

Коллоидный журнал, 2019, T. 81, № 2, стр. 170-174
Молекулярно-динамическое моделирование неустойчивого равновесия сферического зародыша для определения межфазной энергии в двухкомпонентной системе Pb–Cu
В. В. Королев 1, 2, В. М. Самсонов 2, П. В. Проценко 1, *
1 Московский государственный университет им. М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
2 Тверской государственный университет, физико-технический факультет
170002 Тверь, Садовый переулок, 35, Россия
* E-mail: protsenko@colloid.chem.msu.ru
Поступила в редакцию 06.09.2018
После доработки 06.11.2018
Принята к публикации 28.09.2018
Аннотация
Молекулярно-динамический метод расчета межфазной энергии на границе раздела расплав–кристалл, основанный на классической теории зародышеобразования, обобщен на случай бинарных металлических систем. Полученная температурная зависимость (в интервале 1025–1125 K) межфазной энергии для биметаллической системы Pb–Cu хорошо согласуется с результатами статистических расчетов в рамках упрощенной квазикристаллической когерентной модели жидкой фазы и межфазной границы, учитывающей межатомные взаимодействия с ближайшими соседями. Схожая методология может быть использована для определения межфазной энергии широкого круга многокомпонентных металлических систем при наличии достоверных потенциалов межчастичного взаимодействия.
ВВЕДЕНИЕ
Изучение структуры и свойств межфазных границ расплав/кристалл в многокомпонентных металлических системах представляет практический интерес из-за их определяющего влияния на процессы образования и роста кристаллов из жидкой фазы, формирования дендритов [1, 2] и т.д. Однако, несмотря на активное развитие методов рентгеновского рассеяния и просвечивающей электронной микроскопии, непосредственное экспериментальное наблюдение межфазных границ до сих пор крайне затруднительнo [3–6]. По этой причине использование компьютерного моделирования представляется достаточно перспективным.
Благодаря относительно низкому уровню вычислительных затрат и приемлемому воспроизведению разнообразных физических явлений в конденсированных средах молекулярно-динамическое моделирование является на данный момент наиболее популярным методом изучения структуры и свойств межфазных границ в металлических системах [7]. Бурное развитие метода молекулярной динамики в значительной степени связано с ростом доступных вычислительных мощностей и применением графических процессоров для параллельных вычислений. Использование крупных вычислительных кластеров позволило добиться увеличения моделируемых систем до субмикронных размеров [8, 9].
Основная часть исследований межфазных границ в металлах методами молекулярной динамики посвящена однокомпонентным системам [10–16] и лишь незначительная – сплавам [17–19], в то время как именно многокомпонентные системы представляют наибольший практический интерес. Обобщение методологии, применяемой для однокомпонентных систем, на многокомпонентные затрудняется прежде всего отсутствием достоверных гетероатомных потенциалов межчастичного взаимодействия. Межфазная энергия – критически важный параметр, в значительной степени определяющий форму образующихся из расплава кристаллов и их механические свойства. Однако экспериментальное определение этой величины сопряжено с теми же сложностями, что и непосредственное наблюдение за межфазными границами. Помимо того, что рассматриваемые межфазные границы разделяют две конденсированные фазы, абсолютные значения межфазной энергии на порядок меньше типичных значений поверхностной энергии в металлических системах.
К настоящему времени существуют два основных метода определения межфазной энергии в металлических системах с использованием молекулярно-динамического моделирования: метод капиллярных флуктуаций [13, 20, 21] и метод критического зародыша [22, 23]. Первый из них позволяет определить межфазную энергию для различных ориентаций поверхности твердого тела (т.е. анизотропию межфазной энергии). Однако он применим только при температуре плавления (Tm) и чувствителен к размерным эффектам, т.е. получение релевантных данных требует использования модельных ячеек предельно допустимых размеров и, как следствие, значительных вычислительных затрат.
Метод критического зародыша позволяет изучать межфазное равновесие в широком температурном диапазоне, но в силу упомянутых выше причин ранее использовался только для однокомпонентных систем. В последнее время опубликован ряд работ, в которых были определены значения межфазной энергии в таких системах [23, 24]. Полученные значения хорошо согласуются с экспериментальными результатами [16].
В данной работе предлагается обобщение метода критического зародыша на случай многокомпонентных металлических систем на примере бинарной системы Pb–Cu. Выбор этой системы в качестве модели для отработки методологии определения межфазной энергии в бинарной металлической системе обусловлен несколькими причинами. Во-первых, оптимизированный для оценки термодинамических характеристик системы потенциал взаимодействия Pb–Cu имеется в открытом доступе [25]. Во-вторых, термодинамические характеристики границы фаз расплав/кристалл в этой системе подробно исследованы экспериментально [26], что дает возможность верифицировать результаты расчетов.
МЕТОДЫ МОДЕЛИРОВАНИЯ
Выбор подходящего потенциала межчастичного взаимодействия играет определяющую роль при проведении молекулярно-динамического моделирования. Для описания взаимодействий Pb–Pb и Cu–Cu использовались потенциалы внедренного атома (EAM), предложенные в работах [27] и [28] соответственно. Данные потенциалы ранее успешно применялись для моделирования твердых тел, включая свойства дефектов упаковки.
Для описания взаимодействия Pb–Cu также использовался потенциал EAM, оптимизированный путем сравнения доступных экспериментальных и расчетных значений, полученных с его помощью [25]. Параметры модели были подобраны таким образом, чтобы с максимальной точностью воспроизвести значения теплот смешения для серии расплавов Pb–Cu различного состава. Посредством термодинамического интегрирования и моделирования методом Монте-Карло были получены значения Tm чистых компонентов [25], положение линий солидуса и ликвидуса, т.е. воспроизведена фазовая диаграмма рассматриваемой системы. Расчетная фазовая диаграмма содержит область несмешиваемости (в области значений температуры приблизительно на 100 K ниже точки плавления меди), а также характеризуется крайне низкой растворимостью свинца в твердой меди (менее 0.1% при 1000 K), что хорошо согласуется с имеющимися экспериментальными данными.
Так как расплавы Cu–Pb не являются идеальными растворами, активность свинца была рассчитана в работе [25] как функция концентрации и сопоставлена с имеющимися экспериментальными данными. Несмотря на то, что используемый потенциал качественно воспроизводит поведение неидеальных растворов, значения активности Pb оказались существенно ниже полученных экспериментально. Надежные данные по активности меди для системы Cu–Pb в интересующих нас диапазонах температуры и концентрации в литературе отсутствуют, поэтому для расчета межфазной энергии мы использовали в уравнении Фрейндлиха–Оствальда значения концентрации.
Здесь r – радиус исходного зародыша, Vm – молярный объем меди, a – активность меди в растворе, Т – абсолютная температура, R –универсальная газовая постоянная.
Молекулярно-динамическое моделирование проводили, используя программный пакет LAMMPS [29]. Моделировалась кубическая ячейка размером 29.7 × 29.7 × 29.7 нм3 с периодическими граничными условиями. Задавались изобарно-изотермические условия, шаг по времени составлял 1 фс.
Молекулярно-динамический эксперимент включал следующие стадии.
1. Полное заполнение модельной ячейки идеальной кристаллической структурой Pb (при температуре 0 K).
2. Нагрев системы до выбранной температуры, релаксация ячейки (на этом этапе и далее гидростатическое давление поддерживалось равным 0 бар).
3. Замена заданной части атомов Pb на атомы Cu c повторной релаксацией ячейки.
4. Внедрение в центр модельной ячейки кристаллического зародыша заданного радиуса. Поскольку, согласно экспериментальной и расчетной фазовым диаграммам, растворимость свинца в кристаллической меди крайне низка, сферический зародыш не содержал атомов свинца.
5. Непосредственное моделирование процессов растворения/роста сферического зародыша меди в условиях пересыщения относительно равновесного содержания меди в расплаве.
Рост/растворение кристаллического зародыша фиксировали исходя из изменения количества атомов, имеющих локальное окружение, соответствующее ГЦК-решетке. Для определения локального порядка атомов использовали метод анализа локального окружения [30], реализованный в программном пакете OVITO [31]. Данный алгоритм позволяет определять локальное кристаллографическое окружение атома исходя из топологии связей, образованных атомами из его первой координационной сферы. Для каждой пары центральный атом−атом, находящийся от центрального атома на расстоянии, меньшем заданного (в зависимости от предполагаемого типа локального окружения) рассчитываются следующие характеристики:
– число атомов, находящихся на расстоянии меньше заданного от обоих атомов из данной пары (ncn),
– число связей между такими общими соседними атомами (nb),
– число связей в самой длинной цепочке, соединяющей общие соседние атомы (nlcb).
Затем набор полученных триплетов (ncn, nb, nlcb) сопоставляется с наборами, соответствующими различным локальным кристаллографическим окружениям (ГЦК, ГПУ и т.д.)
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Молекулярно-динамическое моделирование проводилось при 1025–1125 K. Нижней естественной границей служила температура монотектики (Tmon), равная 1012 K согласно расчетной фазовой диаграмме, представленной в работе [25]. С повышением температуры содержание меди в равновесном расплаве значительно возрастает и предельно возможное пересыщение снижается. Таким образом, радиус критического зародыша увеличивается, и его величина становится сопоставимой с размером модельной ячейки. При изменении его размера в процессах роста/растворения заметно изменяется состав расплава, и применение метода расчета, основанного на уравнении Фрейндлиха–Оствальда, становится недопустимым. Данная особенность является основным ограничением для применения выбранной методики к составам с предельно высоким содержанием меди.
На рис. 1 представлена зависимость межфазной энергии системы Pb–Cu от температуры, полученная следующим образом. Для каждой температуры проводилась серия модельных расчетов с различными значениями пересыщения при фиксированном начальном радиусе зародыша. Каждый отдельный эксперимент дает представление о направлении процесса, т.е. о том, растет зародыш или растворяется. На рис. 1 показаны только те значения межфазной энергии, рассчитанные по уравнению Фрейндлиха–Оствальда (1), которые соответствуют наименьшему из растущих и наибольшему из растворяющихся зародышей.
Рис. 1.
Зависимость межфазной энергии от приведенной температуры, полученная молекулярно-динамическим моделированием в рамках метода внедренного атома (ЕАМ). 1 – Максимальные растворяющиеся зародыши, 2 – минимальные растущие зародыши, 3 – межфазная энергия в однокомпонентной системе твердая медь/расплав меди [23], сплошная линия – расчет по уравнению (2).
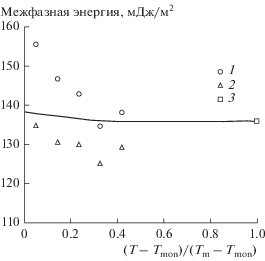
Полученные значения межфазной энергии лежат в диапазоне 130–145 мДж/м2, температурная зависимость σ слабо выражена. Определение межфазной энергии в однокомпонентной системе твердая медь/расплав меди молекулярно-динамическим моделированием, проведенное в работе [23] двумя методами – критического зародыша и капиллярных флуктуаций, дало значения 136 ± 3 и 149 ± 14 мДж/м2 соответственно. Отсутствие заметного влияния свинца на межфазную энергию в системе Cu–Pb на первый взгляд является неожиданным результатом. Известно, что на границе фаз расплав/газ в этой же системе свинец проявляет значительную поверхностную активность [32]. В рамках модели [33] снижение поверхностного натяжения расплава меди в два раза наблюдается при введении в расплав около 2 ат. % Pb.
Насколько нам известно, экспериментальные значения межфазной энергии в системе Pb–Cu для исследованного температурного диапазона отсутствуют. Это объясняется тем, что в данной системе при температуре выше температуры монотектики наблюдается полное растекание расплава по поверхности меди [32] и смачивание расплавом границ зерен [34]. Соответственно, определить значение межфазной энергии расплав/твердая фаза на основании анализа капиллярных равновесий не представляется возможным.
Для независимой оценки адсорбционной активности свинца на межфазной поверхности твердое тело/расплав были проведены статистические термодинамические расчеты в соответствии с методологией, изложенной в [35]. Межфазная поверхность представлялась в виде двух упорядоченных атомных монослоев – жидкого (L) и твердого (S), см. рис. 2. При этом считалось, что атомы жидкости находятся в узлах квазикристаллической решетки, изоморфной и когерентной решетке твердого кристалла. При расчете энергии системы учитывались только взаимодействия в первой координационной сфере. В рамках данной модели межфазная энергия σ вычисляется по уравнению
(2)
$\begin{gathered} {\sigma }{{{\Omega }}_{{\text{м }}}} = y_{{\text{A}}}^{{\text{S}}}{\sigma }_{{\text{A}}}^{{\text{0}}}{{\Omega }_{{\text{м }}}} + y_{{\text{В }}}^{{\text{S}}}{\sigma }_{{\text{В }}}^{{\text{0}}}{{{\Omega }}_{{\text{м }}}} + \\ + \,\,RT\sum\limits_j {\left( {y_{{\text{A}}}^{j}\ln \frac{{y_{{\text{A}}}^{j}}}{{x_{{\text{A}}}^{j}}} + y_{{\text{B}}}^{j}\ln \frac{{y_{{\text{B}}}^{j}}}{{x_{{\text{B}}}^{j}}}} \right)} + \\ + \,\,n\sum\limits_j {{\lambda }_{{{\text{AB}}}}^{j}y_{{\text{A}}}^{j}y_{{\text{B}}}^{j}} + ... + m{\lambda }_{{{\text{AB}}}}^{{\text{L}}}\left( {y_{{\text{A}}}^{{\text{L}}}y_{{\text{B}}}^{{\text{S}}} + y_{{\text{B}}}^{{\text{L}}}y_{{\text{A}}}^{{\text{S}}}} \right) - \\ - \,\,(n + m)\sum\limits_j {{\lambda }_{{{\text{AB}}}}^{j}\left( {y_{{\text{A}}}^{j}x_{{\text{B}}}^{j} + y_{{\text{B}}}^{j}x_{{\text{A}}}^{j} - x_{{\text{A}}}^{j}x_{{\text{B}}}^{j}} \right)} . \\ \end{gathered} $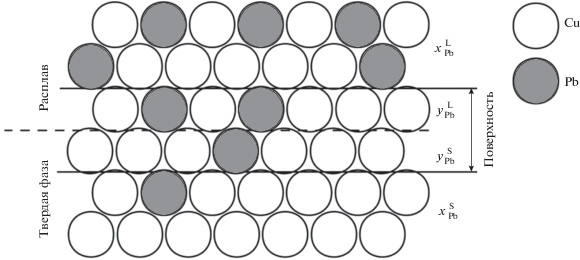
Здесь $x_{k}^{j}$ и $y_{k}^{j}$ – доли компонента k в объеме и на поверхности фазы j, ${\lambda }_{{{\text{AB}}}}^{j}$ – теплота смешения A c B при бесконечном разведении в фазе j, Ωм – молярная площадь, ${\sigma }_{k}^{0}$ – межфазная энергия на границе расплав–кристалл в однокомпонентной системе на основе k, n и m – доли связей атома в плотноупакованной и в прилежащей плоскостях соответственно. Параметры ${\lambda }_{{{\text{AB}}}}^{j}$ и $x_{k}^{j}$ вычисляются на основе бинарной фазовой диаграммы. Величина межфазной энергии минимизируется при варьировании параметров $y_{k}^{j}.$ В работе [35] было показано, что эта модель хорошо описывает зависимость межфазной энергии на границе расплав/твердый кристалл в тройной системе Fe–Pb–Cu от концентрации меди, а также адекватно воспроизводит поверхностную активность третьего компонента в ряде других тройных металлических систем.
Для сопоставления результатов статистического и молекулярно-динамического моделирования значения межфазной энергии были рассчитаны по уравнению (2); при этом задавалось значение ${\sigma }_{{{\text{Cu}}}}^{0} = 136$ мДж/м2, так как оно была получено в работе [23] c использованием методологии, применяемой в нашей работе.
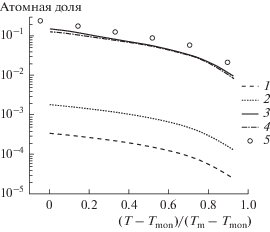
Результаты статистических расчетов, представленные на рис. 1, количественно согласуются со значениями, полученными методом молекулярно-динамического моделирования. Отметим, что в рассмотренном температурном диапазоне межфазная энергия практически не изменяется, что обусловлено отсутствием заметной адсорбции свинца на межфазной поверхности (рис. 3).
Рис. 3.
Зависимость атомной доли свинца от приведенной температуры в объемных фазах, xPb (согласно фазовой диаграмме и по результатам молекулярно-динамического моделирования) и в поверхностных монослоях, yPb (статистические расчеты по уравнению (2)). 1 – $x_{{{\text{Pb}}}}^{{\text{S}}},$ 2 – $y_{{{\text{Pb}}}}^{{\text{S}}},$ 3 – $x_{{{\text{Pb}}}}^{{\text{L}}},$ 4 – $y_{{{\text{Pb}}}}^{{\text{L}}},$ 5 – $x_{{{\text{Pb}}}}^{{\text{L}}}$ [25].
Таким образом, в работе впервые предложена методология определения межфазной энергии на границе расплав–кристалл в двухкомпонентной металлической системе. Значения межфазной энергии, полученные при экстраполяции результатов на температуру плавления чистой меди, хорошо согласуются с величиной, найденной при моделировании однокомпонентной системы. Полученная температурная зависимость межфазной энергии также хорошо согласуется с результатами статистических расчетов в рамках упрощенной квазикристаллической и когерентной модели жидкой фазы и межфазной границы, учитывающей межатомные взаимодействия с ближайшими соседями.
Работа выполнена с использованием оборудования Центра коллективного пользования сверхвысокопроизводительными вычислительными ресурсами МГУ имени М.В. Ломоносова при финансовой поддержке Российского фонда фундамен-тальных исследований (проекты № 17-33-50103_мол_нр и № 18-08-01508_а), а также Министерства науки и высшего образования Российской Федерации в рамках выполнения государственного задания в сфере научной деятельности (проект № 3.5506.2017/БЧ).
Список литературы
Kaplan W.D., Kauffmann Y. // Annu. Rev. Mater. Res. 2006. V. 36. P. 1.
Asta M., Beckermann C., Karma A., Kurz W., Napolitano R., Plapp M., Purdy G., Rappaz M., Trivedi R. // Acta Mater. 2009. V. 57. P. 941.
Hulsman W.J., Peters J.F., Zwanenburg M.J., De Vries S.A., Derry T.E., Abernathy D., Van der Veen J.F. // Nature. 1997. V. 390. P. 379.
Gabrisch H., Dahmen U., Johnson E. // Microsc. Microanal. 1998. V. 4. P. 286.
Reichert H., Klein O., Dosch H., Denk M., Honkima V. // Transition. 2000. V. 408. P. 1998.
Pereiro-López E., Ludwig W., Bellet D., Cloetens P., Lemaignan C. // Phys. Rev. Lett. 2005. V. 95. 215501.
Sosso G.C., Chen J., Cox S.J., Fitzner M., Pedevilla P., Zen A.,Michaelides A. // Chem. Rev. 2016. V. 116. P. 7078.
Shibuta Y., Sakane S., Miyoshi E., Okita S., Takaki T., Ohno M. // Nat. Commun. 2017. V. 8. P. 10.
Shibuta Y., Ohno M., Takaki T. // Adv. Theory Simul. 2018. V. 1. 1800065.
Broughton J.Q., Bonissent A., Abraham F.F. // J. Chem. Phys. 1981. V. 74. P. 4029.
Broughton J.Q., Gilmer G.H. // J. Chem. Phys. 1986. V. 84. P. 5759.
Davidchack R.L, Laird B.B. // J. Chem. Phys. 1998. V. 108. P. 9452.
Hoyt J.J., Asta M., Karma A. // Phys. Rev. Lett. 2001. V. 86. P. 5530.
Davidchack R.L., Laird B.B. // Phys. Rev. Lett. 2005. V. 94. P. 1.
Buta D., Asta M., Hoyt J.J. // Phys. Rev. E. 2008. V. 78. 031605.
Asadi E., Zaeem M.A., Nouranian S., Baskes M.I. // Acta Mater. 2015. V. 86. P. 169.
Asta M., Hoyt J.J., Karma A. // Phys. Rev. B. 2002. V. 66. 1001011.
Becker C.A., Olmsted D., Asta M., Hoyt J.J., Foiles S.M. // Phys. Rev. Lett. 2007. V. 98. P. 98.
Zheng X.Q., Yang Y., Gao Y.F., Hoyt J.J., Asta M., Sun D.Y. // Phys. Rev. E. 2012. V. 85. 041601.
Brown N.T., Martinez E., Qu J. // Acta Mater. 2017. V. 129. P. 83.
Asadi E., Asle Zaeem M. // Acta Mater. 2016. V. 107. P. 337.
Mahata A., Zaeem M.A., Baskes M.I. // Model. Simul. Mater. Sci. Eng. 2018. V. 26. 025007.
Zhou H., Lin X., Wang M., Huang W. // J. Cryst. Growth. 2013. V. 377. P. 107.
Wu L.K., Li Q.L., Xu B., Liu W. // J. Mater. Res. 2016. V. 31. P. 3649.
Hoyt J.J., Garvin J.W., Webb E.B., Asta M. // Model. Simul. Mater. Sci. Eng. 2003. V. 11. P. 287.
Camel D., Eustathopoulos N., Desré P. // Acta Metall. 1980. V. 28. P. 239.
Lim H.S., Ong C.K., Ercolessi F. // Surf. Sci. 1992. V. 269–270. P. 1109.
Foiles S.M., Baskes M.I., Daw M.S. // Phys. Rev. B. 1986. V. 33. P. 7983.
Plimpton S. // J. Comput. Phys. 1995. V. 117. P. 1.
Stukowski A. // Model. Simul. Mater. Sci. Eng. 2012. V. 20. 045021.
Stukowski A. // Model. Simul. Mater. Sci. Eng. 2010. V. 18. 015012.
Timoshenko V., Traskine V., Zhevnenko S., Protsenko P. // J. Phys. Chem. C. 2016. V. 120. P. 7662.
Tanaka T., Hack K., Iida T., Hara S. // Z. Metallkd. 1996. V. 87. P. 380.
Eustathopoulos N. // Int. Met. Rev. 1983. V. 28. P. 189.
Chatain D., Pique D., Coudurier L., Eustathopoulos N. // J. Mater. Sci. 1985. V. 20. P. 2233.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал