Z.P., Chesalov Y.A., Berdnikova P. V, Uslamin E.A., Yushchenko D.Y., Uchenova Y. V, Khlebnikova T.B., Baltakhinov V.P., Kochubey D.I., Bukhtiyarov V.I. // Appl. Catal. A Gen. 2020. V. 604. P. 117786.
Manassero A., Passalia C., Negro A.C., Cassano A.E., Zalazar C.S. // Water Res. 2010. V. 44. № 13. P. 3875.
Morgenstern D.A., Fobian Y.M., Oburn D.S. // Catal. Org. React. 2000. P. 349.
Stern M.K. // Environ. Challenges Chem. Sci. 21st Century. 2003. P. 140.
Pat. US3956370, 1976.
Pat. US3927080, 1975.
Pat. EP0055695, 1982.
Pat. US5068404, 1991.
Мурзин Д.Ю. // Кинетика и катализ. 2020. Т. 61. № 1. С. 85.
Pat. WO9941260A1, 1999.
Pat. RU2641897, 2018.
Pyrjaev P.A., Yushchenko D.Y., Moroz B.L., Pai Z.P., Bukhtiyarov V.I. // Chemistry Select. 2019. V. 4. № 36. P. 10756.
Пыряев П.А., Мороз Б.Л., Зюзин Д.А., Нартаво А.В., Бухтияров В.И. // Кинетика и катализ. 2010. Т. 51. № 9. С. 914.
Bulushev D.A., Yuranov I., Suvorova E.I., Buffat P.A., Kiwi-Minsker L. // J. Catal. 2004. V. 224. № 1. P. 8.
Pat. RU2663905, 2018.
Yushchenko D.Y., Simonov P.A., Khlebnikova T.B., Pai Z.P., Bukhtiyarov V.I. // Catal. Commun. 2019. V. 121. P. 57.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ