

Кинетика и катализ, 2023, T. 64, № 1, стр. 3-20
Окисление графита диоксидом азота при комнатной температуре при участии наночастиц платиновых металлов
М. Ю. Смирнов a, *, А. В. Калинкин a, А. М. Сорокин a, А. Н. Саланов a, В. И. Бухтияров a
a ФГБУН Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: smirnov@catalysis.ru
Поступила в редакцию 22.09.2022
После доработки 03.10.2022
Принята к публикации 03.10.2022
- EDN: KIWSES
- DOI: 10.31857/S0453881123010094
Аннотация
В работе представлен обзор результатов, полученных при исследовании взаимодействии NO2 с модельными системами, приготовленными вакуумным напылением платиновых металлов на поверхность высоко ориентированного пиролитического графита (M/ВОПГ, M = Pt, Pd, Rh), при комнатной температуре и давлении 10–6–10–4 мбар. Особое внимание было сосредоточено на установлении химического состояния частиц нанесенного металла и углеродного носителя с помощью рентгеновской фотоэлектронной спектроскопии (РФЭС). Перед обработкой в NO2 образцы M/ВОПГ были охарактеризованы методами сканирующей туннельной и/или растровой электронной микроскопии (СТМ и РЭМ). При взаимодействии с NO2 нанесенные палладий и родий оставались в металлическом состоянии и при этом проявляли каталитическую активность в окислении графита. Процесс сопровождался разрушением структуры ≥10–15 графеновых слоев с внедрением частиц металла вглубь углеродного носителя. Родий проявлял меньшую активность в окислении графита по сравнению с палладием по причине заполнения его поверхности молекулами NO, возникающими при диссоциации NO2. При обработке в NO2 образцов с нанесенной платиной углеродный носитель претерпевал минимальные изменения без нарушения своей исходной структуры. При этом платина сохраняла металлическое состояние в случае ее нанесения на поверхность графита, отожженного в вакууме, и окислялась до оксидов PtO и PtO2 на поверхности, активированной травлением ионами аргона. На основании полученных результатов был предложен механизм взаимодействия систем M/ВОПГ с NO2 при комнатной температуре.
ВВЕДЕНИЕ
Платиновые металлы в виде наночастиц, нанесенных на оксидные носители, являются важнейшим компонентом каталитических систем, применяемых для нейтрализации выхлопных выбросов дизельных двигателей. Они, в частности, участвуют в процессе окисления сажи, особые надежды в успешной реализации которого связывают с использованием такого сильного окислителя, как диоксид азота [1–3]. NO2 присутствует в выхлопных газах в небольших количествах (∼5% от общего содержания оксидов азота [4]), но его концентрацию можно преднамеренно увеличить. Превращение NO в NO2 способствует не только окислению сажи, но и более полной нейтрализации самих оксидов азота в системах поглощения–восстановления NOx [5] и селективного восстановлении NOx углеводородами или аммиаком в сажевых фильтрах с непрерывной регенерацией [6–8]. На платиновых металлах перевод основного N-содержащего компонента NO в NO2, который далее окисляет сажу, осуществляется легко даже в условиях окислительной реакционной среды [6, 8–10]. Проблема, однако, состоит в том, что при взаимодействии с продуктом окисления NO–NO2 – платиновые металлы могут перейти из активного в данной реакции металлического состояния [6, 11, 12] в неактивное оксидное [6, 8, 10].
С целью выяснения особенностей взаимодействия NO2 с платиновыми металлами, находящимися в контакте с углеродом, нами с использованием метода рентгеновской фотоэлектронной спектроскопии (РФЭС) были исследованы модельные системы, образованные нанесением наночастиц Pt, Pd и Rh методом вакуумного напыления на поверхность высоко ориентированного пиролитического графита (ВОПГ). Ниже приведен обзор полученных результатов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Образцы металлов Pt, Pd и Rh, нанесенных на поверхность ВОПГ, готовили в камере подготовки рентгеновского фотоэлектронного спектрометра SPECS (Германия) методом напыления в условиях сверхвысокого вакуума (табл. 1). Предварительно графит (“HOPG SPI-3 Structure Probe Inc.”, США) отжигали в вакууме при 600°С в течение нескольких часов. Рентгеновские фотоэлектронные (РФЭ) спектры отожженного графита содержали только фото- и Оже-линии углерода, а изображение его поверхности, полученное методом сканирующей туннельной микроскопии (СТМ), имело атомное разрешение. Подготовленный таким образом носитель далее называется исходным ВОПГ. При приготовлении ряда образцов перед напылением металла поверхность графита была предварительно активирована травлением ионами аргона по методике, описанной в работах [18–21] с целью создания на ней дефектов, служащих центрами закрепления частиц металла. Такой графит обозначен как ВОПГ-А. Травление осуществляли ионами Ar+ с кинетической энергией 0.5 кэВ при давлении аргона 5 × × 10–7 мбар. Продолжительность травления для каждого из исследованных образцов указана в табл. 1. Напыление осуществляли с помощью устройства EFM3 (“Omicron”, Германия), в котором предварительно оплавленную проволоку из выбранного металла нагревали электронным пучком; при этом температуру носителя поддерживали вблизи комнатной. В том случае, когда в качестве носителя был использован графит с активированной поверхностью (ВОПГ-А), после напыления металла образцы прогревали в вакууме при 300–400°C в течение 1 ч, обеспечивая тем самым локализацию и стабилизацию частиц на дефектах [18–21].
Таблица 1.
Время травления ВОПГ перед напылением на него платиновых металлов, поверхностные концентрации и размер частиц металла по данным методов СТМ и РЭМ. Для образцов Pt/C-A2 и Pt/C-A3 приведен размер частиц Pt после взаимодействия с NO2
| Образец | Время травления, с | [M]/[C] | dср, нм | (h/d)ср | Ссылки |
|---|---|---|---|---|---|
| Pt/C | 0 | 0.025 | 5.5 | н/о | [13] |
| Pt/C-A1 | 1 | 0.007 | 2.5 | н/о | [13] |
| Pt/C-А2 | 5 | 0.010 | 2.8 | 0.29 | [14] |
| Pt/C-А3 | 5 | 0.015 | 5.1 | 0.32 | [14] |
| Pt/C-А2 + NO2 | 5 | 0.009 | 2.3 | 0.43 | [14] |
| Pt/C-А3 + NO2 | 5 | 0.012 | 5.0 | 0.35 | |
| Pd/C | 0 | 0.0067 | 4 | н/о | [15] |
| Pd/C-A | 10 | 0.0047 | 3 | н/о | [16] |
| Rh/C | 0 | 0.022 | 3 | н/о | [17] |
Для обработки образцов металлов, нанесенных на поверхность графита, диоксидом азота применяли источник, принцип работы которого основан на термическом разложении нитрата свинца [13]. Температура образца в процессе обработки в NO2 была близка к комнатной; давление реакционной газовой смеси регулировали в интервале от 10–6 до 10–4 мбар варьированием температуры нагрева источника. Состав газа контролировали при помощи масс-спектрометра PrismaPlus QMG 220 (“Pfeiffer”, Германия). Принимая во внимание то, что окислительная способность NO2 намного больше, чем у кислорода, и в реакционной смеси его парциальное давление в несколько раз выше, воздействием O2 на исследуемые образцы пренебрегали.
Спектры РФЭС регистрировали спектрометром SPECS, оснащенным 9-канальным детектором и анализатором PHOIBOS-150-MCD-9. В зависимости от поставленной задачи фотоэмиссию возбуждали исходным или монохроматизированным излучением AlKα с энергией квантов hν = = 1486.6 эВ, а при необходимости дополнительно монохроматизированным излучением AgLα (hν = = 2983.4 эВ). Перед проведением измерений спектрометр калибровали по линиям Au4f7/2 и Cu2p3/2, энергии связи (Eсв) которых для фольг соответствующих металлов равны 84.0 и 932.7 эВ. Принимая во внимание хорошую проводимость графита, Eсв определяли без корректировки на возможную зарядку образцов. В исходном ВОПГ энергия связи линии C1s составляла 284.4 эВ, что соответствовало литературным данным для графита [22–28]. Анализ спектральных линий с разложением на отдельные компоненты выполняли с помощью программы XPSPeak [29]. Линию фона задавали функцией Ширли. Атомные отношения [M]/[C] рассчитывали из отношений интенсивностей соответствующих фотоэмиссионных линий (Pt4f, Pd3d, Rh3d, C1s), измеренных в спектрах образцов сразу после их приготовления, с учетом факторов атомной чувствительности, которые опубликованы в [30].
Изображения сканирующей туннельной микроскопии (СТМ) образцов с нанесенными Pt и Pd получали с помощью вакуумного микроскопа GPI-300.02 (НПФ “Сигма Скан”, Россия) с использованием платиновых игл в качестве зонда. В исследованиях с нанесенной Pt дополнительно был задействован микроскоп UHV 7000 VT (“RHK Technology”, США) с применением резаных Pt–Ir игл. СТМ-изображения обрабатывали и анализировали с помощью пакетов программ XPMPro 2.0 и WSxM [31]. Микроскопические изображения частиц палладия и родия в образцах Pd/C и Rh/С были также получены методом растровой электронной микроскопии (РЭМ) на приборе Regulus 8230 (“Hitachi”, Япония) с холодной автоэмиссией при величинах энергии электронов зонда, равных 15 и 20 кэВ, в режиме вторичных электронов. В процессе исследований образцы переносили из вакуумной камеры РФЭ-спектрометра в камеру микроскопа и обратно в естественных условиях; при этом длительность экспозиции образцов на воздухе не превышала 20 мин. За указанное время в РФЭ-спектрах в регионе O1s появлялась линия с энергией связи ∼532.5–533.5 эВ, обусловленная адсорбцией воды и образованием гидроксильных групп на поверхности графита [32]. Величины среднего размера частиц металла (dср), определенные методами СТМ и РЭМ для образцов в исходном состоянии, приведены в табл. 1.
ПРОСТРАНСТВЕННОЕ РАСПРЕДЕЛЕНИЕ ЧАСТИЦ МЕТАЛЛА, НАНЕСЕННЫХ НА ГРАФИТ
Образцы нанесенных на графит Pt, Pd и Rh до проведения реакции с NO2 были охарактеризованы методами СТМ и РЭМ с целью установления характера распределения частиц металла по поверхности носителя и оценки среднего размера частиц. Согласно данным СТМ, в образце Pt/C частицы платины заполняют поверхность исходного ВОПГ неоднородно с преимущественным концентрированием в области ступеней (рис. 1а). Дополнительно на террасах базисной грани графита образовывались агрегаты из частиц, которые имели цепочечную структуру и, вероятно, закреплялись на присутствующих там биографических дефектах. На поверхности активированного графита ВОПГ-А частицы Pt распределялись более равномерно с отсутствием преимущественного концентрирования на ступенях (рис. 1б). В отличие от платины заселение палладием как исходной (рис. 1в), так и активированной поверхности было однородным (рис. 1г), вероятно, вследствие более сильного взаимодействия его частиц с графитом. Однако на изображении, полученном методом РЭМ для образца Pd/C, приготовленного на исходном ВОПГ (рис. 1д), отчетливо видно, что и в данном случае имело место концентрирование частиц в области ступеней. На террасах частицы распределялись равномерно. Тем не менее, и здесь можно было отметить тенденцию к формированию агрегатов из частиц, хотя выраженную и не столь ярко, как в образце Pt/C. В случае образца Rh/C, РЭМ-изображение которого приведено на рис. 1е, также наблюдалось преимущественное заполнение ступеней и формирование агрегатов из частиц Rh на террасах.
Рис. 1.
Изображения образцов Pt/C (а), Pt/C-A1 (б), Pd/C (в, д), Pd/C-A (г) и Rh/A (е), полученные методами СТМ (а–г) и РЭМ (д, е).

Таким образом, пространственное распределение частиц платинового металла по поверхности графита зависит от ее предварительной подготовки и природы металла. В согласии с результатами, опубликованными в литературе [33–38], на исходной поверхности ВОПГ частицы предпочитали локализоваться на ступенях и биографических дефектах террас, собираясь в агрегаты. Дефекты, созданные на поверхности ВОПГ посредством ионного травления, служили центрами закрепления частиц металлов, в результате чего их пространственное распределение становилось более равномерным, а связывание с поверхностью более прочным [36–40]. В работе [41] на примере исследования методами РФЭС и СТМ нанесенного на графит золота было показано, что на исходном ВОПГ частицы металла, локализованные главным образом на ступенях, имели форму, близкую к сферической. На активированной поверхности графита ВОПГ-А частицы равномерно покрывали всю поверхность и при этом приобретали уплощенную форму, так что высота частицы, измеренная в направлении перпендикулярно поверхности, оказывалась меньше ее радиуса, определенного в плоскости поверхности носителя. Примером могут служить результаты СТМ, полученные для образцов Pt/C-A2 и Pt/C-A3 на приборе RHK-UHV 7000 VT (табл. 1) [14]. Уплощенная форма частиц, по-видимому, была следствием их более прочного связывания с поверхностными дефектами, созданными в результате ионного травления.
ИЗМЕНЕНИЕ СОСТОЯНИЯ УГЛЕРОДНОГО НОСИТЕЛЯ В РЕЗУЛЬТАТЕ ВЗАИМОДЕЙСТВИЯ ОБРАЗЦОВ M/C И M/C-A С NO2 ПО ДАННЫМ РФЭС
Состояние углеродного носителя определяли из спектров в регионе C1s. Исходный ВОПГ характеризовался узкой линией с энергией связи 284.4 эВ и асимметричной формой с “хвостом”, вытянутым в направлении бóльших значений энергии связи (рис. 2а, спектр 1) [22, 23, 28, 42, 43]. В области ∼291 эВ обнаруживался пик плазмонных потерь (на рис. 2 данная область не показана), присущий упорядоченной трехмерной структуре графита [22, 23, 25, 28, 44]. После ионного травления ВОПГ в мягких условиях в спектре региона C1s наблюдалось лишь небольшое уширение фотоэмиссионной линии и уменьшение интенсивности пика плазмона на ∼10% (рис. 2б, спектр 1), что указывало на сохранение графитом в значительной степени характерной для него структуры. В процессе взаимодействия NO2 с ВОПГ (рис. 2а, спектр 2) и ВОПГ-А (рис. 2б, спектр 2), а также с образцами Pt/C (рис. 2а, спектр 3) и Pt/C-A3 (рис. 2б, спектр 3) в рассматриваемых условиях существенных изменений в спектрах C1s не обнаружено.
Рис. 2.
Спектры C1s (а) исходного ВОПГ до (1) и после взаимодействия с NO2 (2), а также образцов Pt/C (3), Pd/C (4) и Rh/C (5) после обработки в NO2; (б) активированного графита ВОПГ-А до (1) и после взаимодействия с NO2 (2), а также образцов Pt/C-A3 (3) и Pd/C-A (4) после обработки в NO2.
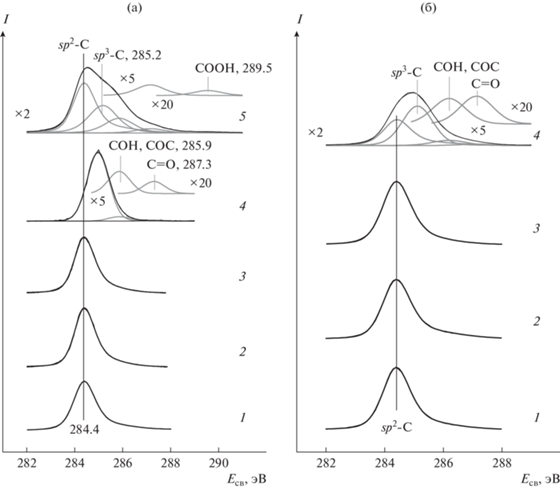
При взаимодействии с NO2 образцов с нанесенным палладием вне зависимости от того, использовался ли для их приготовления в качестве носителя исходный (образец Pd/C, рис. 2а, 4) или активированный графит (образец Pd/C-A, рис. 2б, 4), спектр C1s претерпевал значительные изменения. Спектр смещался в сторону бóльших энергий связи, и наблюдалось появление новых линий, обусловленных состояниями углерода, отличными от sp2-C. Кроме того, из спектра исчезал плазмон при ∼291 эВ (не показано) вследствие нарушения дальнего порядка в сопряженной системе π-связанных sp2-гибридных атомов углерода [45, 46] из-за деструкции поверхностного слоя графита. Спектр образца Pd/C после реакции с NO2 был разложен на три компонента, отнесенных sp3-гибридному углероду, не связанному с атомами кислорода (связи C–C, C–H) (285.0 эВ), атомам углерода в составе гидроксильных (C–O–H), эфирных (C–O–C) групп (285.9 эВ) и карбонильных групп (287.3 эВ) (рис. 2а, спектр 4) [23–25, 28, 32, 47–49]. Пик sp2-гибридного углерода при этом не обнаруживался, что указывало на значительное разрушение структуры графита. В спектре образца Pd/C-A, записанном после взаимодействия с NO2, было выделено четыре линии (рис. 2б, спектр 4). Линия с энергией связи 284.4 эВ принадлежала sp2-углероду в структуре графита, а остальные три были приписаны вышеперечисленным группам со связями C–C и C–H (285.1 эВ), C–O–H и C–O–C (286.2 эВ), C=O (287.1 эВ), образованным на поверхности графита в результате взаимодействия с NO2. Наблюдавшееся в случае образца Pd/C более глубокое превращение углерода, приводившее к полному исчезновению пика от sp2-C, могло быть обусловлено бóльшей поверхностной концентрацией палладия в этом образце (табл. 1).
Как и в случае с палладием, взаимодействие образца Rh/C с NO2 приводило к окислению графита, которое проявлялось в ослаблении линии при 284.4 эВ, принадлежавшей sp2-гибридному углероду и появлении линий от sp3-гибридного углерода (285.2 эВ), гидроксильных, эфирных (285.9 эВ) и карбонильных групп (287.2 эВ) (рис. 2а, спектр 5). Дополнительно регистрировалась линия при 289.5 эВ, отнесенная карбоксильным группам [24, 25, 47, 48, 50–55]. Исчезновение плазмона при ∼291 эВ также служило подтверждением разрушения упорядоченной структуры в поверхностном слое графита.
Из отношения интенсивностей пиков sp2-углерода, измеренных до (I0) и после (I) взаимодействия образцов Pd/C-А и Rh/C с NO2, с помощью выражения:
была оценена толщина разрушенного слоя графита δ. Здесь λ – длина свободного пробега в графите фотоэлектрона, эмитируемого с уровня C1s sp2-углерода под действием излучения AlKα; в соответствии с данными [56] λ = 3.31 нм. θ – угол регистрации фотоэлектронов анализатором спектрометра, измеренный по отношению к нормали к поверхности; в нашем случае θ = 0° (cos θ = 1). В результате расчетов по формуле (1) для обоих образцов были получены величины, попадавшие в интервал от ∼3 до ∼5 нм, что соответствовало ∼10–15 графеновым слоям в структуре графита. Из сравнения спектров C1s, записанных для образцов Pd/C (рис. 2а, спектр 4) и Rh/C (рис. 2а, спектр 5) после реакции с NO2, видно, что в присутствии нанесенного родия разрушение поверхностного слоя графита происходило на меньшую глубину, чем в случае с палладием, даже несмотря на то что атомное отношение [Rh]/[C] = 0.022 в образце Rh/C больше атомного отношения [Pd]/[C] = 0.0067 в образце Pd/C.ИЗМЕНЕНИЕ СОСТОЯНИЯ НАНЕСЕННОГО МЕТАЛЛА В РЕЗУЛЬТАТЕ ВЗАИМОДЕЙСТВИЯ ОБРАЗЦОВ M/C И M/C-A С NO2
Платина
Обработка образца Pt/C в NO2 не вызывала заметных изменений в спектральном регионе Pt4f (рис. 3а). Во всем интервале экспозиций энергия связи линии Pt4f7/2 с хорошей точностью совпадала с величиной, характеризовавшей массивную металлическую платину, Eсв(Pt4f7/2) = 71.2 эВ, откуда следовало, что частицы нанесенной платины сохраняли свое первоначальное металлическое состояние.
Рис. 3.
Спектры Pt4f (а) образца Pt/C, записанные после обработки в NO2 при давлении 3 × 10–6 мбар в течение 0 (1), 20 (2), 1100 (3) и 1500 мин (4); (б) образца Pt/C-A1 в исходном состоянии (1) и образцов Pt/C-A1 (2), Pt/C-A2 (3) и Pt/C-A3 (4) после взаимодействия с NO2 при давлении 3 × 10–6 мбар в течение 1818 (2), 1180 (3) и 1180 мин (4).

В реакции с NO2 были исследованы три образца платины, приготовленные с использованием графита, активированного ионным травлением – Pt/C-А1, Pt/C-А2 и Pt/C-А3. Образцы отличались друг от друга количеством напыленного металла и концентрацией дефектов на поверхности ВОПГ-А, регулируемой длительностью травления (табл. 1). На рис. 3б приведены спектры Pt4f для этих трех образцов после длительного взаимодействия с NO2 (рис. 3б, спектры 2–4), а также для сравнения спектр образца Pt/C-A1, записанный в исходном состоянии (рис. 3б, спектр 1). До начала обработки в NO2 энергия связи уровня Pt4f7/2 в образцах Pt/C-A1, Pt/C-A2 и Pt/C-A3 составляла 71.8, 72.1 и 71.8 эВ соответственно, что характерно для металлических частиц платины нанометрового размера [57, 58]. После обработки в NO2 в спектре Pt4f образца Pt/C-A1 в дополнение к линии металлической платины появлялись две дублетные линии с энергиями связи Eсв(Pt4f7/2) = 72.8 и 74.8 эВ, близкими к значениям для PtO и PtO2, которые обнаруживаются в интервалах 72.2–72.8 и 74.2–74.8 эВ [59–66]. В образцах Pt/C-А2 и Pt/C-А3 происходило полное окисление платины, на что указывало появление в регионе Pt4f только двух дублетных линий с Eсв(Pt4f7/2) = 73.0 и 74.2 эВ, принадлежавших оксидам PtO и PtO2. Из анализа интенсивностей линий платины Pt4f и Pt3d5/2, записанных с использованием излучения AgLα, было установлено, что в образцах Pt/C-A2 и Pt/C-A3 оксид PtO2 располагался во внутренней части окисленной частицы, образуя ее ядро, тогда как оксид PtO находился во внешней части в виде оболочки вокруг ядра [14]. Такое возможно, если при взаимодействии с NO2 частицы платины полностью превращались в PtO2, а PtO возникал в результате восстановления PtO2 под действием рентгеновского излучения в процессе съемки спектра. Хорошо известно, что соединения Pt(IV) обладают высокой чувствительностью к рентгеновскому излучению, проявляя при этом склонность к восстановлению до Pt(II) [67, 68].
Отличие Pt/C-A2 и Pt/C-A3 от образца Pt/C-A1, для которого в аналогичных условиях значительное количество платины оставалось в металлическом состоянии (рис. 3б, спектр 2), по-видимому, обусловлено созданием на поверхности графита перед напылением платины повышенной концентрации дефектов вследствие более продолжительного ионного травления (табл. 1). Можно предположить, что при относительно низкой концентрации дефектов в образце Pt/C-A1 все они оказывались заполненными частицами платины, и при этом часть частиц была вынуждена формироваться на бездефектных участках поверхности графита, где их склонность к окислению существенно ниже. Следует отметить, что при достаточно высокой концентрации дефектов, достигнутой при ионном травлении в течение 5 с, в окисленное состояние полностью переходили не только мелкие (Pt/C-А2), но и более крупные частицы (Pt/C-А3).
Методом СТМ на примере образцов Pt/C-A2 и Pt/C-A3 было показано, что взаимодействие с NO2 не приводило к агломерации частиц нанесенной платины [14]. Более того, было найдено, что в образце Pt/C-A2 средний размер частиц dср даже уменьшался, но одновременно увеличивалось отношение высоты к диаметру, h/d, так что средний объем сохранялся близким к первоначальному (табл. 1).
Палладий
Как следует из спектров, записанных в регионах Pd3d (рис. 4а) и Pd-MVV (рис. 4б), палладий в образцах Pd/C и Pd/C-А после обработки в NO2 при комнатной температуре сохранял свое первоначальное металлическое состояние. Энергия связи линии Pd3d5/2 у образца Pd/C-A в исходном состоянии (рис. 4а, спектр 3) на 0.3 эВ выше, чем у образца Pd/C (рис. 4а, спектр 1). Вероятно, это обусловлено более прочным химическим связыванием с дефектами, сопровождавшимся переносом электронной плотности от частиц палладия к носителю. После взаимодействия Eсв(Pd3d5/2), а также форма линий Pd3d и Pd-MVV оставались характерными для наночастиц металлического палладия в нанесенных системах [69–71]. Наиболее сильно воздействие NO2 на палладий проявлялось в существенном уменьшении интенсивности его спектральных линий; при этом Оже-линия Pd-MVV становилась слабо различимой на уровне фона (рис. 4, спектры 2, 4, 5).
Рис. 4.
Спектры Pd3d (а) и Pd-MNN (б) образцов Pd/C (1, 2) и Pd/C-A (3–5) до (1, 3) и после взаимодействия с NO2 при давлении 10–6 (2, 4) и 10–5 мбар (5) в течение 30 мин.

Для разных стадий обработки образцов Pd/C и Pd/C-A в NO2 было рассчитано нормализованное отношение интенсивностей двух спектральных линий палладия RN, определенное следующим образом:
Здесь ${{I}_{{MVV}}}$ и ${{I}_{{3d}}}$ – интегральные интенсивности линий Pd-MVV и Pd3d образца с нанесенным палладием, а $I_{{MVV}}^{0}$ и $I_{{3d}}^{0}$ – измеренные в тех же условиях интенсивности соответствующих линий массивного металлического палладия. Длина свободного пробега фотоэлектрона, испускаемого с уровня Pd3d, выше, чем у Оже-электрона (λ3d > λMVV) [56]. В этой связи, по мере роста размера частиц интенсивность эмиссии Оже-электронов раньше приобретает значение, соответствующее массивному металлу, чем интенсивность фотоэмиссии. Следовательно, до тех пор пока размер нанесенных наночастиц палладия сравнительно невелик (меньше, чем ∼3λ3d), RN > 1 [72]. С увеличением размера частиц интенсивность фотоэмиссии растет быстрее, чем интенсивность Оже-эмиссии, вследствие чего RN уменьшается, а когда частицы становятся достаточно большими, асимптотически стремится к 1. В нашем случае интенсивности спектральных линий образцов Pd/C и Pd/C-A в исходном состоянии удовлетворяли условию RN > 1. Однако после проведения обработки в NO2 значение RN резко падало и при этом становилось существенно меньше 1, что не могло быть объяснено только лишь укрупнением частиц. По-видимому, такое резкое снижение RN было вызвано внедрением частиц палладия вглубь графита с формированием на их поверхности углеродной пленки, экранирующей сигнал от палладия.Таким образом, с одной стороны, в отличие от нанесенной на активированный ВОПГ-А платины, частицы которой в результате взаимодействия с NO2 превращались в оксиды, палладий оставался в металлическом состоянии. С другой стороны, палладий, нанесенный на графит, как оказалось, проявлял каталитическую активность в окислении углеродного носителя, что также отличало его от платины. В работе [37] сообщалось о том, что на воздухе в присутствии нанесенного палладия активированная поверхность ВОПГ-А окислялась при температуре ∼460°С. С помощью СТМ было показано, что по мере выгорания углерода на одном участке поверхности частица палладия перемещалась на смежный участок, в результате чего в поверхностном слое графита формировался трек. Вероятно, нечто подобное происходило и в нашем случае, где при использовании NO2 как более сильного по сравнению с кислородом окислителя реакция могла протекать уже при комнатной температуре и пониженных давлениях.
Родий
На рис. 5 приведены спектры, записанные для образца Rh/C в регионе Rh3d (рис. 5а) и в регионе, в котором регистрировались Оже-линии родия и углерода (рис. 5б). Спектры 1 характеризовали образец в исходном состоянии. Спектры 2 и 3 были записаны после взаимодействия Rh/C с NO2 при давлении 10–6 (2) и 10–5 мбар (3). Судя по величине энергии связи Rh3d5/2 (Eсв = 307.3 эВ) и форме линий Rh3d и Rh-M45VV, после обработки в NO2 родий сохранял металлическое состояние (рис. 5а, спектры 2 и 3). Изменения в спектре Rh3d сводились лишь к заметному уменьшению интенсивности сигнала, падавшей после обработки при 10–6 и 10–5 мбар соответственно до 0.66 и 0.58 от величины в исходном образце. Интенсивность Оже-линий родия снижалась при этом в гораздо большей степени, чем интенсивность фотоэмиссионной линии Rh3d. Нормированное отношение интенсивностей линий Rh-M45VV и Rh3d, $RN = \left( {{{I}_{{MVV}}}{\text{/}}{{I}_{{3d}}})(I_{{3d}}^{0}{\text{/}}I_{{MVV}}^{0}} \right)$, для исходного образца было равно 1.35, а после взаимодействия с NO2 оно упало до 0.35. На этом основании было высказано предположение о том, что, как и в случае с палладием, обработка Rh/C в NO2 приводила к внедрению частиц Rh в приповерхностный слой графита. В пользу этого свидетельствовали результаты, полученные методом РЭМ. Как оказалось, после такой обработки микроскопическое изображение поверхности Rh/C становилось нечетким, хотя в нем и угадывался характер распределения частиц родия по поверхности, который был таким же, как и в образце до взаимодействия с NO2 [17].
Рис. 5.
Спектры образца Rh/C в регионах Rh3d (а) и Rh-MVV + C-KLL (б) до (1) и после взаимодействия с NO2 при давлении 10–6 (2) и 10–5 мбар (3) в течение 30 мин.

В спектре 1, изображенном на рис. 5б, в той его части, где регистрировалась Оже-линия углерода C-KLL, отчетливо проявлялся пик с Eкин ∼ 270 эВ, отвечавший Оже-процессу с участием π-электронов валентной зоны. Данный Оже-переход характерен для упорядоченной трехмерной структуры графита, образованной параллельно ориентированными слоями графена [73]. После обработки в NO2 пик с Eкин ∼ 270 эВ из спектра исчезал, что подтверждало вывод о сильном разрушении структуры поверхности графита в процессе ее окисления.
ОБРАЗОВАНИЕ ПОВЕРХНОСТНЫХ КИСЛОРОД- И АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ ПРИ ВЗАИМОДЕЙСТВИИ M/C И M/C-A С NO2
В результате взаимодействия ВОПГ и ВОПГ-А, а также образцов M/C и M/C-A с NO2 происходили изменения в регионе O1s, а для M = Pd и Rh также и в регионе N1s, вызванные появлением на поверхности O- и N-содержащих соединений. До взаимодействия азот в образцах не регистрировался, а кислород обнаруживался в количестве, не превышавшем [O]/[C] ∼ 0.001–0.002. В этой связи присутствие данных элементов в составе углеродных носителей при последующем рассмотрении не принимали во внимание. Как показано выше, для образцов с напыленным палладием и родием взаимодействие сопровождалось также появлением новых линий в регионе C1s, отличных от линии, принадлежащей sp2-углероду. В табл. 2 приведены энергии связи Eсв(O1s), Eсв(N1s), Eсв(C1s) и предельные значения атомных отношений θO = [O]/[C], θN = [N]/[C], рассчитанные после больших экспозиций в NO2. Наименьшее значение θO было получено для исходного ВОПГ, что отражало высокую устойчивость sp2-структуры базисной грани графита к воздействию окислителей, в том числе к NO2 [74]. Спектр O1s был разложен на линии с Есв = 532.3 и 533.6 эВ. Для графита, активированного ионным травлением (ВОПГ-А), предельное покрытие по кислороду становилось существенно больше, чем в случае исходного ВОПГ. В его спектре O1s регистрировалась одна широкая линия с Eсв = 532.6 эВ. Линия с Eсв = 532.3–532.6 эВ была отнесена эфирным (C–O–C) и карбонильным (C=O) группам [18, 53, 54, 75, 76], а линия с Eсв = 533.6 эВ – гидроксильным группам (C–O–H) [32, 77].
Таблица 2.
Энергии связи и атомные отношения, полученные из спектров в регионах O1s, N1s и C1s для поверхностных соединений, образующихся после взаимодействия образцов ВОПГ, ВОПГ-А, M/C и M/C-A с NO2
| Образец | dM, нм | [M]/[C] | O1s | N1s | C1s | Поверхностные соединения | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Eсв, эВ | [O]/[C] | [O]/[Ptox] | Eсв, эВ | [N]/[C] | Eсв, эВ | % | ||||
| ВОПГ | Нет металла | 532.3 533.6 |
0.008 | Нет PtOx | Отсутствует | Не определено | C–O–C, C=O C–OH |
|||
| ВОПГ-А | Нет металла | 532.6 | 0.049 | Нет PtOx | Отсутствует | Не определено | C–OH, C–O–C, C=O | |||
| Pt/C | 5.5 | 0.025 | 530.9 532.8 |
0.049 | Нет PtOx | Отсутствует | Не определено | C=O C–OH, C–O–C, |
||
| Pt/C-A1 | 2.5 | 0.007 | 531.5 533.1 |
0.130 | 25.0 | Отсутствует | Не определено | C=O, C–O–C C–OH |
||
| Pt/C-A2 | 2.8 | 0.010 | 531.6 532.9 |
0.136 | 15.4 | Отсутствует | Не определено | C=O, C–O–C C–OH |
||
| Pt/C-A3 | 5.1 | 0.015 | 531.4 532.8 |
0.116 | 9.2 | Отсутствует | Не определено | C=O, C–O–C C-OH |
||
| Pd/C | ∼4 | 0.0067 | 533.2 | 0.007 | Нет PtOx | Отсутствует | 285.9 | 6.0 | C–OH, C–O–C | |
| Отсутствует | 287.3 | 0.8 | C=O | |||||||
| Отсутствует | 400.9 | 0.005 | Не определено | Пиррол, α-пиридон | ||||||
| Pd/C-A | ∼3 | 0.0047 | 533.5 | 0.039 | Нет PtOx | Отсутствует | 285.9 | 8.9 | C–OH, C–O–C | |
| Отсутствует | 287.0 | 3.5 | C=O | |||||||
| Отсутствует | 400.4 | 0.003 | Не определено | Пиррол, α-пиридон | ||||||
| Rh/C | ∼3 | 0.022 | 533.1 | 0.111 | Нет PtOx | Отсутствует | 285.9 | 15.5 | C–OH, C–O–C | |
| Отсутствует | 287.2 | 4.6 | C=O | |||||||
| Отсутствует | 289.5 | 0.5 | COOH | |||||||
| 531.5 | Не определено | Нет PtOx | 400.0 | 0.017 | Не определено | NOад; пиррол, α-пиридон | ||||
У образца Pt/C было достигнуто существенно большее значение θO, чем у ВОПГ (табл. 2). Вероятно, причина состояла в эффективной диссоциации молекул NO2 на поверхности платины [78–80], приводившей к образованию адсорбированных атомов кислорода Оад, которые в дальнейшем перемещались к границе частицы платины с графитом, где вступали в реакцию с углеродом с образованием поверхностных соединений СxOy с различными функциональными группами. В работе [13] была построена зависимость величины атомного отношения [O]/[C] от экспозиции в NO2, из которой следовало, что скорость накопления кислорода быстро спадала во времени. Такой результат позволял предположить, что область, в которой происходило образование CxOy, сосредоточена на границе раздела Pt–графит и не распространяется на гладкие участки поверхности ВОПГ, свободные от нанесенного металла. Ситуация кардинально менялась при переходе к образцам Pt/C-A1–Pt/C-A3, для которых количество кислорода, накопленного на поверхности в результате взаимодействия, увеличивалось по сравнению с ВОПГ-А и Pt/C в ∼2.5 раза. По-видимому, в данном случае достигалась более глубокая степень превращения графита.
Спектр O1s образца Pt/C, записанный после взаимодействия с NO2, был разложен на две линии с энергиями связи 530.9 и 532.8 эВ (табл. 2). Поскольку частицы платины в данном образце сохраняли металлическое состояние (см. рис. 3а), очевидно, что весь кислород находился в составе поверхностных соединений CxOy, образовавшихся при окислении графита, которым и принадлежали наблюдаемые линии O1s. После взаимодействия с NO2 образцов платины Pt/C-A1–Pt/C-A3, нанесенных на ВОПГ-А, появление линий O1s с энергиями связи ∼531.5 и ∼533 эВ обнаруживалось параллельно с превращением металлической платины в оксиды PtO и PtO2, которые должны были давать определенный вклад в спектр O1s. Однако по оценке, выполненной с использованием интенсивностей линий O1s и принадлежащих оксидам линий Pt4f с учетом соответствующих факторов атомной чувствительности, были получены слишком большие значения атомных отношений [O]/[Ptox] (табл. 2), что указывало на незначительный вклад PtO2 и PtO в полученные спектры O1s. Кроме того, известно, что энергия связи линии O1s в оксидах платины составляет ∼530 эВ [63, 81], что существенно ниже величины, наблюдавшейся в наших экспериментах.
Взаимодействие ВОПГ, ВОПГ-А, Pt/C, Pt/C-A1, Pt/C-A2 и Pt/C-A3 c NO2 приводило лишь к незначительным изменениям в спектральном регионе C1s, которое сводилось к небольшому уширению исходной линии, принадлежавшей sp2-углероду, и ослаблению интенсивности плазмона. На этом основании можно утверждать, что, несмотря на появление значительного количества поверхностных O-содержащих соединений, разрушение структуры графита происходило в несущественной степени. Соответствующие этим соединениям линии C1s не удавалось выделить на фоне интенсивной линии sp2-углерода (см. рис. 2). Взаимодействие указанных образцов с NO2 не вело к появлению линий в регионе N1s, и, следовательно, поверхностные азотсодержащие соединения в этом случае не образовывались.
В случае взаимодействия с NO2 образцов Pd/C и Pd/C-A в регионе С1s наблюдались значительные изменения вследствие существенного разрушения структуры графита в поверхностном слое, в том числе появлялись новые линии, которые были отнесены соединениям с кислородсодержащими функциональными группами (рис. 2). В регионе O1s этим соединениям была отнесена линия при 533.2–533.5 эВ [53, 54, 75]. Параллельно в регионе N1s обнаруживалась слабая линия с энергией связи 400.4–400.9 эВ, отнесенная атомам азота, связанным с sp2-углеродом в фрагменты со структурой пиррола [32, 36, 43, 82–85] или α-пиридона [32, 82–84]. Близким значением энергии связи обладает линия N1s молекул оксида азота, адсорбированных на поверхности палладия [86–91], однако в нашем случае в спектре отсутствовала характерная для NOад линия O1s при ∼530.5–531.5 эВ [90, 92–96].
После взаимодействия с NO2 образца Rh/C в спектре O1s появлялись линии c Eсв = 531.5 и 533.1 эВ, а в регионе N1s – линия c Eсв = 400.0 эВ. Определенное из интенсивностей линий C1s и N1s атомное отношение [N]/[C] ∼ 0.017 было значительно больше полученного для образцов Pd/C и Pd/C-A. Величины энергий связи линии N1s и наименее интенсивной из линий O1s (531.5 эВ) позволяли отнести их адсорбированным молекулам NOад [92–98], образованным на поверхности частиц родия в результате диссоциации молекул NO2. Близкой величиной Eсв обладает также линия N1s, принадлежащая связанным с углеродом атомам азота, которые могут возникать в результате встраивания последних в графит [32, 36, 43, 82–85]. В данном случае можно с уверенностью утверждать, что такого типа азотсодержащие поверхностные соединения с углеродом, действительно, вносили определенный вклад в интенсивность линии N1s. В работе [17] было показано, что после прогрева в вакууме при 300°С образца Rh/C, обработанного в NO2, линия O1s с энергией связи 531.5 эВ, отнесенная NOад, исчезала, тогда как линия N1s все еще обнаруживалась, хотя ее интенсивность уменьшалась более чем в 2 раза. Второй пик O1s с бóльшей интенсивностью и энергией связи ∼533 эВ был отнесен кислородсодержащим группам C–OH, C–O–C, C=O и COOH, образованным в результате окисления поверхности графита [50–52, 54, 83, 99].
МЕХАНИЗМ ВЗАИМОДЕЙСТВИЯ NO2 С ГРАФИТОМ ПРИ УЧАСТИИ ЧАСТИЦ ПЛАТИНОВЫХ МЕТАЛЛОВ, НАНЕСЕННЫХ НА ЕГО ПОВЕРХНОСТЬ
Выше было показано, что при взаимодействии образцов M/C и M/C-A с NO2 при комнатной температуре может происходить окисление как графита, так и частиц платинового металла. Направление, в котором протекает реакция, зависит от природы металла и состояния углеродного носителя. После обработки в NO2 частицы Pt, нанесенные на поверхность исходного ВОПГ, сохраняют металлическое состояние, тогда как частицы, которые располагаются на структурных дефектах ВОПГ-А, активированного ионной бомбардировкой, окисляются до PtO и PtO2. При этом углерод вступает во взаимодействие в незначительной степени, в результате чего происходит образование поверхностных кислородсодержащих соединений в области контакта между частицей металла и графитом. Иная картина наблюдается при взаимодействии с NO2 образцов с нанесенным палладием или родием. В этом случае Pd и Rh остаются в металлическом состоянии, а окислению подвергается графит, что приводит к разрушению структуры ∼10–15 верхних графеновых слоев и появлению кислородсодержащих поверхностных соединений различной природы. Процесс сопровождается внедрением частиц металла в приповерхностный слой носителя и/или их инкапсулированием углеродным материалом. Палладий при окислении графита проявляет бóльшую активность, нежели родий. На примере палладия было также показано, что активация поверхности ВОПГ ионным травлением перед нанесением на нее металла не оказывает принципиального влияния на направление, в котором протекает последующее взаимодействие с NO2. Возникает вопрос: почему в реакции с NO2 платина, будучи нанесенной на ВОПГ, превращается в оксиды, тогда как палладий и родий в таких же условиях ведут окисление углерода с разрушением его структуры?
Для ответа на данный вопрос рассмотрим вероятный механизм взаимодействия систем M/C и M/C-A с NO2, представленный в виде совокупности следующих стадий, схематически проиллюстрированных на рис. 6. На начальном этапе молекулы NO2 диссоциируют на поверхности частиц металла с образованием NOад и Oад, подобно тому, как это происходит на массивных металлах [78, 80, 100–102] (рис. 6а). Вследствие параллельно протекающих десорбции и диссоциации молекул NO поверхность Pt и Pd покрывается атомами Oад [102–106], а на поверхности Rh появляются также и атомы азота Nад [101]. На следующем этапе атомы Oад мигрируют из адсорбированного состояния сначала в приповерхностную область, а затем и в объем частицы металла [80, 105, 107] (рис. 6б). Далее внедренные атомы кислорода могут встраиваться в решетку частицы, перетягивая на себя электроны от атомов металла, что приводит к образованию частицы оксида металла, либо перемещаться в направлении границы раздела с графитом, где они вступают в реакцию окисления углерода (рис. 6в).
Рис. 6.
Схематическое представление механизма окисления частиц нанесенного металла в образцах Pt/C-A и углеродного носителя в образцах Pd/C, Pd/C-A и Rh/C.

Очевидно, что оба процесса требуют эффективного проникновения атомов кислорода в приповерхностную область и далее в объем металла. Массивный палладий, как известно, способен поглощать атомы кислорода в процессе его обработки кислородом или оксидами азота [104, 106, 108–116]. В монокристаллах Pd(111), (110) и (100) приповерхностный кислород образуется при взаимодействии с O2 при температурах ≥150°С [109, 115, 117, 118]. Показано, что в Pd(110) приповерхностные атомы кислорода проникают на глубину 1.5–2.0 нм [117, 118]. Для более глубокого внедрения в объем металла требуются более высокие температуры (>300–350°С) [109, 114]. Необходимость использования повышенных температур обусловлена тем, что на монокристаллах процесс проникновения атомов кислорода в приповерхностную область с последующим растворением в объеме при комнатной температуре протекает медленно из-за низкой концентрации дефектов на поверхности [109]. На поликристаллическом палладии как образование приповерхностного кислорода [115], так и растворение атомов O в объеме [115] идет более эффективно. Можно ожидать, что поглощение кислорода будет происходить еще легче при взаимодействии с наночастицами палладия вследствие высокой дефектности их поверхности [102]. Известно, что родий также способен к образованию в нем приповерхностного (растворенного) кислорода [119–122].
Атомы кислорода, появляющиеся при диссоциации молекул NO2 на поверхности платины, менее склонны к растворению в приповерхностной области и в объеме металла по сравнению с Oад на поверхности палладия [104, 106, 108, 109, 113]. Это находит подтверждение в результатах теоретической работы [123], в которой был выполнен расчет энергии активации перемещения атома кислорода из адсорбированного состояния на Pd(111), Rh(111) и Pt(111) в приповерхностный слой. Было найдено, что эта величина имеет существенно бóльшее значение у Pt (191 кДж/моль) и Rh (183 кДж/моль), чем у Pd (155 кДж/моль).
Когда концентрация растворенных атомов кислорода в объеме данного конкретного металла достигает некоторой критической величины, становится возможным образование оксида. Было установлено, что в результате взаимодействия Pd(111) с NO2 при 260°С поверхностный оксид, спектральные характеристики которого соответствуют PdO, начинает формироваться при достижении в приповерхностной области концентрации кислорода >1.4 монослоев (МС) [105, 106]. Можно ожидать, что наночастицы нанесенных металлов при взаимодействии с NO2 будут переходить в оксиды при более низких температурах и давлениях по сравнению с массивными металлами. Однако если растворенные атомы кислорода расходуются на окисление графита, то их концентрация внутри частиц металла оказывается недостаточной для инициирования процесса образования оксидной фазы. Это может происходить в том случае, когда способность металла хорошо растворять атомы кислорода сочетается с подвижностью растворенных атомов в его объеме, так что подвод кислорода к границе с графитом протекает с большой эффективностью. Таким образом, высокие скорости образования приповерхностного кислорода, диффузии растворенных атомов Oр по объему металлической частицы и вступления их в реакцию окисления углерода на границе раздела металл–графит являются ключевыми факторами, определяющими направление взаимодействия NO2 с Pd/ВОПГ и Rh/ВОПГ в пользу окисления графита, а также стабилизации нанесенных частиц в металлическом состоянии. О быстрой диффузии растворенных атомов кислорода в объеме металлического палладия сообщалось работах [114, 115]. Напротив, в системе Pt/ВОПГ, по-видимому, за счет меньшей подвижности растворенных атомов кислорода обеспечивается высокая локальная концентрация Ор в приповерхностном слое частицы, что приводит к формированию оксида платины. Таким образом, в первом случае под воздействием NO2 происходит окисление графита, а во втором – самого металла.
Хотя системы на основе палладия и родия взаимодействуют с NO2 во многом похожим образом, тем не менее, в поведении этих металлов существуют и определенные отличия. Так же, как и на поверхности палладия, диссоциативная адсорбция NO2 на родии протекает при комнатной температуре с высокой скоростью, но образующиеся молекулы NOад десорбируются только после нагрева до ≥150°С [95, 124–126]. В этой связи при комнатной температуре расходование атомов кислорода на окисление графита сопровождается постепенным заполнением поверхности родиевых частиц NOад (рис. 6). Свидетельством тому служит упомянутое выше обнаружение в РФЭ-спектре линий N1s и O1s с энергиями связи 400.0 и 531.4 эВ, принадлежащих NOад (табл. 2). В результате поверхность родия оказывается блокированной молекулами NOад, что препятствует дальнейшей адсорбции на ней молекул NO2. Такая поверхность из-за отсутствия на ней свободных адсорбционных мест, требуемых для размещения атомов кислорода, не способна к диссоциации NO2, ад и NOад, вследствие чего процесс окисления графита в системе Rh/ВОПГ прекращается. Остается открытым вопрос: каким образом формирование углеродной пленки на поверхности частиц родия сочетается с заполнением ее молекулами NOад? Для ответа на него необходимы дополнительные исследования. В настоящий момент можно предположить, что экранирующая пленка не является сплошной, и имеются участки поверхности, доступные для диссоциативной адсорбции NO2 с образованием NOад. Кроме того, возможно, что инкапсулируются углеродом частицы Rh, уже содержащие на своей поверхности прочно удерживаемые ею молекулы NOад.
Другим фактором, который, по-видимому, также способствует повышенной активности палладия и родия по сравнению с платиной в реакции окисления графита, выступает прочная адгезия частиц этих металлов к поверхности ВОПГ. Говоря о роли, которую играет хорошая адгезия частиц активного компонента к углеродному носителю, следует упомянуть работу [127], в которой было показано, что прочное связывание частиц CoO с поверхностью ВОПГ выступало одним из основных факторов, обеспечивавших окисление графита при продолжительных выдержках системы CoO/ВОПГ на атмосфере при комнатной температуре. Сильная связь частиц палладия и родия с графитом обуславливает их более однородное, нежели у частиц платины, распределение по террасам поверхности ВОПГ (рис. 2). В работе [36] высокая плотность частиц Pd, равномерно распределенных по поверхности, была достигнута при нанесении на исходный ВОПГ 3 монослоев палладия. При этом было отмечено, что при такой существенной концентрации палладия на поверхности размер частиц, полученных как на исходном, так и на модифицированном ионным травлением ВОПГ, одинаков, что также было объяснено хорошей адгезией данного металла к графиту. В случае платины, которая взаимодействуют с графитом слабее, при ее нанесении на исходный ВОПГ частицы, заселяющие террасы, собирались в агрегаты с цепочечной структурой (рис. 2а). Образование агрегатов происходило также при заполнении террас ВОПГ частицами родия, хотя для него такой эффект выражен слабее (рис. 2е).
ЗАКЛЮЧЕНИЕ
Платина, палладий и родий традиционно входят в состав каталитических нейтрализаторов выхлопных автомобильных газов. В настоящем обзоре рассмотрено поведение данных металлов, нанесенных в виде наночастиц на поверхность высоко ориентированного пиролитического графита (ВОПГ), при взаимодействии с NO2, протекающем при комнатной температуре. Предполагается, что системы такого типа (M/ВОПГ) могут выступать в качестве модельных объектов для изучения процесса окисления углеродных отложений (сажи), образующихся при эксплуатации дизельных двигателей. Показано, что среди исследованных металлов палладий ведет окисление графита наиболее эффективно, тогда как родий демонстрирует меньшую активность, а платина, будучи практически неактивной в указанной реакции, сама превращается в смесь оксидов. Среди факторов, определяющих высокую активность палладия, следует выделить хорошую растворимость атомов кислорода в объеме частиц металла, высокую скорость диффузии растворенных атомов к границе раздела с графитом, а также прочную адгезию частиц Pd к углероду. Из всего сказанного следует, что палладий может рассматриваться как перспективный катализатор окисления углеродных отложений в нейтрализаторах выхлопных газов дизельных двигателей с использованием NO2 в качестве окислителя.
Список литературы
Setiabudi A., Makkee M., Moulijn J.A. // Appl. Cat. B: Env. 2003. V. 42. P. 35.
Dhal G.C., Dey S., Mohan D., Prasad R. // Cat. Rev. Sci. Eng. 2018. V. 60. P. 437.
Khobragade R., Singh S.K., Shukla P.C., Gupta T., Al-Fatesh A.S., Agarwal A.K., Labhasetwar N.K. // Cat. Rev. Sci. Eng. 2019. V. 61. P. 447.
Zheng Y., Kovarik L., Engelhard M.H., Wang Y., Wang Y., Gao F., Szanyi J. // J. Phys. Chem. C. 2017. V. 121. P. 15793.
Epling W.S., Campbell L.E., Yezerets A., Currier N.W., Parks J.E. // Catal. Rev. 2004. V. 46. P. 163.
Olsson L., Fridell E. // J. Catal. 2002. V. 210. P. 340.
Mulla S.S., Chen N., Delgass W.N., Epling W.S., Ribeiro F.H. // Catal. Lett. 2005. V. 100. P. 267.
Bhatia D., McCabe R.W., Harold M.P., Balakotaiah V. // J. Catal. 2009. V. 266. P. 106.
Mulla S.S., Chen N., Cumaranatunge L., Blau G.E., Zemlyanov D.Y., Delgass W.N., Epling W.S., Ribeiro F.H. // J. Catal. 2006. V. 241. P. 389.
Boubnov A., Dahl S., Johnson E., Molina A.P., Simonsen S.B., Cano F.M., Helveg S., Lemus-Yegres L.J., Grunwaldt J.-D. // Appl. Cat. B: Env. 2012. V. 126. P. 315.
Benard S., Retailleau L., Gaillard F., Vernoux P., Giroir-Fendler A. // Appl. Catal. B: Env. 2005. V. 55 P. 11.
Mirkelamoglu B., Liu M., Ozkan U.S. // Catal. Today. 2010. V. 151. P. 386.
Калинкин А.В., Сорокин А.М., Смирнов М.Ю., Бухтияров В.И. // Кинетика и катализ. 2014. Т. 55. С. 371.
Смирнов М.Ю., Вовк Е.И., Нартова А.В., Калинкин А.В., Бухтияров В.И. // Кинетика и катализ. 2018. Т. 59. С. 631.
Смирнов М.Ю., Калинкин А.В., Сорокин А.М., Бухтияров В.И. // Кинетика и катализ. 2020. Т. 61. С. 893.
Смирнов М.Ю., Калинкин А.В., Сорокин А.М., Бухтияров В.И. // Кинетика и катализ. 2020. Т. 61. С. 568.
Смирнов М.Ю., Калинкин А.В., Саланов А.Н., Сорокин А.М., Бухтияров В.И. // Кинетика и катализ. 2021. Т. 62. С. 619.
Demidov D.V., Prosvirin I.P., Sorokin A.M., Bukhtiyarov V.I. // Catal. Sci. Technol. 2011. V. 1. P. 1432.
Демидов Д.В., Просвирин И.П., Сорокин А.М., Роша Е., Кноп-Герике А., Бухтияров В.И. // Кинетика и катализ. 2011. Т. 52. С. 877.
Lopez-Salido I., Lim D.C., Kim Y.D. // Surf. Sci. 2005. V. 588. P. 6.
Stable A., Eichhorst-Gerner K., Rabe J.P., Gonzalez-Elipe A.R. // Langmuir. 1998. V. 14. P. 7324.
Yang D.-Q., Sacher E. // Surf. Sci. 2002. V. 504. P. 125.
Rousseau B., Estrade-Szwarckopf H., Thomann A.-L., Brault P. // Appl. Phys. A. 2003. V. 77. P. 591.
Utsumi S., Honda H., Hattori Y., Kanoh H., Takahashi K., Sakai H., Abe M., Yudasaka M., Iijima S., Kaneko K. // J. Phys. Chem. C. 2007. V. 111. P. 5572.
Paredes J.I., Martınez-Alonso A., Tascon J.M.D. // Langmuir. 2007. V. 23. P. 8932.
Demoisson F., Raes M., Terryn H., Guillot J., Migeon H.-N., Reniers F. // Surf. Interface Anal. 2008. V. 40. P. 566.
Sandhu J., Chauhan A.K.S., Govind // J. Nanopart. Res. 2011. V. 13. P. 3503.
Blume R., Rosenthal D., Tessonnier J.-P., Li H., Knop-Gericke A., Schlogl R. // ChemCatChem. 2015. V. 7. P. 2871.
http://xpspeak.software.informer.com/4.1/.
Moulder J.F., Stickle W.F., Sobol P.E., Bomben K.D. Handbook of X-ray Photoelectron Spectroscopy. Eden Prairie: Perkin-Elmer Co, 1992.
Horcas I., Fernandez R., Gomez-Rodriquez J.M., Colchero J., Gomez-Herrero J., Baro A.M. // Rev. Sci. Instrum. 2007. V. 78. P. 013705_1.
Figueiredo J.L., Pereira M.F.R. // Catal. Today. 2010. V. 150. P. 2.
Aktary M., Lee C.E., Xing Y., Bergens S.H., McDermott M.T. // Langmuir. 2000. V. 16. P. 5837.
Lu G., Zangari G. // Electrochimica Acta. 2006. V. 51. P. 2531.
Gao J., Guo Q. // Appl. Surf. Sci. 2012. V. 258. P. 5412.
Favaro M., Agnoli S., Perini L., Durante C., Gennaro A., Granozzi G. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 2923.
Yuan Z., Hanf M.C., Stephan R., Dulot F., Denys E., Florentin A., Harbich W., Wetzel P. // Surf. Interface Anal. 2015. V. 47. P. 82.
Kettner M., Stumm C., Schwarz M., Schuschke C., Libuda J. // Surf. Sci. 2019. V. 679. P. 64.
Fiordaliso E.M., Murphy S., Nielsen R.M., Dahl S., Chorkendorff I. // Surf. Sci. 2012. V. 606. P. 263.
Porsgaard S., Ono L.K., Zeuthen H., Knudsen J., Schnadt J., Merte L.R., Chevallier J., Helveg S., Salmeron M., Wendt S., Besenbacher F. // ChemPhysChem. 2013. V. 14. P. 1553.
Smirnov M.Yu., Kalinkin A.V., Bukhtiyarov A.V., Prosvirin I.P., Bukhtiyarov V.I. // J. Phys. Chem. C. 2016. V. 120. P. 10419.
Schnyder B., Alliata D., Kotz R., Siegenthaler H. // Appl. Surf. Sci. 2001. V. 173. P. 221.
Susi T., Pichler T., Ayala P. // Beilstein J. Nanotechnol. 2015. V. 6. P. 177.
Paredes J.I., Martınez-Alonso A., Tascon J.M.D. // Langmuir. 2007. V. 23. P. 8932.
Afanas’ev V.P., Bocharov G.S., Eletskii A.V., Ridzel O.Yu., Kaplya P.S., Koppen M. // J. Vac. Sci. Technol. B. 2017. V. 35. P. 041804.
Zhu C., Hao X., Liu Y., Wu Y., Wang J. // Appl. Surf. Sci. 2018. V. 427. P. 1137.
Wang Z.-M., Kanoh H., Kaneko K., Lu G.Q., Do D. // Carbon. 2002. V. 40. P. 1231.
Fu C., Zhao G., Zhang H., Li S. // Int. J. Electrochem. Sci. 2013. V. 8. P. 6269.
Saravanan M., Girisun T.C.S., Rao S.V. // J. Mater. Chem. C. 2017. V. 5. P. 9929.
Martínez M.T., Callejas M.A., Benito A.M., Cochet M., Seeger T., Anson A., Schreiber J., Gordon C., Marhic C., Chauvet O., Fierro J.L.G., Maser W.K. // Carbon. 2003. V. 41. P. 2247.
Yang D., Velamakanni A., Bozoklu G., Park S., Stoller M., Piner R.D., Stankovich S., Jung I., Field D.A., Ventrice C.A., Ruoff R.S. // Carbon. 2009. V. 47. P. 145.
Hou S., Su S., Kasner M.L., Shah P., Patel K., Madarang C.J. // Chem. Phys. Lett. 2010. V. 501. P. 68.
Ganguly A., Sharma S., Papakonstantinou P., Hamilton J. // J. Phys. Chem. C. 2011. V. 115. P. 17009.
Stobinski L., Lesiak B., Malolepszy A., Mazurkiewicz M., Mierzwa B., Zemek J., Jiricek P., Bieloshapka I. // J. Electron Spectrosc. Related Phenom. 2014. V. 195. P. 145.
Jena G., Vanithakumari S.C., Thinaharan C., George R.P., Mudali U.K. // J. Bio. Tribol. Corrosion. 2018. V. 4. P. 20.
Shinotsuka H., Tanuma S., Powell C.J., Penn D.R. // Surf. Interf. Anal. 2015. V. 47. P. 871.
Huizinga T., Van t’Blik H.F.J., Vis J.C., Prins R. // Surf. Sci. 1983. V. 135. P. 580
Steinruck H.P., Pesty F., Zhang L., Madey T.E. // Phys. Rev. B. 1995. V. 51. P. 2427.
Holl Y., Krill G., Amamou A., Legare P., Hilaire L., Maire G. // Solid State Commun. 1979. V. 32. P. 1189.
Kaushik V.K. // Z. Phys. Chem. 1991. V. 173. P. 105.
Zafeiratos S., Papakonstantinou G., Jacksic M.M., Neophytides S.G. // J. Catal. 2005. V. 232. P. 127.
Huang C.H., Wang I.K., Lin Y.M., Tseng Y.H., Lu C.M. // J. Mol. Catal. A. 2010. V. 316. P. 163.
Peuckert M., Bonzel H.P. // Surf. Sci. 1984. V. 145. P. 239.
Abe Y., Yanagisawa H., Sasaki K. // Jpn. J. Appl. Phys. 1998. V. 37. P. 4482.
Pitchon V., Fritz A. // J. Catal. 1999. V. 186. P. 64.
Despres J., Elsener M., Koebel M., Krocher O., Schnyder B., Wokaun A. // Appl. Catal. B: Env. 2004. V. 50. P. 73.
Katrib A. // J. Electron Spectrosc. Relat. Phenom. 1980. V. 18. P. 275.
Kalinkin A.V., Smirnov M.Yu., Nizovskii A.I., Bukhtiyarov V.I. // J. Electron Spectrosc. Relat. Phenom. 2010. V. 177. P. 15.
Nosova L.V., Stenin M.V., Nogin Yu.N., Ryndin Yu.A. // Appl. Surf. Sci. 1992. V. 55. P. 43.
Aiyer H.N., Vijayakrishnan V., Subbanna G.N., Rao C.N.R. // Surf. Sci. 1994. V. 313. P. 392.
Oktar N., Mitome J., Holmgreen E.M., Ozkan U.S. // J. Mol. Catal. A. 2006. V. 259 P. 171.
Смирнов М.Ю., Калинкин А.В., Клембовский И.О., Бухтияров В.И. // ЖСХ. 2019. Т. 60. С. 1846.
Dementjev A.P., Ivanov K.E., Tsyvkunova E.A. // Appl. Surf. Sci. 2015. V. 357. P. 1434.
Sjovall P., So S.K., Kasemo B., Franchy R., Ho W. // Chem. Phys. Lett. 1990. V. 172. P. 125.
Oh Y.J., Yoo J.J., Kim Y.I., Yoon J.K., Yoon H.N., Kim J.-H., Park S.B. // Electrochimica Acta. 2014. V. 116. P. 118.
Langley L.A., Villanueva D.E., Fairbrother D.H. // Chem. Mater. 2006. V. 18. P. 169.
Zemlyanov D.Y., Hornung A., Weinberg G., Wild U., Schlogl R. // Langmuir. 1998. V. 14. P. 3242.
Morrow B.A., McFarlane R.A., Moran L.E. // J. Phys. Chem. 1985. V. 89. P. 77.
Huang W., Jiang Z., Jiao J., Tan D., Zhai R., Bao X. // Surf. Sci. 2002. V. 506. P. L287.
Jiang Z., Huang W., Tan D., Zhai R., Bao X. // Surf. Sci. 2006. V. 600. P. 4860.
Svintsitskiy D.A., Kibis L.S., Stadnichenko A.I., Koscheev S.V., Zaikovskii V.I., Boronin A.I. // ChemPhysChem. 2015. V. 16. P. 3318.
Biniak S., Szymanski G., Siedlewski J., Swiatkowski A. // Carbon. 1997. V. 35. P. 1799.
Stanczyk K., Dziembaj R., Piwowarsca Z., Witkowski S. // Carbon. 1995. V. 33. P. 1383.
Pels J.R., Kapteijn F., Moulijn J.A., Zhu Q., Thomas K.M. // Carbon. 1995. V. 33. P. 1641.
Ghosh B., Sarma S., Pontsho M., Ray S.C. // Diamond Relat. Mater. 2018. V. 89. P. 35.
Miki H., Nagase H., Nagase T., Kioka T., Sugai S., Kawasaki K. // Appl. Surf. Sci. 1988. V. 33–34. P. 292.
Sugai S., Watanabe H., Kioka T., Miki H., Kawasaki K. // Surf. Sci. 1991. V. 259. P. 109.
Weststrate C.J., Bakker J.W., Rienks E.D.L., Vinod C.P., Matveev A.V., Gorodetskii V.V., Nieuwenhuys B.E. // J. Cat. 2006. V. 242. P. 184.
Weststrate C.J., Bakker J.W., Rienks E.D.L., Vinod C.P., Lizzit S., Petaccia L., Baraldi A., Nieuwenhuys B.E. // Surf. Sci. 2006. V. 600. P. 1991.
Shimada T., Mun B.S., Nakai I.F., Banno A., Abe H., Iwasawa Y., Ohta T., Kondoh H. // J. Phys. Chem. C. 2010. V. 114. P. 17030.
Toyoshima R., Yoshida M., Monya Y., Suzuki K., Amemiya K., Mase K., Mun B.S., Kondoh H. // Surf. Sci. 2013. V. 615. P. 33.
Baird R.J., Ku R.C., Wynblatt P. // Surf. Sci. 1980. V. 97. P. 346.
Baraldi A., Dhanak V.R., Kiskinova M., Rosei R. // Appl. Surf. Sci. 1994. V. 78. P. 445.
Lizzit S., Baraldi A., Cocco D., Comelli G., Paolucci G., Rosei R., Kiskinova M. // Surf. Sci. 1998. V. 410. P. 228.
Bondino F., Comelli G., Baraldi A., Vesselli E., Rosei R., Goldoni A., Lizzit S. // J. Chem. Phys. 2003. V. 119. P. 12534.
Nakamura I., Kobayashi Y., Hamada H., Fujitani T. // Surf. Sci. 2006. V. 600. P. 3235.
Saito T., Esaka F., Furuya K., Kikuchi T., Imamura M., Matsubayashi N., Shimada H. // J. Electron Spectrosc. Related Phenom. 1998. V. 88-91. P. 763.
Requejo F.G., Hebenstreit E.L.D., Ogletree D.F., Salmeron M. // J. Catal. 2004. V. 226. P. 83.
Kostecki R., Schnyder B., Alliata D., Song X., Kinoshita K., Kotz R. // Thin Solid Films. 2001. V. 396. P. 36.
Wickham D.T., Banse B.A., Koel B.E. // Surf. Sci. 1991. V. 243. P. 83.
Jirsak T., Dvorak J., Rodriguez J.A. // Surf. Sci. 1999. V. 436. P. L683.
Desikusumastuti A., Happel M., Qin Z., Staudt T., Lykhach Y., Laurin M., Shaikhutdinov S., Rohr F., Libuda J. // J. Phys. Chem. C. 2009. V. 113. P. 9755.
Uy D., O’Neill A.E., Weber W.H. // Appl. Catal. B: Env. 2002. V. 35. P. 219.
Parker D.H., Koel B.E. // J. Vac. Sci. Technol. A. 1990. V. 8. P. 2585.
Banse B.A., Koel B.E. // Surf. Sci. 1990. V. 232. P. 275.
Zheng G., Altman E.I. // Surf. Sci. 2000. V. 462. P. 151.
Peterlinz K.A., Sibener S.J. // J. Phys. Chem. 1995. V. 99. P. 2817.
Conrad H., Ertl G., Kuppers J., Latta E. // Surf. Sci. 1977. V. 65. P. 245.
Heras J.M., Estiu G., Viscido L. // Thin Solid Films. 1990. V. 188. P. 165.
Stara I., Nehasil V., Matolin V. // Surf. Sci. 1995. V. 331–333. P. 173.
Leisenberger F.P., Koller G., Sock M., Surnev S., Ramsey M.G., Netzer F.P., Klotzer B., Hayek K. // Surf. Sci. 2000. V. 445. P. 380.
Zemlyanov D., Azalos-Kiss B., Kleimenov E., Teschner D., Zafeiratos S., Havecker M., Knop-Gericke A., Schlogl R., Gabasch H., Unterberger W., Hayek K., Klotzer B. // Surf. Sci. 2006. V. 600. P. 983.
Gabasch H., Unterberger W., Hayek K., Klotzer B., Kleimenov E., Teschner D., Zafeiratos S., Havecker M., Knop-Gericke A., Schlogl R., Han J., Ribeiro F.H., Aszalos-Kiss B., Curtin T., Zemlyanov D. // Surf. Sci. 2006. V. 600. P. 2980.
Han J., Zemlyanov D.Y., Ribeiro F.H. // Surf. Sci. 2006. V. 600. P. 2752.
Nagarajan S., Thirunavukkarasu K., Gopinath C.S. // J. Phys. Chem. C. 2009. V. 113. P. 7385.
Супрун Е.А., Саланов А.Н. // Кинетика и катализ. 2017. Т. 58. С. 98.
Titkov A.I., Salanov A.N., Koscheev S.V., Boronin A.I. // React. Kinet. Catal. Lett. 2005. V. 86. P. 371.
Titkov A.I., Salanov A.N., Koscheev S.V., Boronin A.I. // Surf. Sci. 2006. V. 600. P. 4119.
Wider J., Greber T., Wetli E., Kreutz T.J., Schwaller P., Osterwalder J. // Surf. Sci. 1998. V. 417. P. 301.
Monine M.I., Schaak A., Rubinstein B.Y., Imbihl R., Pismen L.M. // Catal. Today. 2001. V. 70. P. 321.
Gibson K.D., Killelea D.R., Sibener S.J. // J. Phys. Chem. C. 2014. V. 118. P. 14977.
Farber R.G., Turano M.E., Oskorep E.C.N., Wands N.T., Juurlink L.B.F., Killelea D.R. // J. Phys.: Condens. Matter. 2017. V. 29. P. 164002.
German E.D., Sheintuch M., Kuznetsov A.M. // J. Phys. Chem. C. 2009. V. 113. P. 15326.
Campbell C.T., White J.M. // Appl. Surf. Sci. 1978. V. 1. P. 347.
Root T.W., Schmidt L.D., Fisher G.B. // Surf. Sci. 1983. V. 134. P. 30
Schmatloch V., Kruse N. // Surf. Sci. 1992. V. 269/270. P. 488.
Morales C., Diaz-Fernandez D., Mossanek R.J.O., Abbate M., Mendez J., Perez-Dieste V., Escudero C., Rubio-Zuazo J., Prieto P., Soriano L. // Appl. Surf. Sci. 2020. V. 509. P. 145118.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ