

Коллоидный журнал, 2019, T. 81, № 2, стр. 190-201
Полипептиды на поверхности липидных мембран – теоретический анализ электрокинетических данных
Р. Ю. Молотковский 1, Т. Р. Галимзянов 1, 2, Ю. А. Ермаков 1, *
1 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, стр. 5, Россия
2 Национальный исследовательский технологический университет “МИСиС”
119049 Москва, Ленинский просп., 4, Россия
* E-mail: yury.a.ermakov@gmail.com
Поступила в редакцию 01.10.2018
После доработки 06.11.2018
Принята к публикации 02.11.2018
Аннотация
В работе рассматривается теоретическая модель, описывающая экспериментальные данные по электрофоретической подвижности липосом из смесей заряженного (кардиолипин) и нейтрального (фосфатидилхолин) липидов с адсорбированными на них молекулами полилизина. Экспериментальные данные показывают зависимость дзета-потенциала от концентрации адсорбированного полилизина. Предложенная модель используется для определения физически измеряемых характеристик, описывающих систему: толщины адсорбированного слоя полимера, доли площади, занятой полимером в условиях насыщении, и константы связывания полимера с поверхностью липосом. Проведенные расчеты показывают, что обратимость адсорбции резко падает при увеличении размеров молекул адсорбируемого полимера. Представленная модель объясняет поведение точки нулевого заряда при варьировании параметров системы. Она не ограничивается конкретным типом полимера и фосфолипидов и потому может быть применена для исследования адсорбции других биологически значимых синтетических поликатионов и полипептидов.
ВВЕДЕНИЕ
Среди биомедицинских технологий в настоящее время одним из наиболее актуальных направлений является разработка поликатионных мембранотропных лекарств и противомикробных препаратов, биоцидов [1]. Они используются в различных областях медицины и технологии, таких, как антимикробная терапия [2], консервирование пищи [3], иммуностимуляция [4] и др. К числу наиболее изученных полимеров следует отнести полиэтиленимин [5], полиалиламин [6] и полилизин (ПЛ) [7, 8]. Судя по литературным данным, адсорбция таких поликатионов на поверхности клеток определяется, прежде всего, электростатическими взаимодействиями, поскольку клеточные стенки в большинстве случаев имеют отрицательный заряд благодаря присутствию анионных фосфолипидов в липидном матриксе. В литературе описано немало примеров, когда поликатионы линейного типа практически не сорбируются на мембранах из незаряженных липидов (например, из фосфатидилхолина), но предпочтительно взаимодействуют с анионными липидами в составе мембран (фосфатидилглицерином, фосфатидилсерином и кардиолипином) [6, 9–16]. Важно отметить, что размеры макромолекул полимера существенно влияют как на процесс адсорбции, так и на вызванные ими изменения в латеральной структуре мембран. В частности, поликатионы способны вызывать агрегацию отрицательно заряженных липидов вблизи места адсорбции [13, 17] и в ряде случаев приводить к миграции отрицательно заряженных липидов из внутреннего монослоя мембран на внешний (флип-флоп) [18], а достаточно большие полипептиды на основе лизина и аргинина нарушают целостность мембран [8]. Согласно данным работы [19], длинные молекулы полилизина в отличие от коротких формируют на поверхности мембран бета-структуры, которые в зависимости от фазового состояния липидного бислоя различным образом влияют на его стабильность. Конечно, участие поликатионов в образовании дефектов мембран во многом зависит от наличия у них гидрофобных фрагментов и боковых углеводородных цепочек, однако характер электростатических взаимодействий с поверхностью мембран остается при этом идентичными для большинства полиэлектролитов [20]. Поэтому для исследования таких взаимодействий особое значение имеют электрокинетические измерения, позволяющие контролировать подвижность клеток и их липидных моделей, липосом, в электрическом поле, когда на их поверхности присутствуют заряженные макромолекулы. К сожалению, получаемые при этом результаты обычно интерпретируются лишь качественно и с большими оговорками, а детальный теоретический анализ равновесной адсорбции больших молекул представляет собой сложную задачу с большим количеством недоступных для экспериментальной проверки параметров (см., например, [21–23]). Более того, чрезвычайно высокая эффективность адсорбции больших поликатионов, обнаруживаемая в электрокинетических измерениях, заставляет сомневаться, что они находятся в равновесии с поверхностью и адсорбируются на ней практически необратимо [9]: это допущение может оказаться зависящим от конкретных условий наблюдения.
В данной работе мы предлагаем достаточно простую теоретическую модель, которая помогает количественно описать опубликованные ранее электрокинетические данные с использованием небольшого числа физически обоснованных параметров системы. Модель позволяет оценить характер заполнения поверхности липосом линейными поликатионами и сделать определенные предсказания относительно геометрических и электрических свойств полимерного слоя, доступные для экспериментальной проверки.
Предлагаемая модель принимает во внимание достоверно установленные факты, обнаруженные ранее при электрокинетическом исследовании суспензий липосом в присутствии полипептидов разной молекулярной массы на основе лизина, результаты которого опубликованы в нескольких работах [9–12].
1. Образование локальных структур в присутствии ПЛ происходит исключительно на отрицательно заряженных доменах фосфолипидов. Этот факт подтвержден методами атомно-силовой микроскопии [24], инфракрасной спектроскопии [25] и электрокинетическими исследованиями [9, 11, 26]. Это указывает на преимущественно кулоновскую природу адсорбции ПЛ на липидных мембранах.
2. Высокомолекулярные ПЛ сорбируются на поверхности мембран из анионных фсофолипидов (фосфатидилсерина, кардиолипина) с чрезвычайно высокой эффективностью. В работе [9] приведен опыт, в котором уменьшение количества липида в измерительной ячейке приводило к пропорциональному уменьшению количества ПЛ, необходимого для перезарядки поверхности вблизи точки нулевого заряда (ТНЗ) и последующего достижения предельного значения дзета-потенциала при полном заполнении поверхности липосом. Таким образом, на положение кривых относительно шкалы концентраций влияет не количество добавленного в ячейку полимера, а площадь поверхности, доступная для его адсорбции. Отсюда сделан вывод, что адсорбционное равновесие полимера вблизи поверхности смещено таким образом, что последовательное заполнение поверхности достигается при практически необратимом связывании ПЛ с липосомами. Дополнительный аргумент в пользу этого вывода был получен авторами этого исследования при попытках удалить ПЛ с поверхности плоских бислойных липидных мембран (БЛМ) непрерывной перфузией ячейки. Высокая стабильность полимерных слоев на поверхности липидного бислоя позволила использовать атомно-силовую микроскопию для изучения геометрических параметров кластеров, образуемых полилизинами [9, 12]. Полученные при этом результаты хорошо согласуются с литературными данными [27] относительно значений средней толщины слоев ПЛ с разной длиной цепи.
3. Некоторое противоречие с предыдущим выводом было обнаружено и не получило объяснения в аналогичных опытах, где снижалась доля анионного липида в составе смеси при фиксированном полном количестве липида в суспензии [11, 12]. При адсорбции ПЛ с малой молекулярной массой положение ТНЗ оставалось неизменным или даже смещалось в сторону бóльших концентраций полимера. Есть основание предполагать, что в этих опытах проявлялась способность полилизинов малого размера к частичной десорбции.
4. Экспериментальные данные указывают на существенное влияние ионной силы электролита на характер адсорбции ПЛ и величину предельного потенциала в области насыщения поверхности. Однако оценка поверхностной плотности заряда, проведенная в рамках классических приближений (модель Гуи–Чепмена и соотношение Смолуховского) дает примерно постоянное значение этой величины как для коротких, так и для длинных полимеров при увеличении ионной силы электрoлита [12]. Этот факт представляется неожиданным, поскольку классические теории полиэлектролитов предполагают заметное изменение состояния ионизации полимеров в результате конденсации противоионов [21]. В частности, согласно данным работы [27], заряд молекул полилизинов, близко расположенных друг к другу на поверхности слюды, оказывается весьма малым из-за конденсации противоионов и не зависит от концентрации электролита, тогда как толщина полимерного слоя определяется длиной полипептида в большей степени, чем его вторичной структурой. Это позволяет нам не учитывать влияния противоионов на геометрические параметры адсорбированных молекул. По всей видимости, в слое адсорбированных на заряженной проверхности мембран поликатионов происходит перераспределение ионов разного знака, и в итоге состояние ионизации внешней поверхности полимерного слоя оказывается слабо зависящим от концентрации фонового электролита. В предлагаемой ниже модели конденсация противоионов учтена введением коэффициента, который понижает плотность заряда в полимерном слое и находится аппроксимацией экспериментальных данных.
Таким образом, адсорбция катионных полимеров является комплексным процессом, влияние на который оказывают как минимум три фактора: липидный состав мембраны, размер молекул полимера и адсорбция противоионов фонового электролита. В силу многофакторности и сложности этого процесса для его описания необходимы упрощающие предположения. При анализе адсорбции полимеров на заряженной мембране требуется учесть электростатическое взаимодействие мембраны и полимера и сформулировать условия для описания равновесного распределения макромолекул в области двойного электрического слоя (ДЭС) вблизи поверхности с помощью изотермы адсорбции. Согласно оценкам, приведенным в обзоре [28], во многих случаях распределение заряда и электрического поля в диффузной части ДЭС с достаточной точностью количественно описывается в рамках простой модели Гуи–Чепмена, дополненной изотермой Ленгмюра в работах Штерна (далее теория ГЧШ) [6, 12, 29]. Однако в рамках модели ГЧШ весьма сложно учесть размер макромолекул при выборе параметров изотермы адсорбции и при описании электрических характеристик процесса адсорбции. Нам известна одна из ранних попыток решить эту проблему в работе [30], в которой адсорбция полимеров предполагается кооперативной, когда образование связей каждым звеном макромолекулы полимера с поверхностью зависит от соседних звеньев. В данной работе предлагается другая модель, которая позволяет адекватно интерпретировать полученные недавно экспериментальные факты [9–12] и учесть установленные в этих работах основные тенденции с учетом размера молекул поликатионов.
ОПИСАНИЕ МОДЕЛИ
При построении модели были использованы экспериментальные данные об адсорбции ПЛ различной длины липосомами из смеси отрицательно заряженного (кардиолипин, КЛ) и нейтрального (диолеоилфосфатидилхолин, ДОФХ) липидов, опубликованные в работе [11]. Измеряемой величиной в этих опытах являлась электрофоретическая подвижность липосом μ, которая была использована для оценки электрического дзета-потенциала, ζ, в предположении, что выполняется соотношение Смолуховского [31]
где η – вязкость водного раствора, ε – его относительная диэлектрическая проницаемость, ε0 – электрическая постоянная. В работе [11] приведены несколько серий электрокинетических данных для зависимости ζ-потенциала от содержания ПЛ разной молекулярной массы (т.е. с разной длиной цепи) в суспензиях липосом с долей заряженного липида в составе мембран 100 и 20% (рис. 1). Здесь и далее цифры после аббревиатуры “ПЛ” обозначают среднее количество звеньев в цепи полипептида, а концентрация ПЛ дана в мольных единицах основания (лизина).Рис. 1.
Зависимости ζ-потенциала липосом, сформированных из КЛ (а) и его смеси (20%) с ДОФХ (б), от концентрации ПЛ с различным средним количеством мономерных звеньев в цепи, указанным на рисунке (а), по данным работы [11]. Здесь и далее в суспензии липосом присутствуют 1 мг (примерно 1 мМ) фосфолипида и фоновый электролит (10 мМ KCl).
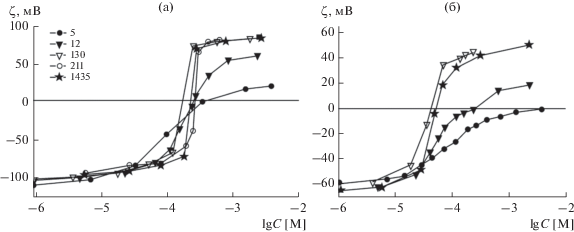
Обращают на себя внимание несколько важных особенностей, наблюдаемых в электрокинетических измерениях в присутствии ПЛ, которые требуется учесть при формулировании аналитической модели. Практически все макромолекулы сорбируются только на отрицательно заряженных поверхностях липосом. Их адсорбция на несколько порядков эффективнее, чем адсорбция молекул лизина. Она наблюдается при крайне низком и примерно одинаковом содержании этих полипептидов в суспензии, приводит к изменению знака заряда липосом и затем достигает предельного значения (насыщения), уровень которого зависит от молекулярной массы (длины) макромолекул. Положение ТНЗ относительно шкалы концентрации, выраженной в мольных единицах основания, оказывается близким к мольной концентрации липида в суспензии, т.е. отражает стехиометрию 1 : 1 между зарядами полимера и липида во внешнем монослое липосом. Положение ТНЗ для липосом из заряженного липида смещается в сторону низких концентраций в присутствии больших молекул ПЛ (рис. 2а) и практически не изменяется при адсорбции небольших молекул ПЛ-12 на липосомах из смеси заряженного и нейтрального липидов (рис. 2б).
Рис. 2.
(а) Зависимости ζ-потенциала липосом, сформированных при содержании КЛ в суспензии 0.2 (1), 1.0 (2) и 2.0 мл/мл (3), от концентрации ПЛ-598 суспензии [9]. (б) Результат контрольных измерений ζ-потенциала липосом, сформированных при варьировании доли КЛ α в смеси с ДОФХ в присутствии фонового электролита. Теоретическая кривая построена в рамках модели ГЧШ с учетом адсорбции ионов калия на отрицательно заряженных молекулах КЛ с константой связывания K * = 1 М–1 и поверхностной плотностью заряда Smax = –150 мКл м–2 [12].
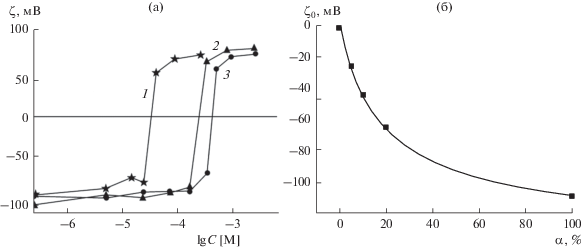
В ряде опытов электрокинетические измерения демонстрируют смещение ТНЗ при варьировании количества заряженного КЛ, использованного для приготовления суспензии липосом [9] (рис. 2а). Важно отметить, что и крутизна экспериментальных кривых, и предельное значение ζ-потенциала при этом оставались неизменными. Отсюда напрашивается вывод о том, что в этих опытах происходит последовательное заполнение поверхности липосом полимером так, как если бы ПЛ адсорбировался практически необратимо.
Для упрощения теоретической модели при описании распределения электрического поля вблизи поверхности липосом можно воспользоваться традиционным способом и использовать модель ДЭС Гуи–Чепмена, дополняя ее несколько измененной изотермой Ленгмюра. Мы принимаем толщину адсорбированного слоя намного меньшей дебаевской длины экранирования в окружающем растворе λ. В простейшем случае можно считать, что плоскость скольжения совпадает с поверхностью мембраны, а величина ζ-потенциала совпадает с потенциалом на поверхности мембраны ϕ. В таком приближении значение ζ-потенциала представляет собой сумму двух вкладов — потенциала адсорбированного слоя полимера, ϕ1, и потенциала оставшейся свободной от полимера части липосомы, ϕ0. Это означает, что величина ϕ является средневзвешенным значением потенциалов ϕ1 и ϕ0 по площади покрытия поверхности полимером:
(2)
$\phi \left( c \right) = {\beta }\left( c \right){{\phi }_{1}} + \left( {1 - {\beta }\left( c \right)} \right){{\phi }_{0}},$Рис. 3.
Схематический вид рассматриваемой системы: 1 – молекулы ПЛ, 2 – молекулы КЛ, 3 – молекулы ДОФХ.
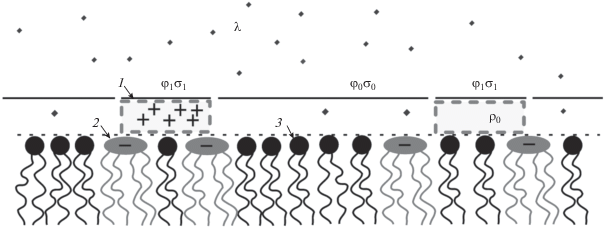
Используя теорию Пуассона–Больцмана, в одномерном приближении можно получить аналитическое выражение для величин ϕ0 и ϕ1. Одномерное приближение в данной задаче обосновано размером липосом, радиус которых ~0.5 мкм, что много больше дебаевской длины в объеме электролита. При этом также предполагается, что заряд КЛ равномерно распределен по области адсорбции на мембране с поверхностной плотностью σ0, а ПЛ равномерно заряжен с объемной плотностью заряда ρ0 и адсорбируется в виде областей со средней толщиной h. Плотность заряда области мембраны с адсорбированным полимером σ1 складывается из плотности заряда исходной мембраны и плотности заряда полимера:
Здесь использовано достаточно естественное допущение, что поверхностная плотность заряда на мембране, не занятой полимером, остается неизменной и равной σ0. Потенциалы на поверхности занятого и свободного участков поверхности липосомы, ϕ0 и ϕ1, находятся по формуле Гуи–Чепмена
(4)
${{\phi }_{i}} = 2\frac{{kT}}{e}\operatorname{arc} {\kern 1pt} {\text{sh}}\left( {\frac{{{{{\sigma }}_{i}}}}{{4e{{c}_{0}}{\lambda }}}} \right),\,\,\,\,i = 0,1,$Значения плотности заряда этих участков, σ0 и σ1, находятся из следующих соображений. Каждая полностью депротонированная молекула КЛ имеет в полярной области две отрицательно заряженные фосфатные группы. Поэтому плотность поверхностного заряда мембраны, состоящей только из КЛ, должна быть равна –2pe/acl, где acl — средняя площадь, приходящаяся на молекулу КЛ в липосоме, p – степень ионизации (депротонирования) КЛ. Результаты контрольных измерений для липосом из смеси КЛ с ДОФХ, проведенных в работе [12], хорошо согласуются с оценками в рамках модели ГЧШ, согласно которым адсорбция катионов приводит к снижению степени ионизации КЛ и величины поверхностного заряда. Для последующего анализа этот факт удобно характеризовать введением поправочного коэффициента K1, отражающего адсорбцию фонового электролита на поверхности мембраны. Таким образом, величина σ0 может быть представлена соотношением
где α – массовая доля КЛ в составе мембраны. Согласно данным работы [12] максимальная плотность заряда мембраны, состоящей только из КЛ, без учета адсорбции ионов составляет Smax = = –150 мКл/м2. Ранее, в работе [32] было показано, что эта величина отличается от максимально возможной, когда плотность заряда рассчитывается с учетом площади молекул липида в бислое при условии полного депротонирования каждой молекулы ПЛ и составляет примерно 230 мКл/м2 (один заряд электрона на каждые 0.6 нм2). В согласии с данными указанной работы мы принимаем пониженное значение плотности заряда, которой соответствует степень депротонирования КЛ p = 0.65. В дальнейшем мы будем пользоваться этим результатом, полагая, что адсорбция ПЛ не влияет на степень депротонирования КЛ ни на свободных от этого полимера участках мембраны, ни на занятых, где в дальнейшем заряд КЛ будет суммироваться с зарядом полимера (см. (7)). Заметим также, что введенный выше поправочный коэффициент K1 можно связать с потенциалом в явном виде (4):(6)
${{K}_{1}} = {\text{sh}}\left( { - \frac{{e{{\phi }_{0}}}}{{kT}}} \right)\frac{{2{{a}_{{{\text{cl}}}}}{{c}_{0}}{\lambda }}}{{{\alpha }p}}.$Это выражение позволяет оценить значения K1 для заданных значений α, используя данные измерений потенциала мембраны в зависимости от поверхностной доли КЛ, приведенные на рис. 2б. В частности, при α = 1 имеем K1 = 0.34, при α = 0.2 K1 = 0.66. Далее, для простоты можно считать, что молекула ПЛ представляет собой цепочку мономеров одинакового объема v0, несущих по одному положительному заряду. Поэтому максимальная объемная плотность заряда внутри слоя ПЛ может быть записана как e/v0. Реальная плотность заряда может отличаться от максимальной за счет адсорбции ионов на полимере и взаимодействия заряженных мономерных звеньев ПЛ друг с другом. Этот факт учитывается поправочным коэффициентом K2, (0 < K2 < 1). В слое полимера минимальной толщины h0, равной примерно 0.5 нм [33], на каждой молекуле КЛ в среднем могут адсорбироваться две структурные единицы ПЛ (т.е. два аминокислотных остатка). Тогда объем v0 может быть записан как aclh0/2. Ниже мы используем примерные значения молярной массы и размера молекул КЛ, для которых справедливы следующие приближенные равенства: acl ≈ 2adopc, Mcl ≈ 2Mdopc, а формула (2) приобретает вид
(7)
${{{\sigma }}_{1}} = \frac{{2e}}{{{{a}_{{{\text{cl}}}}}}}\left( { - {\alpha }{{K}_{1}} + {{K}_{2}}d} \right),$Для формулирования изотермы адсорбции согласно известным экспериментальным данным [11] будем считать, что адсорбция полимера в общем случае обратима, а степень обратимости зависит от размера макромолекул. Кроме того, необходимо учитывать ограниченное количество полимера в измерительной ячейке – адсорбция ПЛ приводит к существенному изменению его концентрации в растворе. Этот факт также учитывался ранее в работах [34–36], где исследовалась адсорбция на поверхности заряженных мембран неорганических многовалентных катионов с высокой афинностью к фосфолипидам. Результат использования модели ГЧШ применительно к этим системам привел к чрезвычайно высоким значениям эффективной константы связывания таких катионов, конкурирующих с катионами фонового раствора за связывание с заряженными группами фосфолипидов. Опираясь на данные молекулярного моделирования систем, аналогичных нашим [6], можно полагать, что в нашем случае, при достаточно низкой ионной силе 10 мМ, противоионы и поликатионы не конкурируют друг с другом за центры связывания на поверхности мембраны. Это позволяет рассматривать адсорбцию полимера независимо от связывания противоионов. Более того, опираясь на экспериментальные факты, указывающие на насыщение поверхности липосом полипептидами, можно полагать, что доля площади, занятой полимером, β(c), в области насыщения не может быть больше некоторого фиксированного значения β0, которое должно зависеть от доли КЛ в составе мембраны. Используя все приведенные выше предположения, можно записать основное кинетическое уравнение адсорбции полипептидов в следующем виде:
(8)
$\frac{{d{\beta }}}{{dt}} = {{K}_{{\text{a}}}}\left( {c - G{\beta }} \right)\left( {{{{\beta }}_{0}} - {\beta }} \right) - {{K}_{{\text{d}}}}{\beta }{\text{.}}$Здесь Ka и Kd — константы адсорбции и десорбции соответственно, а слагаемое Gβ учитывает уменьшение концентрации ПЛ в растворе за счет его адсорбции на мембране. Из общих соображений понятно, что оно должно быть пропорционально доле площади β(c), занятой адсорбированным полимером; G имеет смысл коэффициента пропорциональности между β(c) и уменьшением концентрации. Чтобы определить явный вид G, необходимо найти количество вещества полимера, адсорбированного на поверхности. Оно равно
где Sp — площадь поверхности, занятая полимером. Учитывая соотношение Sp = βStot, где Stot — полная площадь липосом, получим для G следующее выражение: где Clip — суммарное количество липида, образующего липосомы (в мг на мл раствора), Mcl — молярная масса КЛ. Введя константу связывания K, равную Ka/Kd, и рассмотрев стационарное состояние в (8), получим аналитическое выражение для изотермы адсорбции в следующем виде:(11)
${\beta }\left( c \right) = \frac{{1 + cK + G{{{\beta }}_{0}}K - {{{\left\{ {{{{\left( {1 + cK + G{{{\beta }}_{0}}K} \right)}}^{2}} - 4cG{{{\beta }}_{0}}{{K}^{2}}} \right\}}}^{{\frac{1}{2}}}}}}{{2GK}}.$Подстановка формулы (10) в выражение (11) приводит изотерму в явный вид, который используется далее для расчетов.
РЕЗУЛЬТАТЫ РАСЧЕТОВ И ИХ ОБСУЖДЕНИЕ
Рассмотрим экспериментальные зависимости, полученные для случаев, когда массовая доля КЛ в составе липосом равна 100% и 20%, что соответствует значениям α = 1 и 0.2. Целесообразно разделить эти экспериментальные зависимости на три группы исходя из разного числа звеньев N в молекулах ПЛ, условно называемых далее короткими (N = 5), средней длины (N = 12 и N = 20) и длинными (N > 100). Мы объединяем эти зависимости внутри каждой из групп и рассматриваем их вместе.
В цитируемой выше работе [11] и на рис. 4 представлены шесть зависимостей — по три для α = 1 и α = 0.2, которые аппроксимируются с помощью предлагаемой нами модели. В процессе аппроксимации варьируются следующие параметры: K2, K, d, β0 и Clip. Параметр Clip считается отличающимся от известного добавленного количества липида вследствие того, что некоторая часть липида может участвовать в образовании небислойных структур либо в формировании мультислойных липосом, оставаясь недоступной для полимера. Параметры β0 и Clip могут изменяться при варьировании доли заряженного липида α в смеси, т.е. значения ${\beta }_{0}^{{100}}$ и ${\beta }_{0}^{{20}}$ и $C_{{{\text{lip}}}}^{{100}}$ и $C_{{{\text{lip}}}}^{{20}}$ в общем случае могут быть различными. Толщина полимерного слоя d определяется в основном длиной макромолекул, как это следует из литературных данных [27] и наших данных, полученных методом атомно-силовой микроскопии при адсорбции ПЛ разной молекулярной массы на поверхности бислоя с разным содержанием КЛ [9, 12]. В нашей модели рассматриваются три возможных значения толщины слоя, отнесенные к длинному (dL), средней длины (dM) и короткому (dS) полимерам. Те же группы полимеров характеризуются разными значениями константы связывания: KL, KM и KS соответственно. При аппроксимации экспериментальных кривых были фиксированы исходные параметры системы: характеристики липида acl = 1.2 нм2, Mcl = = 1466 г/моль и дебаевская длина экранирования в фоновом электролите (10 мМ KCl) λ = 3 нм.
Рис. 4.
Зависимости потенциала ϕ на мембране от концентрации ПЛ (в расчете на мономерное звено) в растворе c для случаев α = 1 (а) и α = 0.2 (б). Точки – экспериментальные данные. Теоретические кривые соответствуют полимерам большой (1), средней (2) и малой длины (3) и получены при следующих значениях параметров модели, найденных при аппроксимации экспериментальных данных: K2 = 0.24, ${\beta }_{0}^{{100}}$ = 0.73, ${\beta }_{0}^{{20}}$ = 0.46, $C_{{{\text{lip}}}}^{{100}}$ = 0.35 мг/мл, $C_{{{\text{lip}}}}^{{20}}$ = 0.1 мг/мл, KL = = 3 × 106 М–1, KM = 1.8 × 105 М–1, KS = 1.2 × 105 М–1, dL = 2.6, dM = 1.7, dS = 1.
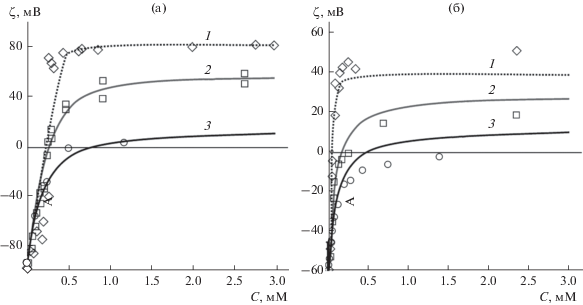
Полученные результаты аппроксимации и найденные при этом параметры модели позволяют сделать ряд важных выводов о характеристиках адсорбционного процесса. Прежде всего, необходимо отметить, что константа связывания длинных молекул, получаемая из аппроксимации данных, является, скорее, оценкой снизу, чем точным ее значением, т.к. увеличение этой константы на порядок не приводит к сколько-нибудь заметному изменению формы кривых адсорбции. Значения констант связывания молекул средней длины и коротких существенно меньше. Их зависимость от размера полимера заметно нелинейна, т.е. отношение констант связывания полимеров средней и малой длины, KM/KS, много меньше, чем отношение констант связывания KL/KM. Это говорит о том, что характер адсорбции существенным образом изменяется при увеличении длины полимера. Если для коротких и средней длины полимеров адсорбцию можно считать заметно обратимой, то адсорбция длинных полимеров становится практически необратимой. Этот вывод хорошо согласуется с экспериментальными данными [9] и, кроме того, подтверждается самим характером экспериментальных зависимостей, представленных на рис. 4. Судя по их форме и с математической точки зрения ясно, что чем больше константа связывания, тем резче кривая адсорбции выходит на насыщение. Именно поэтому в случае длинных молекул ПЛ кривая адсорбции намного круче, чем в случае средних по размеру и коротких молекул этого полимера.
В работе [30] высказано достаточно обоснованное предположение, что по величине константы связывания можно оценить число мономерных звеньев в составе молекулы ПЛ, которые непосредственно связаны с поверхностью мембраны. Авторы этой работы предлагают в качестве гипотезы, что образование каждой связи отдельным звеном полимера приводит к увеличению вероятности появления такой же связи у соседнего звена, что в математическом выражении сводится к перемножению констант связывания отдельных звеньев. Экспериментально определяется эффективное значение константы, равное K(1) = 10 М–1. В рамках данной гипотезы количество связей n между молекулой полимера и мембраной можно оценить, как n = log K. Согласно работе [30] у олигомера пентализина число эффективных связей n = 4. Применение этого формализма к нашей модели дает следующие результаты: для коротких полимеров n = 4.1, для полимеров средней длины n = 4.3, а в случае длинных полимеров n > 5.5. Заметим, что примерно такое же небольшое число связей n отмечается при молекулярно-динамическом моделировании подобных липид-полимерных систем. Так, в работе [6] методами молекулярной динамики было установлено, что общее число сформировавшихся связей между ПЛ и мембраной за все время моделирования (≈6 мкс) составляло 5 ± 2, что гораздо меньше числа аминогрупп в молекуле полимера.
Согласно нашей модели, малое увеличение количества связей с поверхностью при значительном увеличении размеров самой молекулы полимера приводит к росту толщины адсорбированного слоя, тогда как величина n коррелирует только с площадью контакта каждой молекулы полимера с мембраной. Это утверждение согласуется с данными атомно-силовой микроскопии [9]. При небольшой концентрации полимера центры связывания на мембране находятся в избытке, и большая часть из них остаются свободными; как следствие, толщина адсорбированного слоя минимальна и составляет величину h0, а площадь контакта полимерной молекулы с мембраной, наоборот, максимальна. С ростом концентрации полимера, когда он оказывается в суспензии в избытке, площадь контакта уменьшается до некоторого предельного значения, а толщина полимерного слоя растет. Согласно нашим результатам предельное значение площади контакта практически одинаково для молекул ПЛ разной длины. Это означает, что увеличение толщины будет тем больше, чем длиннее полимер. В случае короткого полимера толщина слоя в процессе адсорбции не изменяется и остается равной h0. Именно такой результат следует из данных атомно-силовой микроскопии [9].
Из результатов аппроксимации экспериментальных кривых следует, что при α = 1 отношение β0/αΑ = 0.73, где αΑ — доля площади мембраны, занятая КЛ. В то же время при α = 0.2 имеем β0/αΑ = 2.3. Это означает, что при малом содержании КЛ в составе мембран доля площади, занятой ПЛ, больше, чем суммарная площадь молекул КЛ. Значит, в этом случае под молекулами адсорбированного полимера может находиться не только заряженный, но и электрически нейтральный липид. Этот вывод также подтверждается литературными данными [33], согласно которым в случае октализина при α = 0.1 отношение β0/αΑ = 1.2.
Важнейшей характеристикой рассматриваемой системы является концентрация полимера, при которой отрицательный потенциал липосом полностью компенсируется положительным зарядом полимера в ТНЗ. При малых значениях потенциала его можно линейно связать с количеством поликатионов на поверхности мембраны. Экспериментальные графики на рис. 1 и 2а свидетельствуют о том, что варьирование параметров системы приводит к некоторому смещению ТНЗ. Для определения этого смещения необходимо решить уравнение ϕ(c) = 0 относительно c, используя формулу (2) для потенциала и подставляя в нее выражение (11) для доли площади, занятой ПЛ. Это позволяет получить следующую формулу для положения ТНЗ в шкале концентраций cz:
(12)
${{с }_{{\text{z}}}} = G\frac{{\left( { - {{\phi }_{0}}} \right)}}{{{{\phi }_{1}} - {{\phi }_{0}}}} + \frac{1}{K}\frac{{\left( { - {{\phi }_{0}}} \right)}}{{{{\phi }_{{\max }}}}},$Учитывая явный вид (10) коэффициента G, получаем для cz следующее выражение:
(13)
${{с }_{{\text{z}}}} = {{C}_{{{\text{lip}}}}}\frac{d}{{{{M}_{{{\text{cl}}}}}}}\frac{{\left( { - {{\phi }_{2}}} \right)}}{{{{\phi }_{1}} - {{\phi }_{2}}}}.$В соответствии с выражением (13) положение ТНЗ должно быть прямо пропорционально суммарному количеству липида, из которого приготовлены липосомы, как это и следует из экспериментальных данных (рис. 2а). Этот результат представляется интуитивно очевидным в предположении о необратимой адсорбции полимера, когда все положительно заряженные макромолекулы оказываются связанными с поверхностью мембран в суспензии и для нейтрализации поверхности требуется количество полимера, прямо пропорциональное суммарной поверхности липосом в суспензии, т.е. количеству фосфолипида. Таким образом, в рамках нашей модели (см. выражения (12) и (13)) удается непротиворечивым образом объяснить оба результата, касающихся смещения ТНЗ при варьировании количества заряженного фосфолипида в составе мембраны либо только в суспензии липосом.
Рассмотрим теперь зависимость потенциала ϕ от доли КЛ α в составе липосом при фиксированной концентрации ПЛ. Эта зависимость представляет особый интерес, поскольку именно доля КЛ определяет количество молекул ПЛ, связанных с поверхностью, а значит, и общие характеристики полимерного слоя. Здесь мы учитываем, что предельная доля площади β0, занимаемая ПЛ, является функцией α; определить ее позволяет наша модель при аппроксимации экспериментальных данных. В уже цитированной выше работе [11] содержатся только два значения α: α = 0.2 и α = 1, поэтому необходимо интерполировать зависимость β0(α) с учетом полученных ранее результатов и рассчитать зависимость ϕ(α) для разных значений концентрации полимера c. При этом следует учесть зависимость KL(α) с применением выведенной выше формулы (6), фиксируя длину и другие полученные ранее параметры для ПЛ каждого типа. Результаты расчета указанных теоретических зависимостей пред-ставлены на рис. 5.
Рис. 5.
Зависимости потенциала ϕ(α) для полимеров большой (а), средней (б) и малой длины (в) при фиксированных значениях концентрации c, равных 0.0015 (1), 0.0005 (2) и 0.0003 моль/л (3).
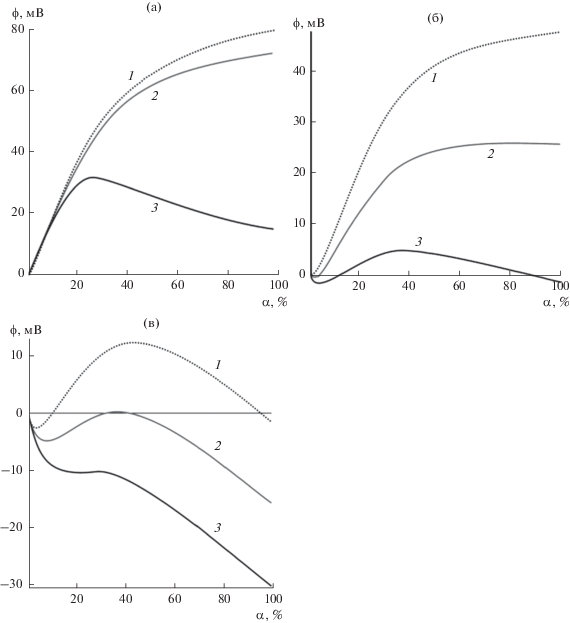
При достаточно большой концентрации полимеров, примерно соответствующей насыщению (c ~ 0.001 М) (кривые 1 и 2 на рис. 5а, 5б), снижение доли заряженного компонента α в составе мембран приводит к монотонному уменьшению потенциала ϕ. Немонотонная зависимость ϕ(α) (кривые 3) наблюдается при небольших концентрациях (примерно соответствующих ТНЗ при c ~ ~ 0.0001 М) для всех ПЛ и становится более выраженной с уменьшением длины полимера. Модель предсказывает разный характер поведения кривых при изменении доли заряженного липида в мембране, т.е. при значениях α вблизи нуля и вблизи единицы.
При малых значениях α потенциал приближенно определяется выражением
(14)
$\phi = \alpha \frac{{kT}}{{e{\lambda }{{c}_{0}}{{a}_{{{\text{cl}}}}}}}\left( {\frac{{cK{{K}_{{\text{2}}}}d}}{{1 + cK}} - {{K}_{1}}} \right).$В этом случае должно выполняться соотношение β0 ~ α. Из выражения (14) следует, что большие значения K и d соответствуют положительным значениям ϕ, а малые — отрицательным. В частности, при обратимой адсорбции молекул ПЛ малого и среднего размеров потенциал в окрестности начала координат на рис. 5б и 5в отрицателен, а при высокоэффективной, почти необратимой адсорбции больших молекул ПЛ — положителен (рис. 5а).
При максимальном содержании в мембране заряженного липида (α ≈ 1) поведение системы в большей степени зависит от концентрации полимера c. При малой концентрации ПЛ (кривые 3 на рис. 5) имеющегося в замкнутой измерительной ячейке количества полимера недостаточно для перезарядки всех липосом, и их средний потенциал остается отрицательным, когда размер молекул ПЛ недостаточно велик. В этом случае (кривые 3) заряженный липид имеется в избытке даже при некотором уменьшении его доли α в составе мембран. Это означает, что в окрестности α = 1 электростатическoе притяжение поликатионов к поверхности несколько понижается, но еще не приводит к существенному уменьшению суммарной площади, занятой полимером. При таких значениях α величина потенциала сдвигается в положительную сторону с увеличением концентрации полимера (производная к соответствующим кривым в этой области имеет отрицательное значение).
При разработке модели мы задавались целью дать наиболее простое количественное описание сложной формы электрокинетических кривых, когда количество параметров модели примерно отражает конечное число ключевых особенностей экспериментальных данных. Упрощение модели стало возможным при использовании ряда очевидных ограничений, которые оправданы только качественно правильным предсказанием основных тенденций в описании достаточно сложного полимер-липидного взаимодействия. Так, толщина адсорбированного слоя h принята много меньшей дебаевской длины экранирования в окружающем фоновом электролите λ. Наши расчеты определяют значение h порядка 1 нм, хотя в описываемых нами экспериментах λ = 3 нм. По этой причине предлагаемая модель не позволяет корректно учесть возможное влияние ионной силы на характеристики полимерного слоя. Более того, в рамках используемого здесь приближения для оценки поверхностного потенциала липосом использовано соотношение Смолуховского (1) и не учтены гидродинамические эффекты и положение плоскости скольжения, которое может смещаться при наличии полимерного слоя на поверхности коллоидных частиц. Представляется очевидным, что чем больше полимера адсорбировано на мембране, тем дальше от ее поверхности должна отстоять плоскость скольжения, что может приводить к уменьшению вклада в потенциал от отрицательного заряда, расположенного на свободных участках мембраны. Для простоты, вместо решения уравнения Пуассона–Больцмана в области занятой полимером, постулируется линейная связь между объемной плотностью заряда внутри полимерного слоя и поверхностным потенциалом на мембране (3). Это допущение приводит к завышенным значениям эффективного заряда полимера и как следствие, приводит к менее реалистичным значениям параметров модели. В частности, согласно нашим расчетам, общее количество липида, принимающее участие в адсорбции полимера, Clip, оказывается в несколько раз меньше использованного в экспериментах (примерно 1 мг/мл). Здесь имеет место сочетание двух факторов: несовершенство модели и особенности проведения экспериментов, в которых участвовали не однослойные, но мультиламеллярные липосомы. И наконец, константа связывания полимера K в рамках данной модели предполагалась независящей от доли заряженного липида α в составе мембраны. Если это не так, а степень адсорбции определяется числом связей молекулы полимера с липидами в мембране, то такая зависимость должна иметь место. Действительно, при уменьшении количества отрицательно заряженных липидов, остается меньше возможности у молекул полимера образовать нужное количество связей для получения измеренной в опыте доли поверхности, заполненной полимером. Адсорбция при этом становится в большей степени обратимой.
ЗАКЛЮЧЕНИЕ
Предлагаемая здесь модель при построении изотермы адсорбции учитывает геометрические размеры адсорбируемых молекул полимера в наиболее простом приближении. Это позволило получить объяснение основных закономерностей, замеченных ранее в работах [9–12] при проведении электрокинетических измерений в суспензии липосом. При этом были получены численные значения параметров модели после аппроксимации экспериментальных данных. Используя подход, предложенный для подобных систем в работе [30], мы пришли к выводу, что даже значительное увеличение размеров полимера приводит к весьма незначительному увеличению числа его связей с мембраной. Столь малое изменение количества связей, может быть, компенсируется увеличением толщины слоя полимера на мембране в области насыщения. Это обстоятельство становится особенно существенным для полимеров большого размера. Мы полагаем, что этот вывод может быть проверен методами молекулярной динамики, прямыми измерениями методом атомно-силовой микроскопии или путем регистрации структуры полимерного слоя другими способами. Экспериментальным проверкам доступны и другие предсказания модели. В частности, варьирование доли отрицательно заряженного липида в составе мембраны при заданной величине концентрации ПЛ в растворе может приводить к сложному и немонотонному изменению потенциала (рис. 5). Такая немонотонность может быть замечена при анализе адсорбции полимеров небольших размеров и концентрациях, примерно соответствующих ТНЗ. Следует отметить, что используемая нами модель не привязана к конкретной химической структуре поликатиона и потому может быть применена для исследования адсорбции других биологически значимых синтетических поликатионов и полипептидов.
Авторы благодарят А.М. Нестеренко, А.А. Ярославова и его сотрудников за ценные обсуждения и рекомендации.
Работа поддержана грантом Российского фонда фундаментальных исследований № 16-04-00556.
Список литературы
Kenawy E.-R., Worley S.D., Broughton R. // Biomacromolecules. 2007. V. 8. P. 1359.
Tyagi P.K., Gupta B., Singh H. // J Macromol. Sci. A. 1993. V. 30. P. 303.
Kenawy E.-R., Abdel-Hay F.I., El-Shanshoury A.E.-R.R., El-Newehy M.H. // J. Polym. Sci., Part A: Polym. Chem. 2002. V. 40. P. 2384.
Wang C., Dong L. // Trends Biotechnol. 2015. V. 33. P. 10.
Vinogradov S.V., Bronich T.K., Kabanov A.V. // Adv. Drug Deliv. Rev. 2002. V. 54. P. 135.
Kostritskii A.Yu., Kondinskaia D.A., Nesterenko A.M., Gurtovenko A.A. // Langmuir. 2016. V. 32. P. 10402.
Horner A., Antonenko Y.N., Pohl P. // Biophys. J. 2009. V. 96. P. 2689.
Reuter M., Schwieger C., Meister A., Karlsson G., Blume A. // Biophys. Chem. 2009. V. 144. P. 27.
Marukovich N., McMurray M., Finogenova O., Neste-renko A., Batishchev O., Ermakov Yu. // In: Advances in Planar Lipid Bilayers and Liposomes. A Tribute to Marin D. Mitov / Ed. by Iglic A., Genova J. Amsterdam: Elsevier, 2013. P. 139.
Марукович Н.И., Нестеренко А.М., Ермаков Ю.А. // Биол. мембраны. 2014. Т. 31. С. 401.
Финогенова О.А., Филинский Д.В., Ермаков Ю.А. // Биол. мембраны. 2008. Т. 25. С. 217.
Финогенова О.А., Батищев О.В., Инденбом А.В., Золотаревский В.И., Ермаков Ю.А. // Биол. мембраны. 2009. Т. 26. С. 429.
Ikeda T., Tazuke S., Watanabe B. // Biochem. Biophys. Acta. 1983. V. 735. P. 380.
Rossi G., Monticelli L. // J. Phys. Condens. Matter. 2014. V. 26. 503101.
Schultz M., Olubummo A., Binder W.H. // Soft Matter. 2012. V. 8. P. 4849.
Ivashkov O.V., Sybachin A.V., Efimova A.A., Pergu-shov D.V., Orlov V.N., Schmalz H., Yaroslavov A.A. // Chem. Phys. Chem. 2015. V. 16. P. 2849.
Yaroslavov A.A., Sitnikova T.A., Rakhnyanskaya A.A., Ermakov Y.A., Burova T.V., Grinberg V.Y., Menger F.M. // Langmuir. 2007. V. 23. P. 7539.
Yaroslavov A.A., Kul’kov V.E., Polinsky A.S., Baibakov B.A., Kabanov V.A. // FEBS Lett. 1994. V. 340. P. 121.
Laroche G., Carrier D., Pezolet M. // Biochemistry. 1988. V. 27. P. 6220.
Февралева И.С., Ермаков Ю.А., Атаулаханов Р.И. // Иммунология. 1986. Т. 1. С. 66.
Hesselink F.T. // J. Colloid Interface Sci. 1977. V. 60. P. 448.
Hill R.J. // Phys. Rev. E. Stat. Nonlin. Soft. Matter Phys. 2004. V. 70. № 5. Pt. 1. P. 051406.
Hill R.J., Saville D.A. // Colloids Surf. A. 2005. V. 267. P. 31.
Mueller H., Butt H.J., Bamberg E. // J. Phys. Chem. B. 2000. V. 104. P. 4552.
Schwieger C., Blume A. // Biomacromolecules. 2009. V. 10. P. 2152.
Yaroslavov A.A., Sitnikova T.A., Rakhnyanskaya A.A., Yaroslavova E.G., Davydov D.A., Burova T.V., Grinberg V.Y., Shi L., Menger F.M. // J. Am. Chem. Soc. 2009. V.131. P. 1666.
Kurihara K. // Adv. Colloid Interface Sci. 2010. V. 158. P. 130.
Ермаков Ю.А. // Коллоид. журн. 2000. Т. 62. С. 437.
Khomich D.A., Nesterenko A.M., Kostrinskii A.Yu., Kondinskia D.A., Ermakov Yu.A., Gurtovenko A.A. // J. Phys.: Conf. Series. 2017. V. 794. 012010.
Kim J., Mosior M., Chung L.A., Wu H., McLaughlin S. // Biophys. J. 1991. V. 60. P. 135.
Hunter R.J. Zeta Potential in Colloid Science. Principles and Applications. London: Academic Press, 1981.
Ermakov Yu.A. // Biochim. Biophys. Acta. 1990. V. 1023. P. 91.
McGeachy A.C., Caudill E.R., Liang D., Cui Q., Pedersen J.A., Geiger F.M. // Chem. Sci. 2018. V. 9. P. 4285.
Ermakov Yu., Kamaraju K., Dunina-Barkovskaya A., Vishnyakova K., Egorov Y., Anishkin A., Sukharev S. // Biochemistry. 2017. V. 56. P. 5457.
Ermakov Y.A., Kamaraju K., Sengupta K., Sukharev S. // Biophys. J. 2010. V. 98. P. 1018.
Ermakov Y.A., Averbakh A.Z., Yusipovich A.I., Sukharev S. // Biophys. J. 2001. V. 80. P. 1851.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал