

Кинетика и катализ, 2021, T. 62, № 6, стр. 678-691
Двухфакторная прогностическая модель антирадикальной активности гидроксибензойных кислот в средах с физиологическим pH
Н. И. Белая a, *, А. В. Белый a, И. Н. Щербаков b, Е. А. Будникова a
a ГОУ ВПО Донецкий национальный университет
283001 Донецк, ул. Университетская, 24, Украина
b ФГАОУ ВО Южный федеральный университет
344006 Ростов-на-Дону, ул. Большая Садовая, 105/42, Россия
* E-mail: nat.iv.belaya@gmail.com
Поступила в редакцию 21.05.2021
После доработки 21.07.2021
Принята к публикации 22.07.2021
Аннотация
Предложен регрессионно-классификационный алгоритм скрининга природных гидроксибензойных кислот по антирадикальной активности в средах с физиологическим рН на основе комбинации двухфакторных линейных регрессий “дескриптор–активность” и специализированной кинетической схемы реакций. Разработанная модель решает проблему одновременного количественного определения активности фенолкарбоновых кислот и разделения их на группы веществ с высокой, средней и низкой реакционной способностью. Ее высокая прогностическая способность доказана путем исследования констант скоростей реакций контрольной группы веществ (гидроксиацетофенонов) с азот- и кислород-центрированными радикалами с расчетом относительной погрешности аппроксимации, величина которой не превышает 12%.
Графический абстракт

ВВЕДЕНИЕ
Гидроксибензойные кислоты относятся к одной из наиболее многочисленных групп растительных фенольных соединений. Их важнейшими структурными элементами являются O–H-группа, соединенная с ароматическим циклом и ответственная за антиоксидантную активность вещества [1, 2], а также карбоксильная НООС-группа, способная в водных средах активировать фенольную группу за счет образования карбоксилат-ионов, более реакционноспособных, чем соответствующие молекулы [3, 4]. Из-за широкой распространенности этих соединений в пищевой, фармацевтической и парфюмерной продукции на растительной основе возникает необходимость в формировании модели для оценки и прогнозирования их активности. К факторам, способствующим развитию прогностических методологий, относятся как возможность “отсеивания” веществ с нежелательными свойствами на ранних этапах доклинических исследований, так и поиск новых антиоксидантов природного происхождения.
Ведущее место занимают полуэмпирические модели прогнозирования антирадикальной и антиоксидантной активностей веществ на основе качественной взаимосвязи “структура–реакционная способность” [5, 6] и количественной взаимосвязи “структура–реакционная способность” (QSAR, Quantitative Structure–Activity Relationships) [7, 8]. При прогнозировании свойств на качественном уровне проводится решение классификационной задачи (например, разделение на группы активных, малоактивных и неактивных антиоксидантов), тогда как при прогнозировании числовых значений свойств – решение регрессионной задачи. Для того чтобы найти соотношение между реакционной способностью (активностью) соединений и их строением, необходимо структурные особенности молекулы представлять в виде численных характеристик, которые получили название дескрипторов молекулярного строения (или просто дескрипторы) [9]. Тогда алгоритм QSAR дает возможность заменить поиск связей “структура–активность” анализом соотношений “дескрипторы–активность”. Подобный подход был реализован ранее в работе [10] применительно к растительным фенолам группы флавоноидов. Настоящее исследование является ее логическим продолжением, где на более многочисленной группе веществ были решены одновременно и регрессионная, и классификационная задачи прогнозирования.
Цель работы – создать новую регрессионно-классификационную модель прогнозирования антирадикальной активности (АРА) природных гидроксибензойных кислот на основе кинетической схемы их реакций с N- и O-центрированными радикалами в средах с физиологическим рН и системы двухфакторных регрессий “дескриптор–активность”.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
В качестве объектов исследования использовали соединения класса растительных фенолкарбоновых кислот (НООСArOH) (I–XX) и гидроксиацетофенонов (AcArOH) (XXI–XXVII) следующего строения:
 |
I – 2-гидроксибензойная (салициловая) кислота | R1,2 = OH, R3,4,5,6 = H; | |
| II – 3-метил-2-гидроксибензойная кислота | R1,2 = OH, R3 = СH3, R6,5,4 = H; | ||
| III – метиловый эфир 3-метил-2-гидроксибензойной кислоты | R2 = OH, R3 = СH3, R1 = ОСH3, R5,6,4 = H; | ||
| IV – 3-гидроксибензойная кислота | R3,1 = OH, R6,5,2,4 = H; | ||
| V – 4-гидроксибензойная кислота | R4,1 = OH, R6,5,2,3 = H; | ||
| VI – этиловый эфир 4-гидроксибензойной кислоты | R4 = OH, R6,5,2,3 = H, R1 = OС2Н5; | ||
| VII – 2,3-дигидроксибензойная (пирокатеховая) кислота | R2,3,1 = OH, R4,5,6 = H; | ||
| VIII – 2,4-дигидроксибензойная (β-резорциловая) кислота | R2,4,1 = OH, R3,5,6 = H; | ||
| IX – метиловый эфир 2,4-дигидроксибензойной кислоты | R2,4 = OH, R1 = ОCH3, R3,5,6 = H; | ||
| X – 2,5-дигидроксибензойная (гентизиновая) кислота | R2,5,1 = OH, R3,4,6 = H; | ||
| XI – 2,6-дигидроксибензойная (γ‑резорциловая) кислота | R2,6,1 = OH, R3,4,5 = H; | ||
| XII – 3,4-дигидроксибензойная (протокатеховая) кислота | R3,4,1 = OH, R2,5,6 = H; | ||
| XIII – 3,5-дигидроксибензойная (α-резорциловая) кислота | R3,5 = OH, R1,2,4,6 = H; | ||
| XIV – 3-метокси-4-гидроксибензойная (ванилиновая) кислота | R4,1 = OH, R3 = ОСН3, R2,5,6 = H; | ||
| XV – 2,3,4-тригидроксибензойная (3-пирогаллолкарбоновая) кислота | R2,3,4,1 = OH, R5,6 = H; | ||
| XVI – 3,4,5-тригидроксибензойная (галловая) кислота | R3,4,5,1 = OH, R2,6 = H; | ||
| XVII – 3,5-диметокси-4-гидроксибензойная (сиреневая) кислота | R3,5 = OСH3, R4,1 = ОH, R2,6 = H; | ||
| XVIII – метиловый эфир 3,4,5-тригидроксибензойной кислоты | R3,4,5 = OH, R2,6 = H, R1 = ОCH3; | ||
| XIX – этиловый эфир 3,4,5-тригидроксибензойной кислоты | R3,4,5 = OH, R2,6 = H, R1 = ОС2H5; | ||
| XX – 2,4,6-тригидроксибензойная (флороглюцинкарбоновая) кислота | R2,4,6,1 = OH, R5,3 = H; | ||
| XXI – 2-гидроксиацетофенон | R1 = СН3, R2 = OH, R3,4,5,6 = H; | ||
| XXII – 3-гидроксиацетофенон | R1 = СН3, R3 = OH, R2,4,5,6 = H; | ||
| XXIII – 4-гидроксиацетофенон | R1 = СН3, R4 = OH, R2,3,5,6 = H; | ||
| XXIV – 2,4-дигидроксиацетофенон | R1 = СН3, R2,4 = OH, R3,5,6 = H; | ||
| XXV – 2,5-дигидроксиацетофенон | R1 = СН3, R2,5 = OH, R3,4,6 = H; | ||
| XXVI – 3,4-дигидроксиацетофенон | R1 = СН3, R3,4 = OH, R2,5,6 = H; | ||
| XXVII – 3-метокси-4-гидроксиацетофенон | R1 = СН3, R4 = OH, R3 = OСН3, R2,5,6 = H. | ||
Гидроксибензойные кислоты (I–XX), гидроксиацетофеноны (XXI–XXVII) и антиоксидант сравнения – тролокс (Тх) (6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновая кислота) (производства “Fluka”, Швейцария; “Merck”, Германия; “Panreac”, Испания) использовали без предварительной очистки.
На этапе эмпирического отбора антиоксидантов (АО) антирадикальную активность соединений (I–XXVII) изучали в реакции с N-центрированным радикалом 2,2'-дифенил-1-пикрилгидразилом (DPPH•) (“Merck”, Германия). Раствор радикала в диметилсульфоксиде (“Merck”, Германия) имеет интенсивный фиолетовый цвет с максимумом поглощения при длине волны 520 нм. В условиях его хранения в темноте интенсивность спектра в максимуме поглощения остается неизменной на протяжении 72 ч. Генератор свободных О-центрированных радикалов – гидрофильное азосоединение 2,2'-азобис(2-амидинопропан) дигидрохлорид (AAPH) (“Merck”, Германия), в качестве активатора свечения использовали родамин 6Ж (Rd6G) (“Merck”, Германия). Для задания рН водных растворов применяли фосфатную (с рН 7.35) буферную систему, приготовленную по методике [11], точное значение рН которой контролировали с помощью иономера И-160МИ (Россия). Диметилсульфоксид (ДМСО) очищали по известной методике [12].
Кинетические исследования
Для изучения кинетики взаимодействия I–XXVII с DPPH• при 293 ± 2 K были использованы ДМСО, буфер с физиологическим рН 7.35 и их смеси в разных соотношениях. Применение ДМСО было необходимо, поскольку радикал DPPH• нерастворим в воде, а также для снижения скорости исследуемой реакции за счет подавления диссоциации фенольных соединений. Реакцию НООСArOH и AcArOH с DPPH• проводили в растворителях, из которых предварительно удаляли кислород путем барботирования аргона в течение 15–20 мин, что позволило исключить возможные реакции фенолов и продуктов их превращения с участием кислорода. Кинетику реакции исследовали при λmax = 520 нм на спектрофотометре Specord S300 UV-VIS (“Carl Zeiss Jena”, Германия) при температуре Т = 293 ± 2 К в интервале начальных концентраций реагирующих веществ 10‒3–10‒5 моль/л. Реагенты смешивали, затем измеряли оптическую плотность смеси и с помощью молярного коэффициента светопоглощения (εДМСО = 1.6 × 103 л моль‒1 мм‒1) по закону Бугера–Ламберта–Бера рассчитывали концентрацию радикала DPPH•. Соотношение фенолокислоты и радикала составляло 1 : 1 при определении как общего порядка (n), так и константы скорости (k) реакции. Для этого кинетические данные обрабатывали нелинейным методом обобщенного приведенного градиента (ОПГ) [13], реализованного в Solver MS Excel. В качестве критерия выбора порядка реакции использовали параметр S, отражающий относительный разброс вычисленного ряда констант:
Порядок реакции определяли тем значением, при котором относительный разброс S константы скорости реакции, вычисленной из исходных данных (концентрация и время) по этому порядку, имеет наименьшее значение. Кинетические кривые, смоделированные на основе констант реакции второго порядка (n = 2), рассчитанных в Solver MS Exсel, хорошо согласуются с экспериментальными данными, отклонения наблюдаются при степени превращения радикала более 80%, что может быть вызвано влиянием вторичных продуктов превращения гидроксибензойных кислот – димерных фенолов [14]. В связи с этим расчет порядков реакции и констант скорости проводили до момента времени, соответствующего 60–70% расходования DPPH•. Частный порядок по DPPH•, равный единице (${{n}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}}}}$ = 1), установлен при 10–20-кратном недостатке радикала.
Реакцию НООСArOH и AcArOH с 2-амидинопропан-2-пероксильными радикалами (APOO•), генерируемыми при термическом распаде AAPH в водной среде, изучали методом хемилюминесценции (ХЛ) [15]. Фенольные соединения растворяли в диметилсульфоксиде, а затем аликвоту этого раствора (0.1 мл) вводили в буфер (4.9 мл), содержащий AAPH и Rd6G. Окисляющуюся смесь помещали в термостатированную кювету хемилюминометра и барботировали воздухом для ее насыщения кислородом и перемешивания. Температура проведения эксперимента Т = 323 ± ± 2 K соответствовала температуре, при которой используемый инициатор распадается на радикалы APOO• с постоянной скоростью. Значения константы скорости реакции распада AAPH на радикалы (kd= 7.1 × 10‒6 c‒1) и выхода радикала из клетки (e = 0.48) при pH 7.4 и T = 323 K взяты из работ [16, 17]. Скорость инициирования (генерирования) пероксирадикалов рассчитывали по формуле ${{W}_{{\text{i}}}} = 2e{{k}_{{\text{d}}}}\left[ {{\text{AAPH}}} \right]$ [16]. Измерения проводили с помощью хемилюминесцентной установки (фотоумножитель ФЭУ-38), принципиальная схема которой описана в работе [15, 18], с цифровой обработкой сигнала посредством АЦП “LCARD” (Россия). Активатор Rd6G усиливает интенсивность хемилюминесценции (ХЛ), но не влияет на кинетику ее снижения.
Квантово-химический расчет
Квантово-химические расчеты выполняли с использованием пакета Gaussian 09 [19] в рамках теории функционала плотности (DFT) с гибридным функционалом B3LYP, выбор которого обусловлен опубликованными данными об успешном его применении для соединений фенольного типа [14, 20]. Поиск стабильных конформеров предварительно осуществляли полуэмпирическим методом PM6. Структуры с минимальной энергией использовали в качестве начального приближения для расчетов на уровне B3LYP/6-311++G(d,p) – для систем с закрытыми оболочками (синглетное основное состояние) и UB3LYP/6-311++G(d,p) – для систем с открытыми оболочками (дублетное основное состояние). Геометрия всех структур реагентов и продуктов реакций была оптимизирована по всем независимым переменным и без ограничений по симметрии для водной среды.
Влияние растворителя учитывали в рамках модели поляризуемого континуума РСМ [21]. Для построения полости растворенного вещества задавали радиусы атомных сфер из модели силового поля UFF [22]. Характер всех стационарных точек определяли расчетом матрицы Гессе. Частоты нормальных колебаний (в гармоническом приближении) и тепловые поправки к свободной энергии (с использованием немасштабированных частот) были рассчитаны теми же методами. Все полученные результаты относятся к нормальным условиям (T = 298 К, Р = 1 атм) в водной среде.
Значения потенциалов ионизации карбоксилат-($P{{I}_{{^{ - }{\text{OOCArOH}}}}}$) и фенолят-ионов ($P{{I}_{{^{ - }{\text{OOCArO}}{{{\text{H}}}^{ - }}}}}$) кислот рассчитывали по формулам:
Расчет методом QSPR
Путем моделирования методом QSPR [23] в программном пакете Marvin 18.14 [24] оценивали рКа соединений по первой и второй ступеням диссоциации (для эфиров кислот и гидроксиацетофенонов – по первой ступени диссоциации), а также вычисляли распределение их молекулярной и ионных форм в зависимости от рН водной среды при 298 К. Модуль для расчета Protonation интегрирован в MarvinSketch и способен предсказывать рКа для органических молекул, смоделированных в MarvinSketch.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В смешанном растворителе ДМСО–буфер в присутствии всех исследуемых НООСArOH (I–XX) происходит расходование DPPH•, которое можно зафиксировать в приемлемом временном интервале при низком содержании буфера, до 20–25 об. %. Видно (рис. 1а), что при добавлении даже небольшого количества буфера в ДМСО, когда полярность среды изменяется незначительно, реакционная способность НООСArOH увеличивается.
Рис. 1.
(а) – Кинетические кривые расходования DРРН• в реакции с 3-пирогаллолкарбоновой кислотой (С = 5 × × 10‒5 моль/л) при Т = 298 ± 2 К в смеси ДМСО с добавками фосфатного буфера с рН 7.35 (об. %): 1 – 5; 2 – 10; 3 – 15; 4 – 20. (б) – Расчет константы скорости реакции в чистом растворителе $\left( {{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}} \right)$ из зависимости (1): ln kДМСО–буфер = (6.56 ± 0.04) + (11.28 ± 0.33)(1 – ωДМСО), где ωДМСО – доля ДМСО в смеси ДМСО–буфер.
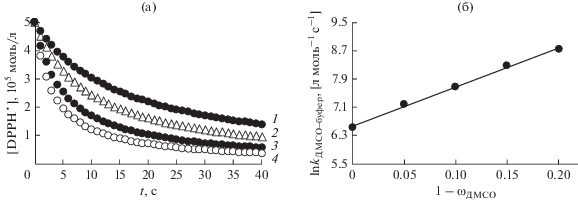
Поскольку гидразильный радикал нерастворим в воде, а скорость исследуемой реакции при большом содержании буфера настолько велика, что не позволяет корректно оценить начальные участки кинетических кривых, то величину антирадикальной активности кислот в чистом буфере (${{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}$) определяли из линейной зависимости (1) ln kДМСО–буфер от объемной доли буфера, вводимого в реакционную смесь:
(1)
$\begin{gathered} \ln {{k}_{{{\text{ДМСО}}--{\text{буфер}}}}} = \\ = \ln {{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{ДМСО}}} \right)}}}{{\omega }_{{{\text{ДМСО}}}}} + \ln {{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH}}\,{\text{7}}} \right)}}}~\left( {1--{{\omega }_{{{\text{ДМСО}}}}}} \right), \\ \end{gathered} $Из углового параметра линейных регрессий (рис. 1б), полученных в координатах уравнения (1), рассчитывали константы скорости реакции ${{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}$ в буфере с физиологическим рН. Отношение константы исследуемого вещества к константе для тролокса $\left( {\frac{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}\left( {{\text{pH\;}}\,7} \right)}}}}}{{k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}\,7} \right)}}^{{{\text{Tx}}}}}}} \right)$ показывает, во сколько раз АРА изученного соединения выше активности реперного АО [25].
Вычисленные значения ${{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}$ (табл. 1) указывают на то, что в нейтральной среде наиболее активны с гидразильным радикалом соединения групп ди- и тригидроксибензойных кислот (VII, X, XV) с тролоксовым эквивалентом ${{Т}_{{{\text{Е}}({\text{DPP}}{{{\text{H}}}^{ \bullet }})}}}~$больше 10. Низкие константы характерны для гидроксибензойных кислот с одной гидроксигруппой или О–Н-группами в мета-положении, а также для эфиров кислот (III, V, VI, IX, XIII, XVIII, XIX).
Таблица 1.
Экспериментальные константы скоростей (k) реакции соединений I–XXVII с DPPH• и АРОО•, а также значения потенциалов ионизации их моно- и биионных форм ($P{{I}_{{^{ - }{\text{OOCArOH\;}}}}}$, $P{{I}_{{^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}$), рассчитанные методом DFT B3LYP/6-311++G(d,p), РСМ
| Соединение | ${{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{ДМСО}}} \right)}}}$, л моль‒1 с‒1 |
${{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}$, л моль‒1 с‒1 |
${{\left( {\frac{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}} \right)}^{{\text{a}}}}$ | ${{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}$, л моль‒1 с‒1 |
${{\left( {\frac{{{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}} \right)}^{{\text{b}}}}$ | рКа1 (рКа2) |
$P{{I}_{{{}_{{}}^{ - }{\text{OOCArOH\;}}}}}$, эВ | $P{{I}_{{{}_{{}}^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}$, эВ |
|---|---|---|---|---|---|---|---|---|
| I | <0.01 | (2.50 ± 0.08) × 104 | 4.17 | (9.5 ± 0.4) × 105 | 9.5 | 2.79 (13.23) | 5.33 | 4.22 |
| II | (1.11 ± 0.05) × 102 | (2.50 ± 0.08) × 104 | 4.17 | (1.39 ± 0.04) × 106 | 14.0 | 2.8 (13.89) | 5.21 | 4.11 |
| III | (1.21 ± 0.05) × 102 | 60.0 ± 2.5 | 0.01 | (2.85 ± 0.15) × 102 | 0.0029 | 10.1 | 6.23с | 4.65d |
| IV | <0.01 | (9.5 ± 0.4) × 102 | 0.16 | (1.70 ± 0.06) × 104 | 0.17 | 3.85 (9.55) | 5.92 | 4.32 |
| V | <0.01 | (5.00 ± 0.21) × 102 | 0.083 | (4.50 ± 0.21) × 103 | 0.045 | 4.38 (9.67) | 5.83 | 4.38 |
| VI | 1.61 ± 0.05 | 12.0 ± 0.6 | 0.0020 | 25 ± 0.9 | 0.00025 | 8.5 | 6.54с | 4.88d |
| VII | (3.00 ± 0.09) × 102 | (9.1 ± 0.4) × 104 | 15.17 | (1.60 ± 0.05) × 106 | 16.0 | 2.56 (9.7) | 5.05 | 3.97 |
| VIII | 58 ± 2 | (3.9 ± 0.18) × 103 | 0.65 | (1.28 ± 0.05) × 105 | 1.28 | 3.1 (9.81) | 5.33 | 4.44 |
| IX | 12 ± 0.5 | 30.0 ± 1.6 | 0.005 | 25 ± 0.9 | 0.00025 | 8.58 | 6.35с | 5.00d |
| X | (5.8 ± 0.16) × 102 | (1.31 ± 0.04) × 105 | 21.88 | (3.51 ± 0.18) × 106 | 35.1 | 2.53 (10.02) | 5.01 | 3.85 |
| XI | (1.51 ± 0.05) × 102 | (1.50 ± 0.04) × 103 | 0.25 | (4.09 ± 0.18) × 104 | 0.41 | 1.65 (13.26) | 5.56 | 4.16 |
| XII | (5.11 ± 0.15) × 102 | (1.90 ± 0.08) × 103 | 0.32 | (8.00 ± 0.4) × 104 | 0.8 | 4.16 (9.4) | 5.64 | 4.26 |
| XIII | (1.18 ± 0.05) × 102 | (5.05 ± 0.21) × 102 | 0.084 | (1.01 ± 0.04) × 104 | 0.10 | 3.61 (9.29) | 5.94 | 4.35 |
| XIV | < 0.01 | (2.43 ± 0.16) × 103 | 0.4 | (7.00 ± 0.28) × 104 | 0.7 | 4.16 (10.14) | 5.57 | 4.16 |
| XV | (2.5 ± 0.09) × 102 | (7.97 ± 0.29) × 104 | 13.28 | (3.00 ± 0.18) × 106 | 30.0 | 2.88 (9.07) | 5.09 | 3.98 |
| XVI | (8.5 ± 0.4) × 102 | (3.40 ± 0.08) × 103 | 0.57 | (1.59 ± 0.07) × 105 | 1.6 | 3.94 (9.04) | 5.64 | 4.18 |
| XVII | (7.5 ± 0.3) × 102 | (8.1 ± 0.3) × 103 | 1.35 | (1.20 ± 0.04) × 105 | 1.2 | 3.93 (9.55) | 5.59 | 4.12 |
| XVIII | (2.31 ± 0.09) × 102 | 80 ± 3 | 0.013 | (1.20 ± 0.04) × 103 | 0.012 | 8.11 | 6.20с | 4.63d |
| XIX | (2.62 ± 0.09) × 102 | 150 ± 8 | 0.025 | (2.00 ± 0.08) × 103 | 0.02 | 8.11 | 6.18с | 4.62d |
| XX | (1.17 ± 0.05) × 102 | (2.80 ± 0.08) × 103 | 0.47 | (1.10 ± 0.04) × 104 | 0.11 | 1.96 (9.94) | 5.40 | 4.53 |
| XXI | 11 ± 0.4 | 92 ± 4 | 0.015 | (1.39 ± 0.04) × 103 | 0.014 | 9.15 | 6.40с | 4.39d |
| XXII | 7.1 ± 0.3 | 67 ± 2 | 0.011 | (2.00 ± 0.08) × 103 | 0.020 | 8.91 | 6.47с | 4.31d |
| XXIII | 6.5 ± 0.3 | 61 ± 2 | 0.010 | (1.15 ± 0.04) × 103 | 0.012 | 7.79 | 6.53с | 4.34d |
| XXIV | 9.6 ± 0.4 | 101 ± 4 | 0.017 | (1.80 ± 0.06) × 103 | 0.018 | 7.88 | 6.39с | 4.36d |
| XXV | 58.5 ± 2.1 | (1.16 ± 0.04) × 103 | 0.193 | (4.63 ± 0.21) × 104 | 0.463 | 9.22 | 5.89с | 4.16d |
| XXVI | 15.5 ± 0.5 | 301 ± 16 | 0.050 | (9.9 ± 0.5) × 103 | 0.099 | 7.91 | 6.23с | 4.19d |
| XXVII | 23.1 ± 1.6 | 270 ± 16 | 0.045 | (3.20 ± 0.08) × 103 | 0.032 | 8.28 | 6.12с | 4.44d |
a Константа скорости реакции тролокса с DPPH• при рН 7.35, Т = 298 К равна $k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{рН}}\,\,7} \right)}}^{{{\text{Tx}}}}~$= (6.00 ± 0.25) × 103 л моль‒1 с‒1. bКонстанта скорости реакции тролокса с АPOO• при рН 7.35, Т = 323 К равна $k_{{{\text{АPO}}{{{\text{O}}}^{ \bullet }}\left( {{\text{рН}}\,\,7} \right)}}^{{{\text{Tx}}}}$ = (1.00 ± 0.04) × 105 л моль‒1 с‒1. cПотенциал ионизации молекулярной формы. dПотенциал ионизации моноионной формы.
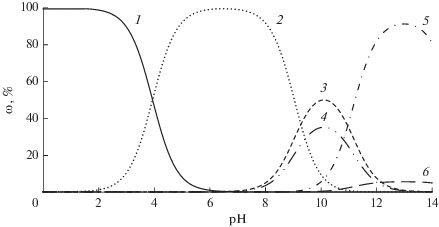
Примечательно, что при переходе от ДМСО к буферному раствору величины констант в среднем возрастают на 1–2 порядка, что обусловлено усилением диссоциации НООСArOH в водной среде. Из распределения молекулярной и ионных форм фенолкарбоновых кислот и их эфиров (рис. 2), рассчитанных методом QSPR в программе Marvin 18.14 на основе величин рКа (табл. 1), видно, что в буферном растворе с рН 7.35 все фенолкарбоновые кислоты присутствуют в виде активных карбоксилат- и фенолят-ионов (эфиры находятся только в молекулярной и моноионной формах), способных реагировать со свободными радикалами [25]:
(I)
$\begin{gathered} {\text{НООСArOH}} + {{{\text{H}}}_{2}}{\text{O}}~\,\, \rightleftarrows \,{{\,}^{ - }}{\text{ООСArOH}} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }}, \\ ^{ - }{\text{ООСArOH}} + {{{\text{H}}}_{2}}{\text{O}}\,\,~ \rightleftarrows \,\,{{~}^{ - }}{\text{ООСAr}}{{{\text{O}}}^{ - }} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }}, \\ ^{ - }{\text{ООСAr}}{{{\text{O}}}^{ - }}\left( {^{ - }{\text{ООСArOH}}} \right) + \\ + \,\,{\text{DPP}}{{{\text{H}}}^{ \bullet }}~\xrightarrow{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{\centerdot }}\left( {{\text{pH\;}}7} \right)~~~~}}}}}\,{{\,}^{ - }}{\text{ООСAr}}{{{\text{O}}}^{\centerdot }} + {\text{DPP}}{{{\text{H}}}^{--}}. \\ \end{gathered} $Рис. 2.
Распределение доли ионных форм (ω) галловой кислоты в зависимости от рН среды: 1 – НООСArOH; 2 – ‒ООСArOH; 3 – ‒ООСAr-4-O‒; 4 – ‒ООСAr-3-O‒; 5– ‒ООСAr-3,5-O‒; 6 – ‒ООСAr-3,4-O‒. Расчеты проведены методом QSPR в программе Marvin 18.14, Т = 298 К, растворитель – вода.
Антирадикальная активность фенолокислот, установленная в реакции со стабильным N-центрированным радикалом, была подтверждена при взаимодействии с лабильными О-центрированными радикалами, имитирующими пероксильные радикалы органических субстратов.
Согласно классическим представлениям метода хемилюминесценции [15], в реакции рекомбинации пероксирадикалов (АРOO•), генерируемых при распаде азосоединения (в настоящей работе гидрофильный ААРН), может возникать ХЛ, которая в случае необходимости усиливается активатором свечения Rd6G:
(II)
$\begin{gathered} {\text{ААРН}}\xrightarrow{{{{k}_{{\text{d}}}}}}{\text{\;\;}}2{\text{А}}{{{\text{Р}}}^{\centerdot }}{\text{\;}}; \\ {\text{А}}{{{\text{Р}}}^{ \bullet }} + {{{\text{O}}}_{2}}\xrightarrow{{{{k}_{i}}}}{\text{\;АРO}}{{{\text{O}}}^{ \bullet }}{\text{\;}}; \\ {\text{АРO}}{{{\text{O}}}^{ \bullet }} + {\text{АРO}}{{{\text{O}}}^{ \bullet }}\xrightarrow{{{{k}_{6}}}}{\text{М}}*; \\ ~{\text{М*}} + {\text{Rd}}6{\text{G}} \to {\text{М}} + {\text{Rd}}6{\text{G*}}; \\ {\text{Rd}}6{\text{G*}} \to {\text{Rd}}6{\text{G}} + h\nu , \\ \end{gathered} $Если считать, что ХЛ возбуждается лишь в реакции рекомбинации ${\text{АРO}}{{{\text{O}}}^{\centerdot }}$, то ее интенсивность (I0) должна быть пропорциональна квадрату концентрации перекисных радикалов: I0 ~ [APOO•]2. При добавлении в систему НООСArOH как акцепторов пероксирадикалов наблюдается уменьшение интенсивности ХЛ-свечения (I) по реакции:
(III)
$\begin{gathered} ^{ - }{\text{ООСAr}}{{{\text{O}}}^{ - }}\left( {^{ - }{\text{ООСArOH}}} \right) + \\ + \,\,{\text{APO}}{{{\text{O}}}^{ \bullet }}~\xrightarrow{{~~~~{{k}_{7}}~~}}\,{{\,}^{ - }}{\text{ООСAr}}{{{\text{O}}}^{\centerdot }} + {\text{APO}}{{{\text{O}}}^{--}}. \\ \end{gathered} $При этом ХЛ в присутствии АО не ослабляется до нуля. Это, вероятно, обусловлено возбуждением ХЛ в каких-либо нерадикальных (молекулярных) реакциях (Iмол), на которые перехватчик радикалов НООСArOH не влияет. В отдельной серии экспериментов было установлено, что появление молекулярной ХЛ связано с воздействием активатора свечения. Относительную интенсивность Iмол определяли, когда в реакционной смеси присутствовал только растворитель и активатор свечения, при этом ее величина не превышала 0.18.
Константы скорости реакции (III) ${{k}_{7}}$ находили в фосфатном буфере при рН 7.35 методом, предложенным в работе [15], при условии низких скоростей инициирования (${{W}_{{\text{i}}}}$) и достаточно высоких концентрациях антиоксиданта. Метод расчета констант ${{k}_{7}}$ позволяет исключить влияние на кинетику реакции продуктов превращения НООСArOH, а также не требует знания величины константы скорости ${{k}_{6}}$, определение которой возможно только после детального изучения ХЛ реакций самого инициатора и образующихся при его распаде радикалов.
Расчет ${{k}_{7}}$ проводили в начальный момент времени, когда концентрация $^{ - }{\text{ООСAr}}{{{\text{O}}}^{\centerdot }}$ крайне мала и реакциями (VI), (V) можно пренебречь:
(IV)
${\text{АРO}}{{{\text{O}}}^{ \bullet }}{{ + }^{ - }}{\text{ООСAr}}{{{\text{O}}}^{\centerdot }}\xrightarrow{{~~{{k}_{8}}}}\,\,~{\text{продукты}};$(V)
$^{ - }{\text{ООСAr}}{{{\text{O}}}^{\centerdot }} + \,{{\,}^{ - }}{\text{ООСAr}}{{{\text{O}}}^{\centerdot }}\xrightarrow{{~{{k}_{9}}~}}\,\,~{\text{продукты}}.$С учетом реакций (II), (III), (IV) изменение концентрации перекисных радикалов описывается дифференциальным уравнением:
(2)
$\begin{gathered} \frac{{{\text{d}}\left[ {{\text{APO}}{{{\text{O}}}^{ \bullet }}} \right]}}{{{\text{d}}t}} = {{W}_{{\text{i}}}} - {{k}_{6}}{{\left[ {{\text{APO}}{{{\text{O}}}^{ \bullet }}} \right]}^{2}} - \\ - \,\,{{k}_{7}}\left[ {{\text{APO}}{{{\text{O}}}^{ \bullet }}} \right]{{\left[ {{\text{НООСArOH}}} \right]}_{0}} - ~ \\ - \,\,{{k}_{8}}\left[ {{\text{APO}}{{{\text{O}}}^{ \bullet }}} \right]\left[ {^{ - }{\text{ООСAr}}{{{\text{O}}}^{\centerdot }}} \right]. \\ \end{gathered} $В начальный момент, когда $\left[ {^{ - }{\text{ООСAr}}{{{\text{O}}}^{\centerdot }}} \right] \to 0$, последним членом в уравнении (2) можно пренебречь. Тогда его решение при условии ${{k}_{7}}{{\left[ {{\text{НООСArOH}}} \right]}_{0}} \gg \sqrt {{{k}_{6}}{{W}_{{\text{i}}}}} $ и после преобразований будет иметь вид [15]:
(3)
${\text{ln}}\sqrt {\frac{{{{I}_{0}}}}{I}} = {{k}_{7}}{{\left[ {{\text{НООСArOH}}} \right]}_{0}}t~{\kern 1pt} ,$Начальный участок ХЛ-кривой (рис. 3а) спрямляли (рис. 3б) в полулогарифмических координатах кинетического уравнения (3) первого порядка. С использованием линейного регрессионного анализа, проведенного в системе Statistica Demo 6.0 [26], из величины k' рассчитывали значения ${{k}_{7}}$, а затем по полученным в координатах уравнения (1) зависимостям вычисляли константы скорости реакции ${{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}$ в буфере с физиологическим рН (рис. 3в).
Рис. 3.
(а) – Кинетика относительной интенсивности ХЛ при термолизе ААРН в присутствии галловой кислоты (C = = 1 × 10‒6 моль/л) в фосфатно-цитратном буфере с рН 7.35 с добавками ДМСО (об. %): 1 – 75; 2 – 70; 3 – 65; 4 – 60. (б) – Полулогарифмические анаморфозы кинетических кривых ХЛ. (в) – Расчет константы $\left( {{{k}_{{{\text{APO}}{{{\text{O}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}} \right)$ в чистом растворителе из зависимости (1): ln kДМСО–буфер = (7.45 ± 0.12) + (11.98 ± 0.4)(1 – ωДМСО). ${{W}_{{\text{i}}}}$ = 5 × 10‒9 моль л‒1 c‒1, [${\text{Rd}}6{\text{G}}$] = 3 × 10‒3 моль/л. Т = 323 К. ${{W}_{{\text{i}}}}$ – скорость инициирования пероксирадикалов.

В отличие от реакции с DPPH•, АРА относительно тролокса ${{Т}_{{{\text{Е}}({\text{АРО}}{{{\text{О}}}^{ \bullet }})}}}$ всех исследованных веществ I–XX при взаимодействии с АPOO• явно возрастает (табл. 1). Сравнительный анализ $\frac{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}$ и $\frac{{{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}$ свидетельствует о том, что модельная реакция с DPPH• позволяет провести первичный отбор веществ с антирадикальными свойствами и выявить самые эффективные АО. Более чувствительна в этом плане реакция с цианоизопропанпероксилами, по величинам констант скоростей которой можно определить вещества с меньшей реакционной способностью. Так (табл. 1), с гидразильным радикалом активность выше или как у тролокса $\left( {\frac{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}} \geqslant 1} \right)$ проявили соединения I, II, VII, X, XV и XVII (эффективные антиоксиданты), а в реакции с АPOO•$\left( {\frac{{{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}} \geqslant 1} \right)$ к указанным веществам добавились еще VIII и XVI (вещества со средней АРА). Для оставшихся фенолкарбоновых кислот в обоих случаях $\frac{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}$ и $\frac{{{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}$ меньше единицы (вещества с низкой АРА).
Таким образом, исследование кинетики реакции НООСArOH с N- и О-центрированными радикалами позволило сформировать кинетическую схему (реакции (I), (III)), позволяющую классифицировать природные фенолы на группы веществ с высокой, средней и низкой антирадикальной активностью.
Следующий этап разработки прогностической модели был направлен на выбор неэмпирической составляющей, то есть молекулярных дескрипторов фенолов, связанных с лимитирующей стадией механизма реакции.
Рассматривая гидроксибензойные как АО с фенольной функциональной группой, можно выделить несколько известных механизмов их антирадикального действия. В водных средах имеет место последовательный перенос электрона и протона. Если потеря протона предшествует лимитирующей стадии переноса электрона, то механизм именуется SPLET (Sequential Proton Loss–Electron Transfer) [25, 27 ], и в реакции непосредственно участвует ионизированная форма антиоксиданта ($^{ - }{\text{ООСAr}}{{{\text{O}}}^{ - }}$). Это характерно для щелочных сред c рН ≥ 7. Если потеря протона следует за медленной стадией переноса электрона, то имеет место механизм ET–PT (Electron Transfer–Proton Transfer) (также упоминается как SET–PT или SEPT) [27]. Тогда в реакции с радикалом принимает участие молекулярная форма фенольного антиоксиданта (НООСArOH и $^{ - }{\text{ООСArOH}}$), что характерно для водных сред с низким значением рН ≤ 3. В нейтральных средах, где присутствуют и молекулярная, и ионная формы антиоксиданта, будут реализовываться оба механизма, на что указывают исследования, проводимые в области изучения механизмов действия фенольных антиоксидантов [27, 28].
ET–PT- и SPLET-зависимым дескриптором является, как правило, потенциал ионизации молекулярной и ионной форм фенольного антиоксиданта соответственно [27, 28]. Поиск и расчет дескрипторов осуществляли путем комбинирования двух методов. Вначале методом QSPR в программе Marvin 18.14 определяли ионы, доля которых в системе при заданном рН достаточно велика, чтобы влиять на скорость лимитирующей стадии. Затем для выбранных ионов методом DFT рассчитывали значения потенциалов ионизации карбоксилат- ($P{{I}_{{^{ - }{\text{OOCArOH}}}}}$) и фенолят-ионов ($P{{I}_{{^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}$) кислот.
Между вычисленными SPLЕT- и ЕТ–РТ-зависимыми дескрипторами (табл. 1) и экспериментальными константами скоростей (${{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)~}}}$ и ${{k}_{{{\text{APO}}{{{\text{O}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)~}}}$) устанавливали одно- и двухфакторные линейные регрессионные зависимости (уравнения (4)–(7)), по коэффициентам корреляции которых видно, что влияние ${\text{\;}}P{{I}_{{^{ - }{\text{OOCArOH}}}}}$ (как ЕТ–РТ-зависимого дескриптора) соизмеримо с $P{{I}_{{^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}{\text{\;}}$ (как SPLЕT-зависимого дескриптора), а значит, в системе перенос электрона с фенолкарбоновой кислоты на радикал, вероятно, представляет собой комбинацию SPLET- и ЕТ–РТ-механизмов:
(4)
$\begin{gathered} {\text{ln\;}}{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}~ = \\ = \left( {40.2 \pm 1.8} \right)--\left( {5.8 \pm 0.3} \right)P{{I}_{{^{ - }{\text{OOCArOH}}}}}, \\ n = 20;\,\,\,\,\tilde {r} = 0.975;\,\,~\,\,{{{\tilde {r}}}^{2}} = 0.950;\,\,\,\,F = 343; \\ p < 0.00000;\,\,\,\,{{S}_{{{\text{est}}}}} = 0.62; \\ \end{gathered} $(5)
$\begin{gathered} {\text{ln\;}}{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}~ = \left( {43 \pm 4} \right)--\left( {8.1 \pm 0.8} \right)P{{I}_{{{}_{{}}^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}, \\ n = 20;\,\,\,\,\tilde {r} = 0.915;~\,\,\,\,{{{\tilde {r}}}^{2}} = 0.837;\,\,\,\,F = 93; \\ p < 0.00000;\,\,\,\,{{S}_{{{\text{est}}}}} = 1.12; \\ \end{gathered} $(6)
$\begin{gathered} {\text{ln\;}}{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}~ = \left( {52 \pm 3} \right)--\left( {7.4 \pm 0.6} \right)P{{I}_{{{}_{{}}^{ - }{\text{OOCArOH}}}}}, \\ n = 20;\,\,\,\,\tilde {r} = 0.946;\,\,\,\,{{{\tilde {r}}}^{2}}~ = 0.896;\,\,\,\,F = 155; \\ p < 0.00000;\,\,\,\,{{S}_{{{\text{est}}}}} = 1.18; \\ \end{gathered} $(7)
$\begin{gathered} {\text{ln\;}}{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}~ = \left( {58 \pm 4} \right)--\left( {11.1 \pm 0.9} \right)P{{I}_{{{}_{{}}^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}, \\ n = 20;\,\,\,\,\tilde {r} = 0.944;~\,\,\,\,{{{\tilde {r}}}^{2}} = 0.892;\,\,\,\,F = 148; \\ p < 0.00000;\,\,\,\,{{S}_{{{\text{est}}}}} = 1.2, \\ \end{gathered} $Поскольку линейная связь между значениями потенциалов ионизации карбоксилат- и фенолят-ионов (табл. 1) не превышает 0.9 ($\tilde {r}{\text{\;}}$= 0.868), то разрешить сложившуюся двойственную ситуацию можно путем установления двухфакторных линейных регрессионных зависимостей АРА фенолов в реакции с DPPH• и АРОО• одновременно со SPLЕT- и ЕТ–РТ-зависимыми дескрипторами:
(8)
$\begin{gathered} {\text{ln\;}}{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}~ = \left( {42.9 \pm 1.6} \right)-- \\ - \,\,\left( {4.3 \pm 0.5} \right)P{{I}_{{{}_{{}}^{ - }{\text{OOCArOH}}}}} - \left( {2.5{\text{ }} \pm {\text{ }}0.8} \right)P{{I}_{{{}_{{}}^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}, \\ n = 20,\,\,\,\,\tilde {r} = 0.984,\,\,\,\,{{{\tilde {r}}}^{2}}~ = 0.969,\,\,\,\,F = 268, \\ p < 0.00000,\,\,\,\,{{S}_{{{\text{est}}}}} = 0.5, \\ {{r}_{{{\text{part}}\left( {P{{I}_{{{}_{{}}^{ - }{\text{OOCArOH}}}}}} \right)}}} = 0.901,\,\,\,\,{{r}_{{{\text{part}}\left( {{}_{{}}^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}} \right)}}} = 0.619; \\ \end{gathered} $(9)
$\begin{gathered} {\text{ln\;}}{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}~ = \left( {58.4 \pm 2.6} \right)-- \\ - \,\left( {4.0{\text{ }} \pm {\text{ }}0.8} \right)P{{I}_{{{}_{{}}^{ - }{\text{OOCArOH}}}}} - \left( {5.8 \pm 1.2} \right)P{{I}_{{{}_{{}}^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}, \\ n = 20,\,\,\,\,\tilde {r} = 0.978,~\,\,\,\,{{{\tilde {r}}}^{2}} = 0.957,\,\,\,\,F = 188, \\ p < 0.00000,\,\,\,\,{{S}_{{{\text{est}}}}} = 0.78, \\ {{r}_{{{\text{part}}\left( {P{{I}_{{{}_{{}}^{ - }{\text{OOCArOH}}}}}} \right)}}} = 0.776,\,\,\,\,{{r}_{{{\text{part}}\left( {{}_{{}}^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}} \right)}}} = 0.765, \\ \end{gathered} $Как видно (уравнения (8), (9)) из значений множественного коэффициента корреляции ($\tilde {r}{\text{\;}}$ = = 0.984 и 0.978 ) и детерминации (${{\tilde {r}}^{2}}$ = 0.969 и 0.957), выбранная линейная модель с двумя факторами $P{{I}_{{{\text{ArOH}}\left( {^{ - }{\text{OOCArOH}}} \right)}}}$ и $P{{I}_{{{\text{Ar}}{{{\text{O}}}^{ - }}\left( {^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}} \right)}}}$ более адекватно описывает экспериментальные данные в отличие от однофакторных регрессий, и вариация ${\text{ln\;}}{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}$ на 96.9 и 95.7% может быть обусловлена вариацией независимых переменных. Критерий Фишера F проверки значимости линейной модели регрессии высокий при уровне значимости p < 0.0000.
Обязательным элементом многофакторного анализа является вычисление частных коэффициентов корреляции ${{r}_{{{\text{part}}}}}$, отражающих степень линейной взаимосвязи между двумя переменными (зависимой и независимой), рассчитанную после устранения влияния всех других независимых переменных [26]. Установлено, что частный коэффициент корреляции уменьшается по сравнению с полным парным коэффициентом корреляции (уравнения (4), (5), (8) и (6), (7), (9)), а следовательно, один аргумент, который исключили из регрессии, не маскирует истинную взаимосвязь между функцией и вторым аргументом, оставшимся в линейной зависимости. Для выбранной и статистически аргументированной модели построен трехмерный график зависимости между параметром АРА фенолов и их квантово-химическими дескрипторами (рис. 4).
Рис. 4.
3D-график зависимости ${\text{ln\;}}{\kern 1pt} {{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{рН\;}}7} \right)}}}$ от $P{{I}_{{^{ - }{\text{OOCArOH}}}}}$, $P{{I}_{{^{ - }{\text{OOCAr}}{{{\text{O}}}^{ - }}}}}$, построенный в системе Statistica Demo 6.

На этапе формирования прогностической модели двухфакторные регрессии объединяли в систему уравнений, связанную с кинетической схемой, которая реализуется в заданной реакционной среде. Полученная комплексная взаимосвязь в общем виде представлена на схеме 1 :
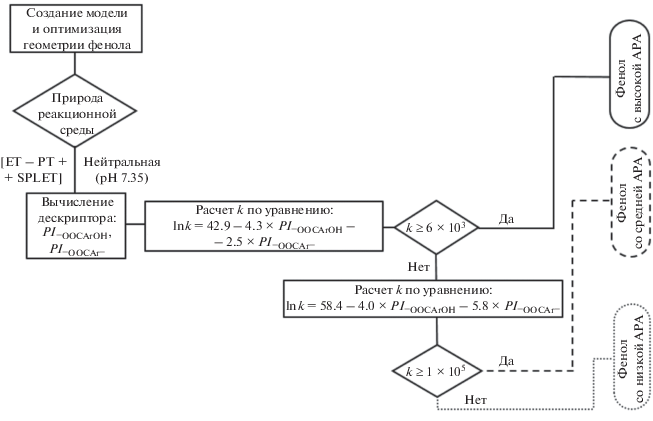
Схема 1 . Алгоритм скрининга природных фенольных антиоксидантов в средах с физиологическим рН на основе прогностической регрессионно-классификационной модели “дескриптор–активность”.
Такой подход в области исследования природных фенольных антиоксидантов был реализован впервые и позволил сформировать новую регрессионно-классификационную модель скрининга фенолкарбоновых кислот по АРА.
Рассмотрим принцип отбора антиоксидантов в рамках предложенной регрессионно-классификационной модели. Вначале проводят неэмпирический расчет химической структуры фенольного антиоксиданта в нейтральной водной среде. Затем по вычисленному механизм-зависимому дескриптору с помощью уравнений регрессии прогнозируют константу скорости реакции с различными радикалами. На первом этапе оценки применяют уравнение для расчета константы скорости реакции фенолов с радикалом ${\text{DPP}}{{{\text{H}}}^{ \bullet }}$. Если прогнозируемая величина константы больше, чем у эталонного АО тролокса, то исследуемое соединение относится к группе веществ с высокой активностью. На втором этапе антирадикальные свойства оставшихся соединений проверяют с О-центрированным радикалом ${\text{АРО}}{{{\text{О}}}^{\centerdot }}$ и по результатам сравнения с тролоксом фенолы разбивают еще на две группы – вещества со средней и низкой АРА.
Прогностическую способность предложенной модели проверяли на контрольной группе веществ, коими являются гидроксиацетофеноны – растительные фенолы, которые имеют близкую с фенолкарбоновыми кислотами структуру, не относятся к известным антиоксидантам и не входят в исследованную ранее группу веществ.
Следуя логике алгоритма (схема 1 ), по рассчитанным SPLET- и ET–PT-зависимым дескрипторам с помощью двухфакторного уравнения (8) были вычислены константы $k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{расч}}}}$ скорости реакции AcArOH с ${\text{DPP}}{{{\text{H}}}^{ \bullet }}$ (табл. 2).
Таблица 2.
Прогнозируемые величины тролоксового эквивалента и константы скорости реакции гидроксиацетофенонов с ${\text{DPP}}{{{\text{H}}}^{ \bullet }}$ и ${\text{NPO}}{{{\text{O}}}^{ \bullet }}$, рассчитанные по уравнению (8) и (9)
| Соединение | $k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{расч}}}}$, л моль‒1 с‒1 |
${{\left( {\frac{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}} \right)}_{{{\text{расч}}}}}~$ | ${{\Delta }_{{k({\text{DPP}}{{{\text{H}}}^{ \bullet }})}}}$a, % | $k_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{расч}}}}$, л моль‒1 с‒1 |
${{\left( {\frac{{{{k}_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}} \right)}_{{{\text{расч}}}}}$ | ${{\Delta }_{{k({\text{АРО}}{{{\text{О}}}^{ \bullet }})}}}~$a, % |
|---|---|---|---|---|---|---|
| XXI | 83 | 0.014 | 9.8 | 1.6 × 103 | 0.016 | 11.2 |
| XXII | 74 | 0.012 | 10.8 | 1.9 × 103 | 0.019 | 7.2 |
| XXIII | 54 | 0.009 | 12.0 | 1.2 × 103 | 0.012 | 7.4 |
| XXIV | 90 | 0.015 | 10.7 | 1.9 × 103 | 0.019 | 3.7 |
| XXV | 1.3 × 103 | 0.22 | 11.8 | 4.5 × 104 | 0.45 | 3.5 |
| XXVI | 275 | 0.046 | 8.5 | 9.5 × 103 | 0.095 | 3.3 |
| XXVII | 243 | 0.041 | 10.0 | 3.6 × 103 | 0.036 | 11.1 |
a Относительная погрешность расчета ${{\Delta }_{k}} = \left( {{{\left| {{{k}_{{\left( {{\text{эксп}}} \right)}}} - {{k}_{{\left( {{\text{расч}}} \right)}}}} \right|} \mathord{\left/ {\vphantom {{\left| {{{k}_{{\left( {{\text{эксп}}} \right)}}} - {{k}_{{\left( {{\text{расч}}} \right)}}}} \right|} {{{k}_{{\left( {{\text{эксп}}} \right)}}}}}} \right. \kern-0em} {{{k}_{{\left( {{\text{эксп}}} \right)}}}}}} \right) \times 100{\text{\% }}$.
Для всех изученных AcArOH значения констант $k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{расч}}}}$ низкие, они менее активны, чем эталонный антиоксидант – тролокс $\left( {{{{\left( {\frac{{{{k}_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}}}}{{k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{Tx}}}}}}} \right)}}_{{{\text{расч}}}}} < 1} \right)$, а значит, среди них нет фенолов с высокой АРА.
Тогда, согласно предложенной модели, исследуемая выборка в полном объеме переходит на второй этап скрининга в реакции с ${\text{АРO}}{{{\text{O}}}^{ \bullet }}$ (табл. 2). В этом случае прогнозируемая константа $k_{{{\text{АРО}}{{{\text{О}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{расч}}}}$ значительно возрастает по сравнению с константой для гидразильного радикала $k_{{{\text{DPP}}{{{\text{H}}}^{ \bullet }}\left( {{\text{pH\;}}7} \right)}}^{{{\text{расч}}}}$, но по-прежнему ни один из представленных AcArOH по своей АРА не превышает таковую у тролокса. Только одно соединение XXV (2,5-гидроксиацетофенон) потенциально может проявить активность в два раза ниже, чем у тролокса. Результаты прогноза таковы, что с большой долей вероятности в средах с физиологическим рН гидроксиацетофеноны будут обладать низкой активностью в реакции со свободными радикалами. Примечательно, что прогностическая (табл. 2) и эмпирическая (табл. 1) оценки совпадают. Хорошие сходимости предсказанных и экспериментальных констант, а также низкие погрешности аппроксимации, не выше 10–12%, подтверждают высокую прогностическую способность предложенной регрессионно-классификационной модели.
ЗАКЛЮЧЕНИЕ
В настоящей работе путем комбинирования методов DFT и QSPR предложены алгоритмы поиска и расчета механизм-зависимых дескрипторов молекулярных и ионных форм фенолкарбоновых кислот. Эмпирический параметр активности вещества, то есть константы скоростей реакций фенолов с радикалами, определяли по специально выбранным методологиям, повышающим точность измеряемых величин. Впервые получены готовые системы полуэмпирических уравнений, связанные с кинетическими схемами и позволяющие одновременно количественно оценить АРА природных фенолов и разделить их на группы веществ с разной реакционной способностью. При создании новой регрессионно-классификационной модели с высокой прогностической способностью использовали комплексный подход “среда–механизм–дескриптор–активность”, что позволяет решить одну из главных проблем методологии QSAR – учет данных о механизме проявления активности вещества, а также найти новые направления в области традиционного моделирования “структура–активность” и улучшить прогностическую способность получаемых моделей.
Список литературы
Перевозкина М.Г. Тестирование антиоксидантной активности полифункциональных соединений кинетическими методами. Новосибирск: СибАК, 2014. 240 с.
Меньщикова Е.Б., Ланкин В.З., Зенков Н.К., Бондарь И.А., Круговых Н.Ф., Труфакин В.А. Окислительный стресс. Прооксиданты и антиоксиданты. Москва: Слово, 2006. 556 с.
Galano A., Perez-Gonzalez A. // Theor. Chem. Accounts. 2012. V. 131. № 9. Art. 1265.
Marković Z., Ðorović J., Dimitrić Marković J.M., Živić M., Amić D. // Monatsh. Chem. 2014. V. 145. № 6. P. 953.
Xiao Z., Wang Y., Wang J., Li P., Ma F. // Food Chem. 2019. V. 275. P. 354.
Chen J., Yang J., Ma L., Li J., Shahzad N., Kim C.K. // Sci. Rep. 2020. V. 10. Art. 2611.
Habibpour E., Ahmadi S. // Curr. Comput.-Aid. Drug. 2017. V. 13. № 2. P. 143.
Jorge E.G., Rayar A.M., Barigye S.J., Rodríguez M.E.J., Veitía M.S.-I. // Int. J. Mol. Sci. 2016. V. 17. № 6. Art. 881.
Todeschini R., Consonni V. Molecular descriptors for chemoinformatics. Weinheim: Wiley-VCH, 2009. 967 p.
Белая Н.И., Белый А.В., Щербаков И.Н. // Кинетика и катализ. 2020. Т. 61. № 3. С. 334. (Belaya N.I., Belyi A.V., Shcherbakov I.N. // Kinet. catal. 2020. V. 61. № 3. Р. 360.)
Рабинович В.А., Хавин З.Я. Краткий химический справочник. Ленинград: Химия, 1991. 432 с.
Armarego W.L.F., Chai C.L.L. Purification of Laboratory Chemicals. Burlington: Elsevier Science, 2003. 608 p.
Сухарев А.Г., Тимохов А.В., Федоров В.В. Курс методов оптимизации. Москва: ФИЗМАТЛИТ, 2005. 368 с.
Белая Н.И., Белый А.В., Заречная О.М., Щербаков И.Н., Помещенко А.И., Горбань О.А. // Кинетика и катализ. 2019. Т. 60. № 1. С. 33. (Belaya N.I., Belyi A.V., Zarechnaya O.M., Shcherbakov I.N., Pomeshchenko A.I., Gorban’ O.A. // Kinet. Catal. 2019. V. 60. № 1. Р. 28.)
Шляпинтох В.Я., Карпухин О.Н., Постников Л.М., Захаров И.П., Вичутинский А.А., Цепалов В.Ф. Хемилюминесцентные методы исследования медленных химических процессов. Москва: Наука, 1966. 300 с.
Stefek M., Kyselova Z., Rackova L., Krizanova L. // Biochim. Biophys. Acta. 2005. V. 1741. P. 183.
Rackova L., Stefek M., Majekova M. // Redox Report. 2002. V. 7. P. 207.
Беляков В.А., Васильев Р.Ф., Федорова Г.Ф. // Кинетика и катализ. 2004. Т. 45. № 3. С. 355.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., X.Li, Hratchian H.P., Izmaylov A.F. et al. Gaussian 09, Revision B.01 Gaussian, Inc., Wallingford CT, 2010.
Weinberg D.R., Gagliardi C.J., Hull J.F., Murphy C.F., Kent C.A., Westlake B., Paul A., Ess D.H., McCafferty G.D., Meyer T.J. // Chem. Rev. 2007. V. 107. № 11. P. 5004.
Tomasi J., Mennucci B., Cammi R. // Chem. Rev. 2005. V. 105. № 8. P. 2999.
Rappe A.K., Casewit C.J., Colwell K.S., Goddard W.A., Skiff W.M. // J. Am. Chem. Soc. 1992. V. 114. № 25. P. 10024.
Elyashberg M., Williams A., Blinov K. Contemporary Computer-Assisted Approaches to Molecular Structure Elucidation. Cambridge: RSCPublishing, 2012. 469 p.
Marvin 18.14.0. ChemAxon (https://www.chemaxon.com).
Tošović J., Marković S. // Food Chem. 2019. V. 278. P. 469.
Боровиков В.П. Популярное введение в современный анализ данных в системе STATISTICA. Москва: Горячая линия–Телеком, 2013. 288 с.
Galano A., Alvarez-Idaboy J.R. // Int. J. Quantum. Chem. 2019. V. 119. № 2. Art. e25665.
Belaya N.I., Belyi A.V., Zarechnaya O.M., Shcherbakov I.N., Doroshkevich V.S. // Russ. J. Gen. Chem. 2020. V. 90. № 1. P. 23.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ