

Коллоидный журнал, 2019, T. 81, № 1, стр. 86-100
Зародышеобразование конденсированной фазы в парах воды на наноструктурированной поверхности кристалла β-AgI. 1. Пространственная организация
С. В. Шевкунов *
Санкт-Петербургский политехнический университет Петра Великого
195251 Санкт-Петербург, ул. Политехническая, 29, Россия
* E-mail: shevk54@mail.ru
Поступила в редакцию 15.06.2018
Аннотация
Методом Монте-Карло выполнено компьютерное моделирование зародышей конденсированной фазы воды на наноструктурированной базовой грани кристалла йодистого серебра при температуре 260 и 400 K. Сравнение рассчитанных пространственных корреляционных функций молекул на наноструктурированной и гладкой подложках свидетельствует о разрушительном действии электрического поля наноструктуры на молекулярные кластеры. Удержание молекул пара на наноструктурированной поверхности на начальной стадии поглощения происходит без образования между молекулами связей, при этом прочность сцепления с подложкой первых поглощенных молекул существенно выше, чем с гладкой поверхностью, где в их удержании существенна роль межмолекулярных взаимодействий. Электрическое поле наноструктурированной поверхности оказывает разрушительное действие на водородные связи между молекулами. Температурная зависимость количества водородных связей демонстрирует обратный ход, при котором повышение температуры в некоторой области условий сопровождается не разрушением, а, наоборот, восстановлением части связей. Предложена упрощенная модель, объясняющая этот эффект.
1. ВВЕДЕНИЕ
Формирование поверхностей с заданными гидрофильными свойствами, влияющих на микроконденсацию, представляет интерес для решения множества практических задач, от борьбы с обледенением [1, 2] и обеспечения прозрачности оптических стекол [3] до стимулирования нуклеации пересыщенных паров воды в качестве инструмента управления атмосферными осадками [4, 5].
В камере Вильсона [6] заряженные частицы вызывают образование капель тумана. Относительно быстро движущаяся частица успевает покинуть индуцированное ее полем скопление молекул пара, а образованные ею ионы успевают рекомбинировать до распада молекулярного сгустка. Если размер сформировавшегося таким образом гомогенного кластера превышает критический размер зародыша, он самостоятельно растет до макроскопических размеров. Выросшие капли тумана выстраиваются в треки и рассеивают свет, делая траектории заряженных частиц видимыми в оптическом диапазоне. Конденсация пара обеспечивается здесь относительно быстрым движением заряженной частицы так, что последняя “подталкивает” образование капли и успевает вовремя покинуть растущий сгусток, не препятствуя его дальнейшему росту, а сильное пересыщение пара, получаемое быстрым адиабатическим расширением, позволяет работать в области малых критических размеров зародышей.
В отличие от быстро движущихся частиц в камере Вильсона молекулярные ионы, образующиеся в атмосфере на фоне естественного ионизирующего излучения, вовлечены в относительно медленное тепловое движение. Частицы атомарных размеров хотя и удерживают в своем электрическом поле от нескольких до нескольких десятков молекул пара [7–15], не способны запустить механизм лавинообразного роста микрокапель до макроскопических размеров [16]. Причина в том, что их сильно неоднородное и локализованное в слишком узкой области электрическое поле препятствует неограниченному росту гидратных оболочек, а состояния относительно слабо пересыщенного пара в естественных условиях делают критический размер слишком большим [17] по сравнению с размером устойчивых гидратных оболочек таких частиц. В результате образуются ион-гидратные комплексы, устойчивые как к распаду, так и росту. Хотя кластерные ионы всегда присутствуют в воздушной среде в концентрации от тысяч до сотен тысяч частиц на кубический сантиметр [18], они из-за своих экстремально малых размеров не могут служить центрами гетерогенной нуклеации. Это позволяет переохлажденным парам воды даже при наличии в них электрических зарядов в течение продолжительного времени пребывать в условиях естественной воздушной среды в метастабильных состояниях и не конденсироваться.
Чтобы обеспечить переход нуклеации в лавинообразную конденсацию, центры зародышеобразования должны быть достаточно крупными [19–21]. В то же время, с увеличением размера таких частиц уменьшается площадь их поверхности в расчете на единицу объема, а вместе с этим показателем падает удельная адсорбционная способность материала и его эффективность в качестве агента нуклеации. Поскольку суммарная площадь поверхности частиц растет при дроблении кристаллического образца обратно пропорционально линейным размерам частиц, разделение макроскопического кристаллического образца с линейными размерами порядка 0.1 м на наночастицы с линейными размерами порядка 100 нм сопровождается увеличением активной поверхности системы в миллион раз. Оптимальными являются размеры, измеряемые десятками нанометров, однако столь мелкодисперсный аэрозоль приготовить в больших количествах технически сложно. Компромиссное решение состоит в применении аэрозольных частиц с размерами, измеряемыми микронами, но с наноскопическими кристаллическими дефектами на их поверхности. Дефекты образуются естественным образом в процессе синтеза аэрозоля, а их характеристики можно контролировать, варьируя условия приготовления. Поверхность становится наноструктурированной при наличии множественных дефектов.
По всей видимости, высокая активность аэрозоля в качестве агента нуклеации не в последнюю очередь определяется способностью частиц удержать на своей поверхности зародыши конденсированной фазы на начальной стадии их роста, еще до замерзания. Поверхностные дефекты могут играть здесь решающую роль. Механизм их воздействия на адсорбционную способность поверхности требует специального изучения [22, 23]. Задачу целесообразно разделить на две части: исследование влияния дефектов на структуру зародышей и их роли в энергетике зародышеобразования.
Сильная зависимость адсорбционной способности поверхностей от температуры указывает на решающую роль термических флуктуаций, делая неизбежным применение к таким системам методов статистической механики. Наноскопические размеры поверхностных дефектов не позволяют использовать приближения теории капиллярности [24, 25]. Дискретная, молекулярная природа адсорбируемого на поверхности вещества должна учитываться здесь в явной форме. Попытки найти какие-либо универсальные закономерности для наноструктурированных поверхностей в приближении теории сплошных сред [26–28] сталкиваются с существенными трудностями из-за сильной зависимости механизма сцепления от пространственных характеристик поверхностных дефектов и молекулярной структуры связей, ответственных за удерживание адсорбированного вещества на поверхности. Данные, полученные компьютерным моделированием [29], указывают на то, что наноструктурированность поверхности может как усиливать, так и уменьшать ее смачиваемость в зависимости от конкретных характеристик межмолекулярных сил.
В первом приближении различают два предельных случая контакта между крупными каплями и шероховатой поверхностью: контакт, при котором молекулы жидкости проникают в полости на структурированной поверхности, полностью их заполняя (смачивание по Венцелю [30]), и контакт, при котором молекулы капли в полости не проникают, и полости заполняются паром (смачивание по Касси−Бакстеру [31, 32]). В обоих случаях учет влияния структурированности поверхности на угол смачивания производится с помощью поправочного множителя к косинусу угла смачивания, представляющего собой увеличение площади поверхности за счет ее шероховатости. В случае зародышей, размеры которых сравнимы с размерами наноструктурных элементов и молекулярными размерами, приходится иметь дело с частичным заполнением полостей, а понятие площади контакта с поверхностью становится неопределенным, так что представляется невозможным отнести такой контакт к первому или второму предельному случаю.
Количественное описание смачиваемости в терминах угла смачивания и поверхностного натяжения теории капиллярности имеет смысл только для капель, размеры которых велики по сравнению с характерными размерами наноструктурных элементов поверхности. Такое описание невозможно на наиболее важной начальной стадии зародышеобразования. В условиях послойной адсорбции толщина молекулярной пленки, покрывающей поверхность, может быть сравнима с толщиной переходного слоя на межфазной границе, делая бессмысленными понятие поверхности раздела, а вместе с ним понятия угла смачивания и поверхностного натяжения и уравнение Юнга, связывающее эти характеристики [33].
В данной статье представлены полученные статистико-механическим моделированием результаты, касающиеся структуры конденсата воды на наноструктурированной поверхности наиболее активного из известных материалов, применяемых в качестве стимулятора нуклеации пара.
2. УСЛОВИЯ МОДЕЛИРОВАНИЯ
2.1. Рельеф поверхности
В представленной работе, в отличие от предыдущих исследований, изучается влияние относительно крупных элементов наноструктурированной поверхности базовой грани кристалла β-AgI на пространственную организацию зародышей конденсированной фазы воды. Каждый структурный элемент представляет собой правильную пирамиду, составленную из 6 кристаллографических слоев, параллельных базовой грани: 3-х слоев анионов йода размерами 20 × 20, 16 × 16, 12 × × 12 ионов и 3-х слоев катионов серебра таких же размеров. Слои анионов йода чередуются со слоями катионов серебра. Ионы, составляющиe пирамидальный элемент на поверхности кристалла, жестко фиксированы в тех позициях, в которых они находились бы в составе объемного кристаллического образца. Релаксационные смещения положения ионов на несущей поверхности и в пирамиде не производились. Предварительные расчеты показали, и данные моделирования, полученные независимыми авторами, подтверждают [34], что гладкая базовая грань с катионами серебра в поверхностном слое обладает большей адсорбционной способностью по отношению к парам воды, чем грань с анионами йода. Поэтому в данной работе моделируется базовая грань, несущая в поверхностном слое именно катионы серебра.
2.2. Область моделирования
Моделирование выполнено методом Монте-Карло в каноническом статистическом ансамбле [35] с применением периодических граничных условий [36], так что пирамидальный элемент регулярным образом повторялся в плоскости базовой грани, а поверхность подложки в целом представляла собой наноструктурированную поверхность бесконечных размеров. Размер ячейки периодичности составлял 95.19 Å × 109.92 Å (24 × × 24 иона). Область моделирования в плоскости адсорбции задавалась размером ячейки периодичности, а в перпендикулярном направлении ограничивалась двумя параллельными плоскостями – одной на 3.5 Å ниже ионов первого кристаллографического слоя несущей поверхности под основанием пирамиды и другой на 10 Å выше ионов поверхностного кристаллографического слоя, находящегося на ее вершине. Положение верхней ограничивающей плоскости выбрано с таким расчетом, чтобы при плотности насыщающего пара среднее количество его молекул в области моделирования над адсорбирующей поверхностью было пренебрежимо мало по сравнению с адсорбированной массой. Таким образом, все находящиеся в области моделирования молекулы можно было считать принадлежащими к адсорбированной компоненте, и в то же время прямой контакт адсорбированной компоненты с этой плоскостью отсутствовал.
2.3. Межмолекулярные взаимодействия и статистические характеристики
Короткодействующая часть взаимодействий в системе рассчитывалась методом ближайшего образа [36]. В явном виде учитывались взаимодействия молекул с шестью кристаллографическими слоями ниже несущей поверхности. Из соображений преемственности применена модель межмолекулярных взаимодействий, предложенная в [37] и развитая в [38–43]. Модель включает электростатические, дисперсионные и непарные поляризационные взаимодействия, обмен и водородные связи. По сравнению с моделью [37] она дополнена учетом поляризационных и дисперсионных взаимодействий на дальних расстояниях. Дальние электростатические и поляризационные взаимодействия с ионами бесконечной в плоскости базовой грани подложки и адсорбированными молекулами суммировались двумерным методом Эвальда, представляющим собой разложение электростатического поля в ряд Фурье в длинноволновой области [41]. В виде поправок учитывались и поляризационные взаимодействия за пределами ячейки периодичности [40]. Детальное описание модели с численными значениями параметров представлено в [40–42].
Для расчета равновесных средних генерировались случайные последовательности молекулярных конфигураций длиной в 15 млн шагов, из которых первые 3 млн использовались для термализации. Применялась процедура последовательного наращивания количества частиц в пределах ячейки периодичности от сеанса к сеансу моделирования. Максимальный сдвиг молекул на каждом шаге составлял 0.25 Å, а угол поворота 18°. Частота принятия новых конфигураций изменялась в пределах 50−60%.
2.4. Кристаллическая структура подложки
Кристаллическая структура регулярно повторяющегося пирамидального элемента, лежащего в основе рельефа наноструктурированной поверхности, определяется кристаллической решеткой β-AgI, устойчивой при нормальном давлении при температуре ниже 147°C [43]. Решетка β-AgI обладает осью симметрии шестого порядка и представляет собой две гексагональные подрешетки анионов йода и катионов серебра, вставленные одна в другую со сдвигом вдоль оси симметрии на 4.684 Å (рис. 1а). В направлении оси симметрии слои ионов йода чередуются со слоями ионов серебра. Период каждой из подрешеток в направлении оси симметрии в основном состоянии составляет 7.494 Å [44], а слои подрешетки с относительным параллельным сдвигом в плоскости базовой грани располагаются на расстоянии 7.494/2 = 3.747 Å друг от друга. Таким образом, минимальное расстояние между двумя соседними слоями ионов йода и ионов серебра оказывается относительно малым и составляет 4.684 Å – 3.747 Å = = 0.937 Å (рис. 1а). При этом ионы соседних слоев располагаются не напротив друг друга, а в промежутках между ионами противоположного слоя, рис. 1б, 1в. Это позволяет атомам водорода молекул воды проникать к анионам второго кристаллографического слоя между ионами первого слоя и образовывать с ними относительно прочные водородные связи. За счет этих связей и удерживается мономолекулярная пленка воды на бездефектной поверхности базовой грани кристалла.
Рис. 1.
Внешний вид кристаллической решетки β-AgI. (а) Фрагмент элементарной ячейки, демонстрирующий относительное положение подрешеток катионов Ag+ и анионов I– (межионные расстояния указаны в ангстремах); (б) плоский фрагмент толщиной в шесть кристаллографических слоев, параллельных базовой грани, размером 5 × 5 узлов каждый; (в) вид на тот же фрагмент под острым углом к базовой грани.
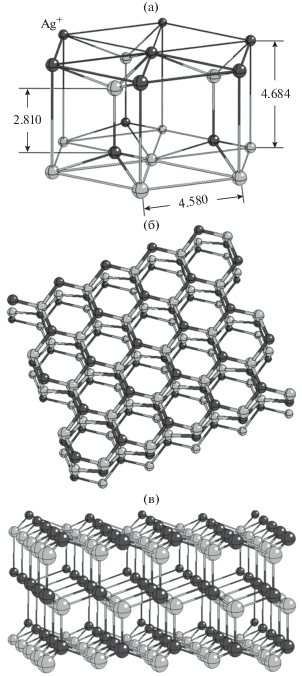
Со стороны базовой грани, а значит, на несущей поверхности и на верхней площадке пирамидального элемента кристаллическая решетка β-AgI демонстрирует выраженную гексагональную ячеистую структуру с пространственными параметрами, близкими к параметрам кристаллической решетки льда Ih, рис. 2а. В то же время на сколах со стороны боковых граней элементарной ячейки гексагональная структура на поверхности себя не проявляет, рис. 2б, 2в. В зависимости от того, в каком направлении сделан боковой скол (см. горизонтальные и вертикальные пунктирные направляющие на рис. 2а), расположение ионов на поверхности оказывается различным, однако общим для боковых сколов является линейное расположение ионов: ионы одного знака заряда выстраиваются на боковой поверхности в цепочки, параллельные базовой грани. Цепочки ионов одного знака заряда чередуются с цепочками ионов противоположного знака, рис. 2б, 2в. Очевидно, при таком расположении зарядов силовые линии электрического поля направлены поперек цепочек ионов, вдоль оси вращательной симметрии кристаллической решетки – от вершины к основанию пирамиды. Этому же направлению силовых линий способствует и схожесть поля пирамиды с полем плоского конденсатора: роль положительно заряженной пластины на вершине играет поверхностный кристаллографический слой катионов серебра, а роль отрицательно заряженной пластины в основании – слой анионов йода.
Рис. 2.
Тот же фрагмент, что и на рис. 1: вид в направлении, перпендикулярном базовой грани (а), а также вдоль базовой грани в направлении вертикальных (б) и горизонтальных пунктирных линий (в) на рисунке (а).
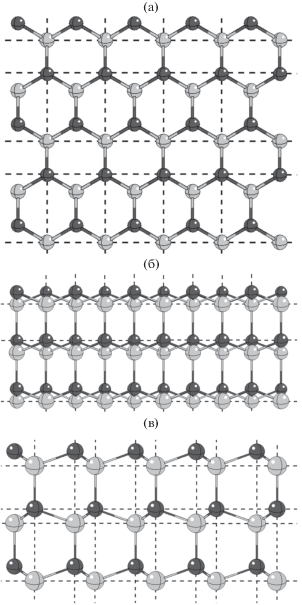
Следует ожидать, что электрическое поле пирамидального элемента может оказать существенное влияние на пространственную организацию адсорбированного вещества. Количественная оценка этого влияния произведена компьютерным моделированием.
3. РЕЗУЛЬТАТЫ РАСЧЕТОВ
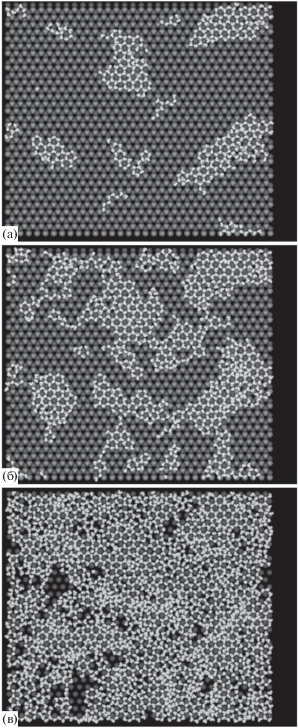
На рис. 3 представлены компьютерные изображения поверхности кристалла на различных стадиях адсорбции пара при температуре 260 K. Поглощение молекул в поле кристаллического дефекта начинается в парах воды с плотностью на 5−6 порядков меньшей, чем плотность насыщенных паров над плоской поверхностью раздела. Как можно видеть на рис. 3а, 3б, первые молекулы располагаются по краям слоев, составляющих пирамидальную конструкцию, причем нижние слои оказываются более предпочтительными. Присоединение молекул к кристаллу происходит в наиболее благоприятных местах, точечно. Относительно большое расстояние между точками присоединения не позволяет образоваться связям между соседними молекулами, чтобы инициировать кластеризацию. На начальной стадии молекулы удерживаются на поверхности исключительно за счет прямых взаимодействий с кристаллом.
Рис. 3.
Компьютерные изображения наноструктурированной поверхности на различных стадиях адсорбции молекул пара при температуре 260 K. Представлен элемент регулярной структуры поверхности в форме усеченной пирамиды высотой в шесть кристаллографических слоев с различным числом адсорбированных молекул $N$ в пределах ячейки периодичности наноскопической структуры размером 95.19 Å × 109.92 Å: 83 (а, б), 307 (в, г), 701 (д ), 2152 (е).

С ростом плотности пара количество его захваченных молекул обратимым образом увеличивается. Присоединение молекул воды по периметру пирамиды осуществляется неравномерно. Захват молекул ступенями пирамиды происходит, прежде всего, на краях, заканчивающихся ионами серебра. Наиболее отчетливо это видно при взгляде сверху, на рис. 3г: на этой стадии на краях с ионами серебра молекулы воды почти полностью покрывают соответствующую террасу, в то время как на противоположной стороне, у края с ионами йода молекулы отсутствуют. На двух других (боковых) краях этого уступа одиночные молекулы воды прочно удерживаются в своих точках присоединения с шагом, определяемым периодом кристаллической решетки, однако “промежуточные” молекулы здесь не присоединяются.
С ростом плотности пара все террасы заполняются молекулами воды, рис. 3д, и только после этого адсорбция происходит на вершине пирамиды и несущей поверхности около ее основания, рис. 3е. Таким образом, адсорбция в присутствие дефектов проходит несколько стадий: сначала захват молекул воды происходит на уступах, затем молекулами покрывается вершина пирамиды и область вокруг основания.
Ступенчатый механизм адсорбции в поле кристаллического дефекта устойчив к нагреву до температуры, превышающей точку кипения воды. Адсорбат сохраняет термодинамическую устойчивость и при температуре 400 K, хотя для удержания молекул с той же поверхностной плотностью требуется плотность пара, на порядки более высокая, чем при 260 K. На компьютерных изображениях, полученных при повышенной температуре (рис. 4), наблюдается характерное асимметричное распределение молекул по краям террас, составляющих пирамиду, при этом надо иметь в виду, что изображения на рис. 4 получены в другом ракурсе – относительно изображения на рис. 3 картинка повернута в плоскости адсорбции на 90°. Как и в условиях низкой температуры, наблюдается повышенная адсорбционная способность воды на ступенях и селективность в отношении различных краев одного и того же кристаллографического слоя. В то же время, повышение температуры приводит к заметному нарушению строгого порядка в расположении первых захваченных молекул. При одном и том же общем количестве адсорбированных молекул воды первые молекулы на вершине пирамиды появляются при повышенной температуре несколько раньше, чем в условиях низкой температуры − сравните рис. 3б−3г и 4б−4г. Следует констатировать, что повышение температуры не меняет ступенчатый механизм адсорбции качественно, но делает различия между ее стадиями менее контрастными.
Рис. 4.
То же, что на рис. 3, при температуре 400 K и различном числе адсорбированных молекул $N$: 83 (а), 307 (б, в), 701 (г), 1622 (д).

Чтобы оценить степень влияния поверхностного дефекта на механизм поглощения пара, на рис. 5 представлены компьютерные изображения молекул воды, адсорбированных на гладкой поверхности β-AgI. В отсутствие поверхностных дефектов структура адсорбата качественно иная: с повышением давления пара над подложкой на ней образуются пятна мономолекулярной пленки, которые растут и сливаются. Следующий слой формируется не раньше, чем вся поверхность кристалла покроется сплошным мономолекулярным слоем воды. Слой имеет гексагональную структуру, напоминающую пчелиные соты. Детальное описание структуры слоя и механизм его удерживания на поверхности представлены в [45, 46].
Рис. 5.
То же, что на рис. 3, для бездефектной поверхности. Число молекул в пределах ячейки периодичности в плоскости адсорбции размером 126.92 Å × × 146.56 Å: 403 (а), 925 (б), 2152 (в).
Особенности сценария поглощения на поверхности с дефектами приводят к низкой степени кластеризации молекул. Если на гладкой поверхности двумерные кластеры в виде пятен мономолекулярной пленки образуются на самой ранней стадии адсорбции, и молекулы удерживаются за счет не только прямых взаимодействий с кристаллом, но и межмолекулярных взаимодействий, то в поле дефекта образование кластеров с резко очерченными границами на ранней стадии адсорбции заторможено, что неизбежно отражается на термодинамическом поведении системы.
Влияние наноструктурированности поверхности на кластеризацию можно оценить из сравнения координационных чисел молекул, полученных для гладкой (рис. 6а) и наноструктурированной (рис. 6б) поверхностей. Представленные на рис. 6 координационные числа имеют смысл среднего количества молекул воды, расположенных в пределах сферы радиуса $r$, описанной вокруг атома кислорода произвольно выбранной молекулы. Ступенчатая форма полученных кривых свидетельствует о выраженной слоистой структуре гидратных оболочек молекул. Кривые, полученные для наноструктурированной поверхности (рис. 6б), отличаются от аналогичных кривых для гладкой поверхности нулевыми значениями в интервале расстояний 2.5 Å < $r$ < 4.0 Å. Этот интервал расстояний отвечает образованию прямой водородной связи с ближайшим соседом и для наноструктурированной поверхности на начальной стадии адсорбции пара остается полностью свободным от молекул. На гладкой поверхности (рис. 6а) молекулы на этих расстояниях появляются прежде, чем на других расстояниях.
Рис. 6.
Координационные числа молекул воды, адсорбированных на гладкой (а) и наноструктурированной (б) базовых гранях кристалла β-AgI при температуре 260 K. Среднее число молекул $\left\langle N \right\rangle $ в пределах ячейки периодичности: 1 – 5.7, 2 – 9.6, 3 – 20.4 (а) и 1 – 5.1, 2 – 9.4, 3 – 20.5 (б).
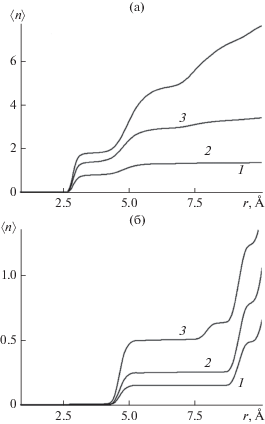
Из приведенных зависимостей следует, что на гладкой поверхности уже при экстремально низкой поверхностной плотности адсорбированные молекулы формируют димеры, которые с ростом давления пара над поверхностью последовательно превращаются в тримеры и более крупные кластеры. На наноструктурированной поверхности димеры на ранней стадии не образуются. Как можно видеть на рис. 3а и 4а, молекулы захватываются подложкой в точках присоединения к боковой поверхности пирамиды, оставаясь на расстояниях, превышающих 4 Å.
При сопоставлении данных, представленных на рис. 6, следует обратить внимание на следующее обстоятельство. Зависимости на обоих рисунках получены при почти одинаковых значениях усредненной поверхностной плотности молекул, при этом абсолютные значения координационного числа на них сильно различаются (обратите внимание на масштаб по вертикальной оси). Значения, отвечающие наноструктурированной поверхности, почти на порядок ниже соответствующих значений для гладкой поверхности. В обоих случаях зависимости на больших расстояниях, за пределами диапазона, представленного на рисунках, выходят на одни и те же предельные значения, определяемые средней плотностью молекул, а пониженные значения на малых расстояниях означают отсутствие кластеров. Следует констатировать, что, разрывая водородные связи между соседними молекулами, поле кристаллических дефектов разрушает молекулярные кластеры и таким образом тормозит образование зародышей конденсированной фазы. Конденсация получает развитие только при более высокой поверхностной плотности молекул − как на рис. 3д и 4д. Таким образом, хотя поглощение молекул наноструктурированной поверхностью начинается при гораздо более низких давлениях, нуклеация двумерного газа молекул на поверхности подложки как начальная стадия формирования конденсированной фазы оказывается здесь сильно заторможенной.
Детальное представление о механизме разрыва водородных связей между молекулами на поверхности с кристаллическими дефектами дают пространственные корреляционные функции кислород−водород молекул воды ${{{\rho }}_{{{\text{OH}}}}}(r)$ на рис. 7. Функции имеют смысл объемной плотности атомов кислорода других молекул на расстоянии r от атома водорода произвольно выбранной молекулы и нормированы следующим образом: $\int_0^R {{{\rho }_{{{\text{OH}}}}}(r)4\pi {{r}^{2}}dr} = $ $\left\langle {{{n}_{{{\text{OH}}}}}(R)} \right\rangle ,$ где $\left\langle {{{n}_{{{\text{OH}}}}}(R)} \right\rangle $ − усредненное по тепловым флуктуациям количество атомов кислорода других молекул, заключенных внутри сферы радиуса $R$, описанной вокруг одного из атомов водорода произвольно выбранной исходной молекулы. Первый максимум на зависимости ${{\rho }_{{{\text{OH}}}}}(r)$ отвечает водородной связи между молекулами. Его положение соответствует длине связи, а ширина максимума позволяет оценить величину ее флуктуаций. Площадь под максимумом $\int_0^{{{R}_{{\min }}}} {{{\rho }_{{{\text{OH}}}}}(r)4\pi {{r}^{2}}dr} ,$ где ${{R}_{{\min }}}$ − положение первого минимума, дает оценку среднего количества водородных связей в расчете на один атом водорода. Следующий максимум соответствует расстоянию до атома кислорода другой молекулы − удерживаемой вторым атомом водорода.
Рис. 7.
Пространственные бинарные корреляционные функции кислород–водород молекул воды, адсорбированных на бездефектной (а) и наноструктурированной (б, в) базовых гранях кристалла β-AgI при температуре 260 K (а, б) и 400 K (в) на различных стадиях адсорбции. Среднее число молекул $\left\langle N \right\rangle $ в пределах ячейки периодичности: 1 – 4.6, 2 – 7.6, 3 – 8.5, 4 – 25.6 (а); 1 – 4.1, 2 – 7.4, 3 – 8.2, 4 – 25.6 (б); 1 – 4.6, 2 – 7.4, 3 – 8.6, 4 – 25.5 (в).
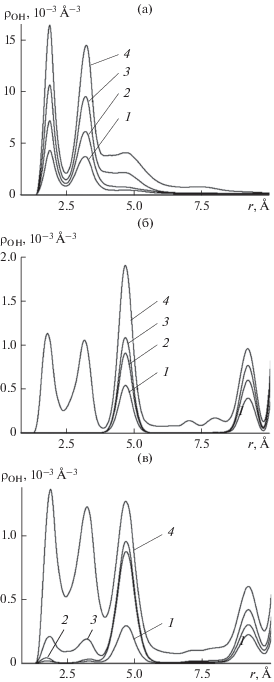
Как следует из зависимостей, представленных на рис. 7а, длина водородной связи на гладкой подложке (положение первого минимума) совпадает с длиной водородной связи в объемной жидкой фазе воды (${{r}_{{{\text{OH}}}}} = 2.04\,\,{{\AA}}$) и остается такой же на наноструктурированной поверхности (рис. 7б), причем с увеличением температуры до 400 K заметных изменений в ее длине не наблюдается (рис. 7в). Из сравнения зависимостей на рис.7а и 7б видно, что в полном соответствии с данными на рис. 6 максимум, отвечающий водородной связи, вместе с сопряженным с ним вторым максимумом в случае наноструктурированной поверхности на кривой 4 на порядок ниже, чем в случае гладкой, а на кривых 1−3 для наноструктурированной поверхности они вообще отсутствуют. В меньшей, но все еще значительной степени этот эффект проявляет себя в третьем (r = 4.8 Å) максимуме, который отвечает положению молекулы, связанной с исходной через одного посредника. В области четвертого максимума и дальше он затухает. Указанные соотношения высот максимумов отражают разрыв водородных связей в поле кристаллических дефектов и разрушение молекулярных кластеров.
Неожиданный характер поведения этих максимумов наблюдается при варьировании температуры. Естественно ожидать, что усиление термических флуктуаций с повышением температуры должно сопровождаться разрывом части водородных связей и уменьшением их среднего равновесного количества в системе. Однако в случае наноструктурированной поверхности наблюдается противоположная картина. Из сравнения данных, приведенных на рис. 7б (260 K) и рис. 7в (400 K), следует, что первый и второй максимумы на кривой 4 с нагреванием не понижаются, а наоборот, растут. Более того, если на рис. 7б кривые 1−3 демонстрируют в этой области расстояний нулевые значения, то после повышения температуры (рис. 7в) они отрываются от нулевого уровня, и на них появляются два характерных максимума, первый из которых отвечает водородной связи между молекулами. Таким образом, при нагревании происходит восстановление части водородных связей, разорванных полем кристаллического дефекта.
Чтобы убедиться, что данный эффект не является артефактом ограниченного объема статистики, накапливаемой в компьютерном эксперименте, сравнительные расчеты при 260 и 400 K были многократно повторены в этой области поверхностной плотности для различных начальных конфигураций молекул. В каждом случае был получен тот же результат. На рис. 8 представлены данные, полученные для одной из таких пар сравнительных расчетов, выполненных для более высокой поверхностной плотности молекул, чем для кривой 3 на рис. 7б. Кривая 1 на рис. 8 получена при низкой температуре, а кривая 2 на том же рисунке при повышенной температуре. На первой кривой нет следов образования водородных связей, а на второй в области контактных межмолекулярных расстояний имеется пара характерных максимумов, свидетельствующих о восстановлении части водородных связей при нагревании. Более того, несмотря на то, что кривая 3 на рис. 7б получена при несколько более низкой поверхностной плотности, чем кривая 2 на рис. 8, на последней в области малых межмолекулярных расстояний имеются два характерных максимума, а на первой их нет. Для образования водородных связей повышенная температура оказалась здесь более сильным стимулирующим фактором, чем фактор пониженной плотности, работающий в противоположном направлении.
Рис. 8.
Пространственные бинарные корреляционные функции кислород–водород молекул воды, адсорбированных на наноструктурированной базовой грани кристалла β-AgI, при температуре 260 (1) и 400 K (2). Среднее число молекул воды $\left\langle N \right\rangle $ в пределах ячейки периодичности 9.1 (1) и 9.6 (2).
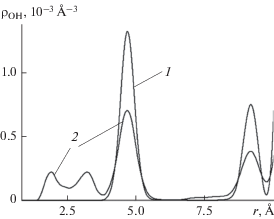
Следует обратить внимание также и на поведение третьего максимума, расположенного в районе r = 4.8 Å. С ростом температуры он существенно понижается, очевидно, вследствие перемещения части молекул с этих расстояний на контактные расстояния, где происходит восстановление водородных связей – часть молекул, захваченных боковыми гранями кристаллического дефекта, высвобождается и перемещается к соседним молекулам, образуя с ними димеры. Таким образом, с повышением температуры степень кластеризации в данном случае не уменьшается, как можно было бы ожидать, а, наоборот, растет.
Эффект обратной температурной зависимости числа водородных связей наблюдается на наноструктурированной поверхности и отсутствует как на гладкой поверхности, так и в условиях без подложки. Причина этого явления лежит в специфической конфигурации электрического поля наноструктурированной поверхности. На поверхности с дефектами существуют два конкурирующих между собой термодинамических состояния: первое характеризуется превалированием связей непосредственно с подложкой, а второе – связей между молекулами. Состояние 1 исключает состояние 2 в силу пространственного расположения точек, наиболее благоприятных для присоединения к поверхности кристаллического дефекта. Соседние точки расположены на слишком больших расстояниях так, что размещение в них молекул сопряжено с разрывом водородных связей между последними. В условиях термодинамического равновесия реализуется сосуществование двух состояний и флуктуационных переходов между ними. Вероятности обнаружить молекулы в одном из этих состояний зависят от свободной энергии каждого из них экспоненциально, $\sim {\kern 1pt} \exp \left( {{{ - F} \mathord{\left/ {\vphantom {{ - F} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right),$ где $F = {{F}_{1}}$ для первого и $F = {{F}_{2}}$ для второго состояния. Если величины внутренней энергии ${{E}_{1}}$ и ${{E}_{2}},$ а также значения энтропии ${{S}_{1}}$ и ${{S}_{2}}$ этих состояний различаются, то какое из них имеет меньшее значение свободной энергии $F = E - TS$ определяется температурой. Если состояние, которое характеризуется превалированием прямых связей с подложкой и разрывом водородных связей, обладает меньшей энергией и, одновременно, меньшей энтропией, то при повышении температуры разница в величинах свободной энергии между состояниями может увеличиваться в пользу более низкого значения для состояния с превалирующими водородными связями. То есть система будет переходить в состояние с более высокой энтропией, и количество водородных связей в системе будет увеличиваться. Детальная иллюстрация этого эффекта на примере простейшей модели, близкой к обсуждаемым условиям, представлена в разделе 5.
Особенности механизма сцепления молекул с подложкой, обусловленные наличием поверхностного кристаллического дефекта, отражаются в форме пространственных корреляций между ионами йода кристаллической решетки и атомами водорода молекул. Соответствующие пространственные корреляционные функции, рассчитанные для гладкой и наноструктурированной поверхностей, представлены на рис. 9. Кривые имеют сложную форму, причем синхронная повторяемость максимумов и минимумов на кривых, полученных независимо, в различных сеансах моделирования, свидетельствует о том, что они не являются следствием статистических погрешностей. Более структурированная на больших расстояниях форма кривых на рис. 9а, по сравнению с кривыми на рис. 9б, отражает более упорядоченное расположение молекул на поверхности без дефектов, рис. 1. На контактных расстояниях кривые для гладкой и наноструктурированной поверхностей имеют схожую форму. Первый максимум на расстоянии r = 2.45 Å свидетельствует об образовании водородной связи между молекулой воды и ионом йода кристаллической решетки.
Рис. 9.
Пространственные бинарные корреляционные функции ион йода кристаллической решетки–атом водорода молекулы воды, полученные для молекул, адсорбированных на гладкой (а) и наноструктурированной (б, в) базовых гранях кристалла β-AgI при температуре 260 K (а, б) и 400 K (в). Среднее число молекул $\left\langle N \right\rangle $ в пределах ячейки периодичности: 1 – 5.5, 2 – 20.5, 3 – 82.5, 4 – 178.3 (а); 1 – 5.2, 2 – 20.8, 3 – 82.8, 4 – 178.5 (б); 1 – 5.6, 2 – 20.5, 3 – 82.4, 4 – 178.4 (в).
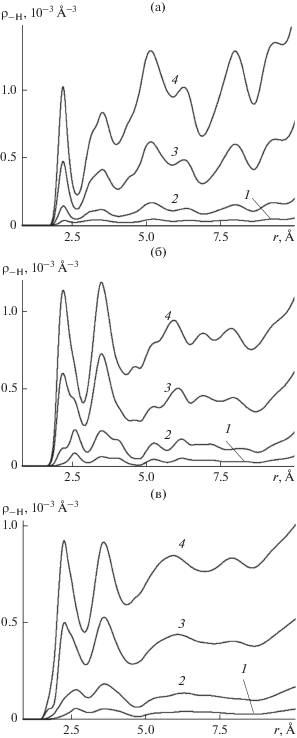
На поверхности с ионами серебра в первом кристаллографическом слое атомы водорода молекул воды проникают между ионами серебра к ионам йода и устанавливают с ними связи, которые играют основную роль в удержании молекул на подложке. Наличие первого одинаково мощного максимума на кривых рис. 9а и 9б свидетельствует о схожих механизмах удерживания молекул в области средних значений поверхностной плотности. Существенное различие наблюдается в области экстремально низких поверхностных плотностей: первый максимум на кривых 1, 2 рис. 9а располагается на расстоянии r = 2.45 Å и свидетельствует об образовании водородной связи с ионами йода, в то время как на кривых 1, 2 рис. 9б первый максимум располагается на большем расстоянии − правее точки 2.5 Å. Другое расположение этого максимума свидетельствует о том, что в области экстремально низких плотностей удерживание молекул в присутствии кристаллических дефектов происходит не только за счет водородных связей с ионами йода. Максимум формируется благодаря молекулам, удерживаемым в точках, наиболее благоприятных для присоединения к боковым поверхностям кристаллического дефекта, как это можно видеть на рис. 3а, 3б. Консолидирующая роль ионов серебра здесь гораздо более существенна.
Тот же эффект можно наблюдать и при повышенной температуре: на рис. 9в первый максимум на кривых 1, 2 расположен на расстояниях, превышающих длину водородной связи между молекулой воды и ионом йода. Только на следующих кривых 3, 4 появляется мощный максимум на расстоянии r = 2.45 Å, отвечающий за водородную связь молекулы с ионом. Вклад в этот максимум дают молекулы воды, покрывающие фрагменты базовой грани – вершину пирамиды, а также горизонтальные поверхности между пирамидами и на их террасах, как на рис. 4г, 4д.
4. ЯЧЕЕЧНАЯ МОДЕЛЬ
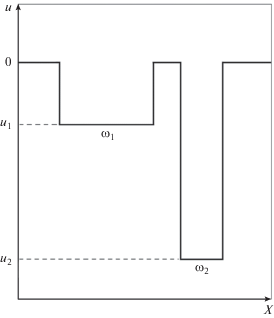
Уяснить главные факторы, являющиеся причиной эффекта обратной температурной зависимости образования водородных связей между молекулами воды, помогает простейшая модель, представленная ниже. Рассмотрим модельную систему $N$ сферических взаимодействующих одинаковых частиц, которая состоит из $M = {N \mathord{\left/ {\vphantom {N 2}} \right. \kern-0em} 2}$ одинаковых ячеек, заключающих в себе по две такие частицы, рис. 10. При этом взаимодействия между частицами разных ячеек отсутствуют, а взаимодействие между частицами внутри ячейки сводится к трем состояниям: в первом из них энергия взаимодействия равна ${{u}_{1}},$ во втором равна ${{u}_{2}},$ а в третьем ${{u}_{3}} = 0.$ Первому состоянию отвечает конфигурационный объем ${{\omega }_{1}},$ а второму ${{\omega }_{2}},$ имеющие размерность длины в шестой степени. Остальной объем $\omega $ относится к третьему состоянию. Схематично эту ситуацию иллюстрирует рис. 11, на котором ось X символизирует набор из шести декартовых координат двух частиц.
Рис. 10.
Модельная система, состоящая из одинаковых ячеек, содержащих по паре взаимодействующих частиц. Частицы из разных ячеек не взаимодействуют.

Рис. 11.
Схема энергетической поверхности пары взаимодействующих частиц в пределах одной ячейки модельной системы: ω1 и ω2 – конфигурационные объемы макросостояний, отвечающих энергиям u1 и u2 соответственно.
Статистическая сумма системы $Q(T)$ в отсутствие взаимодействий между ячейками трансформируется в произведение $M$ идентичных статистических сумм $q(T)$ ячеек $Q(T) = {{(q(T))}^{M}}.$ В свою очередь, статистическая сумма каждой ячейки равна
(1)
$\begin{gathered} q(T) = {{(2!)}^{{ - 1}}}\lambda _{0}^{{ - 6}}(\exp ({{ - {{u}_{1}}} \mathord{\left/ {\vphantom {{ - {{u}_{1}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{1}} + \\ + \,\,\exp (\exp ({{ - {{u}_{2}}} \mathord{\left/ {\vphantom {{ - {{u}_{2}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{2}} + \omega ), \\ \end{gathered} $(2)
$\begin{gathered} F(T) = - {{k}_{{\text{B}}}}T\ln (Q(T)) = \\ = - M{{k}_{{\text{B}}}}T\ln \left( {q(T)} \right) = E - TS, \\ \end{gathered} $Статистическая сумма макросостояния 1, отвечающая конфигурационному объему ${{\omega }_{{{\kern 1pt} 1}}}$, равна ${{q}_{1}}(T) = {{(2!)}^{{ - 1}}}\lambda _{0}^{{ - 6}}\left( {\exp ({{ - {{u}_{1}}} \mathord{\left/ {\vphantom {{ - {{u}_{1}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{1}}} \right),$ а соответствующая ей свободная энергия
(3)
$\begin{gathered} {{f}_{1}}(T) = - {{k}_{{\text{B}}}}T\ln ({{q}_{1}}(t)) = \\ = {{u}_{1}} - {{k}_{{\text{B}}}}T\ln \left( {{{{(2!)}}^{{ - 1}}}\lambda _{0}^{{ - 6}}{{\omega }_{1}}} \right), \\ \end{gathered} $Аналогичное выражение справедливо для состояния 2.
Данная система может служить простейшей моделью образования и разрыва водородных связей в неоднородном электростатическом поле поверхности кристалла. Пусть одно из состояний, например, состояние 1 отвечает образованию между молекулами водородной связи, а другое – их разрыву с одновременным захватом молекул электрическим полем кристалла. Предполагается, что структура этого поля такова, что две молекулы, объединенные водородной связью, не вписываются в геометрию поля на поверхности адсорбции: когда молекулы захватываются полем кристалла, водородная связь между молекулами разрушается.
Поскольку частицы различных ячеек не взаимодействуют, флуктуационные переходы внутри ячеек происходят независимо. Вероятность состояния 1 ячейки равна отношению соответствующих статистических сумм:
(4)
$\begin{gathered} {{w}_{1}}(T) = {{{{q}_{1}}(T)} \mathord{\left/ {\vphantom {{{{q}_{1}}(T)} {q(T)}}} \right. \kern-0em} {q(T)}} = \\ = \exp ( - ({{f}_{1}}(T) - {{f(T))} \mathord{\left/ {\vphantom {{f(T))} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}). \\ \end{gathered} $Используя выражение (3) для первого состояния и аналогичное выражение для второго состояния, можно связать вероятность образования водородной связи с геометрическими параметрами соответствующей ей потенциальной ямы:
(5)
$\begin{gathered} {{w}_{{\text{H}}}}(T) = {{w}_{1}}(T) = \\ = \frac{{\exp ({{ - {{u}_{1}}} \mathord{\left/ {\vphantom {{ - {{u}_{1}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{{\text{1}}}}}}{{\exp ({{ - {{u}_{1}}} \mathord{\left/ {\vphantom {{ - {{u}_{1}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{1}} + \exp (\exp ({{ - {{u}_{2}}} \mathord{\left/ {\vphantom {{ - {{u}_{2}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{{\text{2}}}} + {{\omega }_{{\text{3}}}}}}. \\ \end{gathered} $Состояния 2 и 3 отвечают разрыву водородной связи, вероятность которого равна
(6)
$\begin{gathered} {{w}_{{\text{D}}}}(T) = 1 - {{w}_{1}}(T) = \\ = \frac{{\exp ({{ - {{u}_{2}}} \mathord{\left/ {\vphantom {{ - {{u}_{2}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{{\text{2}}}} + {{\omega }_{{\text{3}}}}}}{{\exp ({{ - {{u}_{1}}} \mathord{\left/ {\vphantom {{ - {{u}_{1}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{{\text{1}}}} + \exp (\exp ({{ - {{u}_{2}}} \mathord{\left/ {\vphantom {{ - {{u}_{2}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{{\text{2}}}} + {{\omega }_{{\text{3}}}}}}. \\ \end{gathered} $Отношение вероятностей образования и разрыва водородных связей равно
(7)
${{{{w}_{{\text{H}}}}(T)} \mathord{\left/ {\vphantom {{{{w}_{{\text{H}}}}(T)} {{{w}_{{\text{D}}}}(T)}}} \right. \kern-0em} {{{w}_{{\text{D}}}}(T)}} = \frac{{\exp ({{ - {{u}_{1}}} \mathord{\left/ {\vphantom {{ - {{u}_{1}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{{\text{1}}}}}}{{\exp ( - {{{{u}_{2}}} \mathord{\left/ {\vphantom {{{{u}_{2}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}){{\omega }_{{\text{2}}}} + {{\omega }_{{\text{3}}}}}}.$Проанализируем, как соотношения между параметрами ${{u}_{1}},$ ${{u}_{2}},$ ${{\omega }_{1}},$ ${{\omega }_{2}}$ и ${{\omega }_{{{\kern 1pt} 3}}}$ влияют на температурную зависимость образования водородных связей. Если ${{u}_{1}} > {{u}_{2}},$ ${{\omega }_{{{\kern 1pt} 1}}} > {{\omega }_{{{\kern 1pt} 2}}}$ (водородной связи соответствует менее глубокий, но более широкий минимум энергии), и при этом ${{\omega }_{3}} \ll $ $ \ll \exp ({{ - {{u}_{2}}} \mathord{\left/ {\vphantom {{ - {{u}_{2}}} {{{k}_{{\text{B}}}}T)}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T)}}{{\omega }_{2}},$ слагаемым ${{\omega }_{{{\kern 1pt} 3}}}$ в знаменателе (7) можно пренебречь, отношение экспонент в (7) меньше единицы, а отношение конфигурационных объемов больше единицы. Величина выражения (7) при таком соотношении параметров существенно зависит от температуры, причем при повышении температуры отношение экспонент увеличивается, приближаясь к единице, и выражение (7) в целом растет. Рост (7) означает образование водородных связей.
Таким образом, если минимум, отвечающий ${{\omega }_{{{\kern 1pt} 2}}}$ (захвату молекул полем кристалла), более глубокий, чем минимум, отвечающий водородной связи, но достаточно узкий (связь жесткая), нагревание может вызвать высвобождение молекул из “ловушек” на поверхности кристалла и образование между ними водородных связей. Именно такой (обратный) ход температурной зависимости наблюдается в компьютерном моделировании адсорбции молекул пара на поверхности кристалла β-AgI. Обратный температурный ход обусловлен здесь соотношением между энергиями состояний, а жесткая связь молекул с поверхностью усиливает этот эффект. При ${{\omega }_{{{\kern 1pt} 1}}} < {{\omega }_{{{\kern 1pt} 2}}}$ образование водородных связей имеет ту же температурную направленность, но может быть существенно заторможено.
Понижение поверхностной плотности молекул соответствует в данной модели увеличению размеров ячейки, содержащей пару частиц, что, в свою очередь, соответствует увеличению числа минимумов, отвечающих состоянию 2 внутри ячейки, что равносильно простому увеличению суммарного объема ${{\omega }_{{{\kern 1pt} 2}}}$. При этом величина (7) уменьшается – водородные связи разрушаются. В соответствие с этим, в компьютерном моделировании образование водородных связей происходит только после достижения определенной поверхностной плотности молекул.
В режиме слишком высокой поверхностной плотности молекул отношение конфигурационных объемов ${{{{\omega }_{1}}} \mathord{\left/ {\vphantom {{{{\omega }_{1}}} {{{\omega }_{2}}}}} \right. \kern-0em} {{{\omega }_{2}}}}$ велико настолько, что отношение экспонент в (7) не способно его скомпенсировать – вероятность образования водородных связей остается высокой и нечувствительной к вариациям температуры. В соответствие с этим, в компьютерном эксперименте с повышением поверхностной плотности молекул наблюдается быстрое ослабление температурной зависимости образования водородных связей.
В терминах внутренней энергии и энтропии эти закономерности получают следующую интерпретацию. Соотношение (7) в терминах свободной энергии имеет вид:
(8)
$\begin{gathered} {{{{w}_{{\text{H}}}}(T)} \mathord{\left/ {\vphantom {{{{w}_{{\text{H}}}}(T)} {{{w}_{{\text{D}}}}(T)}}} \right. \kern-0em} {{{w}_{{\text{D}}}}(T)}} = \\ = \frac{{\exp ({{ - {{f}_{1}}(T)} \mathord{\left/ {\vphantom {{ - {{f}_{1}}(T)} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}})}}{{\exp ({{ - {{f}_{2}}} \mathord{\left/ {\vphantom {{ - {{f}_{2}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}) + \exp \left( {{{ - {\kern 1pt} {{f}_{3}}(T)} \mathord{\left/ {\vphantom {{ - {\kern 1pt} {{f}_{3}}(T)} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right)}}, \\ \end{gathered} $(10)
${{s}_{n}} = {{({{e}_{n}} - {{f}_{n}})} \mathord{\left/ {\vphantom {{({{e}_{n}} - {{f}_{n}})} T}} \right. \kern-0em} T} = {{k}_{{\text{B}}}}[\ln \left( {{{{(2!)}}^{{ - 1}}}{\lambda }_{0}^{{ - 6}}{{\omega }_{n}}} \right) + 3].$В условиях, когда все молекулы связаны либо с поверхностью, либо посредствам водородных связей с другими молекулами, состояние 3 имеет пренебрежимо малый статистический вес, и выражение (8) с учетом того, что ${{e}_{1}} - {{e}_{2}} = $ ${{u}_{1}} - {{u}_{2}},$ принимает следующий вид:
(11)
$\begin{gathered} {{{{w}_{{\text{H}}}}(T)} \mathord{\left/ {\vphantom {{{{w}_{{\text{H}}}}(T)} {{{w}_{{\text{D}}}}(T)}}} \right. \kern-0em} {{{w}_{{\text{D}}}}(T)}} \approx \\ \approx \exp \left( { - {{\left( {{{u}_{1}} - {{u}_{2}}} \right)} \mathord{\left/ {\vphantom {{\left( {{{u}_{1}} - {{u}_{2}}} \right)} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right)\exp \left( {{{\left( {{{s}_{1}} - {{s}_{2}}} \right)} \mathord{\left/ {\vphantom {{\left( {{{s}_{1}} - {{s}_{2}}} \right)} {{{k}_{{\text{B}}}}}}} \right. \kern-0em} {{{k}_{{\text{B}}}}}}} \right). \\ \end{gathered} $Как видно из (10), энтропия является растущей функцией соответствующего конфигурационного объема ${{\omega }_{{{\kern 1pt} n}}}$. Вся температурная зависимость (11) сосредоточена в первой экспоненте. В условиях, когда водородной связи соответствует менее глубокий минимум, чем на поверхности кристалла, ${{u}_{1}} - {{u}_{2}} > 0,$ первая экспонента в (11) растет с ростом температуры, что означает формирование водородных связей.
Таким образом, эффект обратного температурного хода обеспечивается меньшей энергией молекулы на поверхности по сравнению с энергией водородной связи. Температурная зависимость (11) усиливается в области низких температур, однако с понижением температуры уменьшается и сама величина (11), делая количество водородных связей ничтожно малым. Компенсировать эти малые значения первой экспоненты в (11) может вторая экспонента при условии, что ${{s}_{1}} - {{s}_{2}} > 0,$ то есть, с учетом (10), если ${{\omega }_{1}} > {{\omega }_{2}}$ – минимум, отвечающий водородной связи между молекулами, должен быть существенно шире потенциальной ямы, в которой молекула удерживается на поверхности кристалла.
Из представленной модели видно, что обратный температурный ход в образовании водородных связей обеспечивается низкими значениями энергии и энтропии адсорбированных на поверхности молекул по сравнению с соответствующими значениями для водородных связей. Именно такие условия реализуются на боковых сколах кристалла, где поле у поверхности гораздо более неоднородно, чем на базовых гранях. Следует ожидать, что наблюдаемый эффект носит универсальный характер. Чем более неоднородно поле у поверхности кристалла, тем более выражен эффект образования водородных связей при нагревании и их разрыва при охлаждении, что в определенных условиях может существенно влиять на ход реакций поверхностного катализа и гетерогенной нуклеации на поверхности микрокристаллов.
5. ЗАКЛЮЧЕНИЕ
Компьютерное моделирование нуклеации паров воды на наноструктурированной кристаллической поверхности c относительно крупными элементами в форме пирамид выявило ряд особенностей в пространственной организации адсорбированного вещества, которые могут иметь универсальный характер. В отличие от мелкомасштабных регулярных структур с характерными размерами элементов в 2–3 межатомных расстояния, которые изучались в предыдущих работах, относительно крупные структурные элементы сами обладают атомарной структурой и вследствие этого могут быть источниками полей сложной геометрии. В механизме удерживания молекул каждым таким элементом растет роль коллективных эффектов, и конкуренция между различными макросостояними становится более контрастной.
Полученные в представленной работе данные для относительно крупных кристаллических структурных элементов позволяют уверенно наблюдать крайне неравномерное поглощение молекул их гранями. Несмотря на близость структуры базовых граней к структуре льда, захват молекул начинается не на них, а на краях слоев, из которых сложен каждый пирамидальный элемент, причем в первую очередь поглощение происходит на краях, содержащих катионы серебра. Эта избирательность в отношении к ионам разного сорта обусловлена асимметрией пространственного распределения электрического заряда в молекуле воды и узкой направленностью водородных связей.
Главным последствием наноструктурированности поверхности является резкое понижение степени кластеризации адсорбированных молекул, которое сопровождается разрывом части водородных связей между ними. В поле наноструктурированной поверхности возможно наблюдать эффект обратной температурной зависимости числа водородных связей между адсорбированными молекулами, при котором повышение температуры сопровождается не разрывом, а частичным восстановлением водородных связей. Эффект обусловлен более высокой энтропией водородных связей по сравнению с энтропией прямых взаимодействий с поверхностью и наблюдается на начальной стадии поглощения.
Наноструктурированность приводит к существенному подавлению коллективных эффектов. Механизм удерживания адсорбированного вещества смещается от взаимодействий между молекулами и молекул с подложкой в сторону только прямых взаимодействий каждой отдельной молекулы с ионами кристаллической решетки.
Индивидуальный характер удерживания молекул на наноструктурированной поверхности отражается на пространственной организации зародышей конденсированной фазы, особенно, на начальной стадии их формирования. Вместо пятен однородной мономолекулярной пленки молекулы воды образуют на адсорбирующей поверхности сильно неоднородный слой с пустотами и сгустками молекул, в котором не обнаруживаются элементы гексагональной ячеистой структуры, характерные для молекулярного слоя, растущего на бездефектной поверхности.
Работа выполнена при поддержке Министерства образования и науки РФ, проект № 3.4808.2017/6.7 и Российского фонда фундаментальных исследований, проект № 18-03-00011.
Список литературы
Varanasi K.K., Deng T., Smith J.D., Hsu M., Bhate N. // Appl. Phys. Lett. 2010. V. 97. P. 234 102.
Meuler A.J., McKinley G.H., Cohen R.E. // ACS Nano. 2010. V. 4. P. 7048.
Deng X., Mammen L., Butt H.-J., Vollmer D. // Science. 2012. V. 335. P. 67.
Мейсон Б.Дж. Физика облаков. Л.: Гидрометеоиздат, 1962.
Качурин Л.Г. Физические основы воздействия на атмосферные процессы. Л.: Гидрометеоиздат, 1990.
Вильсон Дж. Камера Вильсона. M.: Иностранная литература, 1954.
Lukyanov S.I., Zidi Z.S., Shevkunov S.V. // J. Mol. Struct. (THEOCHEM). 2005. V. 725. P. 191.
Шевкунов С.В. // Электрохимия. 1998. Т. 34. С. 860.
Шевкунов С.В. // Журн. физ. химии. 2009. Т. 83. С. 1108.
Шевкунов С.В. // Коллоид. журн. 2009. Т. 71. С. 404.
Шевкунов С.В. // Коллоид. журн. 2010. Т. 72. С. 92.
Шевкунов С.В. // Коллоид. журн. 2011. Т. 73. С. 123.
Шевкунов С.В. // Коллоид. журн. 2011. Т. 73. С. 267.
Шевкунов С.В. // Журн. физ. химии. 2011. Т. 85. С. 1702.
Куни Ф.М., Щекин А.К., Русанов А.И. // Коллоид. журн. 1982. Т. 44. С. 1062.
Амелин А.М. Теоретические основы образования тумана при конденсации пара, М.: Химия, 1966. С. 18.
Галимзянов Б.Н., Мокшин А.В. // Коллоид. журн. 2017. Т. 79. С. 16.
Тверской П.Н. Атмосферное электричество. Л.: Гидрометеоиздат, 1949.
Шишкова И.Н., Ястребов А.К. // Коллоид. журн. 2015. Т. 77. С. 669.
Шевкунов С.В. // ЖЭТФ. 2009 . Т. 135. С. 510.
Шевкунов С.В. // ЖЭТФ. 2009. Т. 136. С. 282.
Миронов Е.П., Марченко И.В., Артемов В.В., Букреева Т.В. // Коллоид. журн. 2017. Т. 79. С. 304.
Сизова А.А., Сизов В.В., Бродская Е.Н. // Коллоид. журн. 2015. Т. 77. С. 90.
Singha S.K., Das P.K., Maiti B. // J. Chem. Phys. 2015. V. 143. P. 204703.
Xu D., Johnson W.L. // Phys. Rev. B. 2005. V. 72. P. 052101.
Lazaridis M. // J. Colloid Interface Sci. 1993. V. 155. P. 386.
Iwamatsu M. // Langmuir. 2015. V. 31. P. 3861.
Bottiglione F., Carbone G., Persson B.N.J. // J. Chem. Phys. 2015. V. 143. P. 134705.
Svoboda M., Malijevský A., Lísal M. // J. Chem. Phys. 2015. V. 143. P. 104701.
Wenzel R.N. // Ind. Eng. Chem. 1936. V. 28. P. 988.
Cassie A.B.D., Baxter S. // Trans. Faraday Soc. 1944. V. 40. P. 546.
Cassie A.B.D. // Discuss. Faraday Soc. 1948. V. 3. P. 11.
Young Y. // Philos. Trans. R. Soc. London. 1805. V. 95. P. 65.
Fraux G., Doye J.P.K. // J. Chem. Phys. 2014. V. 141. P. 216 101.
Хилл Т. Статистическая механика. М.: Иностранная литература, 1960.
Замалин В.М., Норман Г.Э., Филинов В.С. Метод Монте-Карло в статистической термодинамике, М.: Наука, 1977.
Hale B.N., Kiefer J. // J. Chem. Phys. 1980. V. 73. P. 923.
Шевкунов С.В. // Журн. физ. химии. 2005. Т. 79. С. 1860.
Шевкунов С.В. // Коллоид. журн. 2005. Т. 67. С. 548.
Шевкунов С.В. // Коллоид. журн. 2006. Т. 68. С. 391.
Шевкунов С.В. // Коллоид. журн. 2006. Т. 68. С. 691.
Шевкунов С.В. // ЖЭТФ. 2008. Т. 134. С. 1130.
Burley G. // Am. Mineralogist. 1963. V. 48. P. 1266.
Справочник химика. 2-е изд. Т. 2 / Под ред. Никольского Б.П. и др. М.−Л.: Химия, 1966, С. 404.
Шевкунов С.В. // Коллоид. журн. 2018. Т. 80. С. 224.
Шевкунов С.В. // Коллоид. журн. 2018. Т. 80. С. 241.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал