

Коллоидный журнал, 2019, T. 81, № 2, стр. 212-223
Регулируемое оксифторированием изменение смачиваемости поверхности полимерных пленок
В. Г. Назаров 1, Ф. А. Доронин 1, А. Г. Евдокимов 1, Г. О. Рытиков 1, В. П. Столяров 1, *
1 Московский политехнический университет
107023 Москва, Большая Семеновская ул., 38, Россия
* E-mail: 110505n@gmail.com
Поступила в редакцию 07.08.2018
После доработки 06.11.2018
Принята к публикации 31.08.2018
Аннотация
Изучена модификация поверхности полимеров смесями фтора с кислородом при варьировании в широких диапазонах продолжительности процесса и процентного соотношения этих газов в смеси. Такая модификация приводит к росту гидрофильности поверхности полимеров, тем большему, чем выше доля кислорода в смеси газов и чем больше время оксифторирования. Варьируя условия оксифторирования, можно в достаточно широких пределах управлять такими физико-химическими свойствами полимера как смачиваемость, поверхностная энергия и адгезия. В случае полиолефинов, например, угол смачивания водой изменяется от 78°−87° для исходных полимеров до 49°−60° для модифицированных. Для гетероцепных полимеров этот интервал может быть еще шире и составляет, например, для ПЭТФ от 67° до 4°, т.е. достигается практически полное растекание воды по поверхности полимера. Рассмотрен вклад шероховатости поверхности полимера до и после его химической обработки в наблюдаемые значения краевого угла воды. Показано, что увеличение смачиваемости поверхности полимера в результате оксифторирования может быть использовано для получения полимерных пленок со смачиваемостью, варьируемой путем их последующей деформации растяжением.
ВВЕДЕНИЕ
Известно, что смачивание твердого тела жидкостью зависит как от химического состава поверхности, так и от ее шероховатости [1]. Информация о свойствах поверхностей полимеров, таких как смачиваемость, поверхностная энергия, адгезия, имеет большое значение при разработке широкого спектра технологий, в том числе, нанесения красок и покрытий, получения биоматериалов для трансплантологии, в микрофлюидикe и т.д. [2–13].
Характеристики поверхности полимеров играют важную роль в решении эколого-технологической проблемы биостойкости и биоразлагаемости различных полимерных материалов. В первом случае решаются вопросы защиты материалов от негативного воздействия на них микроорганизмов (ресурсосбережение) и улучшения качества среды обитания человека, так как многие бактерии и грибы (активные биодеграданты) являются условно-патогенными организмами, способными вызывать серьезные заболевания [11, 12]. Во втором − устранения бытовых отходов и отходов промышленных производств. Это связано с тем, что процессы биодеградации начинаются с поверхности полимера и определяются, в том числе, его гидрофильностью и распределением конденсата влаги на поверхности материала [11]. Кроме того показано [13], что биоразрушение полимеров значительно ускоряется при действии на них УФ-излучения или окислительных реагентов. Можно предположить, что негативное влияние этих факторов может быть скорректировано одновременно с регулированием смачиваемости полимерного материала. Поверхностные характеристики полимерных материалов учитываются при разработке нанотехнологий для создания устройств на гибкой основе [14, 15]. Так, например, в ряде работ показано [16–20], что поверхностные характеристики подложки существенным образом влияют на формирование кольцевых осадков при испарении капель дисперсий (coffee ring effect).
Исследования в области поверхностных свойств направлены как на повышение гидрофобности и уменьшение смачиваемости полимеров, так и, наоборот, на повышение их гидрофильности и улучшение смачиваемости. В последнем случае используется широкий набор физических, физико-химических и химических методов воздействия на поверхность полимера [9]. Наиболее универсальными из них являются плазмохимическая обработка [16–19] и модификация поверхности полимера химическими реагентами [9, 25–28]. Применение плазмохимической обработки, особенно в присутствии кислорода, приводит к уменьшению угла смачивания водой $\left( {{{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}} \right)$ в 1.5−2.0 раза таких полимеров как полипропилен (ПП), поликарбонат, полиэтилентерефталат (ПЭТФ), полиэтилен (ПЭ) низкой плотности (ПЭНП), поливинилиденфторид и в 5−6 раз полистирола и полиимида [24, 29]. Однако достигаемый эффект уменьшения угла смачивания не всегда стабилен во времени, а сам этот метод требует использования сложного оборудования. Такая нестабильность связана, по-видимому, с особенностями плазмохимического процесса, первичным актом которого является разрыв химических связей и образование в структуре полимера свободных радикалов (в том числе, долгоживущих). Эти радикалы со временем претерпевают химические (в том числе, окислительные) и рекомбинационные превращения, приводящие к сшиванию и деструкции полимера, образованию ненасыщенных связей и т.д. [24].
Этого недостатка практически лишены методы химической модификации поверхности полимеров, при использовании которых в оптимальных условиях процессы трансформации полимера протекают достаточно целенаправленно (кинетика совокупности соответствующих химических реакций становится более прогнозируемой) и с минимальной долей побочных реакций. В контексте данной статьи необходимо обратить внимание на следующее: сульфирование ПЭ позволяет регулируемо изменять его поверхностную энергию (γ) от 27 до 60 мН/м, однако применение этого метода ограничивается практически только полиолефинами и их галогенсодержащими аналогами [9, 25]. Более универсален метод поверхностного фторирования. На большом количестве примеров показано, что с его помощью можно направленно изменять смачиваемость и адгезионные свойства полимеров [9, 25–28, 30], в том числе, в случае, когда в газовой смеси наряду с фтором присутствует кислород [9, 28, 31–34]. При действии смесей фтора с кислородом на поверхности полимера в результате фторирования образуются группы –CHF– и –CF2–, а за счет окисления – высокополярные группы –C(=O)F и ‒C(=O)–. Фторангидридные группы при контакте с атмосферной влагой быстро превращаются в еще более полярные группы –C(=O)OH [9]. В результате появления в поверхностном слое полимера при его оксифторировании фрагментов, содержащих кислород, значительно возрастают значения γ и ее полярной составляющей (γP). Так, согласно данным работы [25], при обработке поверхности ПЭ смесью 15% F2 + 85% He значения γ и γP сначала возрастают с увеличением времени фторирования примерно на 30%, а затем уменьшаются. Также показано, что обработка ПЭ смесями фтора с кислородом в определенных условиях увеличивает γ почти до 72 мН/м [34]. Для ПП при обработке его смесью 10% F2 + 90% O2 изменения γ и γP также значительно более существенны, чем при обработке смесью 10% F2 + 90% N2: γP возрастает с ≈2 мН/м до 35−39 мН/м, а γ − с ≈ 30.5 до ≈56.5 мН/м [35, 36]. Приведенные примеры показывают, что модификация поверхности полимеров путем обработки смесью фтора с кислородом может быть эффективным методом направленного изменения их смачиваемости и увеличения поверхностной энергии. Однако взаимосвязь условий оксифторирования с достигаемыми значениями угла смачивания и свободной поверхностной энергии не исследована.
Целью нашей работы является изучение кинетики модификации поверхности полимеров смесями фтора и кислорода (оксифторирования) для определения условий регулируемого изменения их смачиваемости.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
При экспериментальном исследовании закономерностей формирования модифицированного поверхностного слоя полимера под действием смесей фтора и кислорода при различных соотношениях этих компонентов и условиях модификации использовали пленки гомо- и гетероцепных полимеров: ПЭНП (толщиной 185 мкм), ПП (50 мкм) и ПЭТФ (20 мкм). Кроме того, для изучения влияния деформации растяжения на смачиваемость модифицированного полимера использовали пленки предельно ориентированного жесткоэластичного ПП (ПП-ЖЭ) толщиной 30 мкм, которые могут быть обратимо деформированы.
Оксифторирование полимерных пленок осуществляли по разработанной ранее методике [37] в реакторе из нержавеющей стали объемом 300 мл, конструкция которого позволяет проводить его вакуумирование до и после проведения этого процесса. Такой небольшой объем реактора был выбран для минимизации времени его заполнения газовой смесью при малых временах модификации. Для выявления закономерностей оксифторирования полимеров использовали смеси фтора и кислорода в буферных газах (гелии и азоте) следующих составов: 7.5 об. % F2 и 0.25 об. % O2 (данная концентрация кислорода определяется его содержанием в промышленно производимом фторе), 7.5% F2 и 6% O2, 7.5% F2 и 10% O2). Содержание кислорода, равное 6 и 10%, задавали подачей в реактор осушенного воздуха и контролировали с помощью вакуумметра с точностью 0.1 атм (содержание кислорода в воздухе принимали равным 21 об. %) [38]. Время обработки полимерных образцов составляло 1, 2, 5, 30, 60 и 180 мин. Процесс оксифторирования контролировали гравиметрически по увеличению массы модифицированного образца, отнесенной к площади его поверхности ($C_{{\text{A}}}^{{\text{M}}},$ кг/м2 – степень модификации).
Физико-химические свойства образцов полимеров в значительной степени определяются свободной поверхностной энергией, зависящей от химического состава и микрорельефа их поверхностей. Поэтому проводили анализ атомарного состава поверхностных слоев и их микрорельефа, а также измеряли углы смачивания пленок полярными и неполярными жидкостями, позволяющие оценить значения свободной энергии в целом, ее полярной и неполярной (дисперсионной) составляющих [40].
Для определения углов смачивания полимерных пленок дистиллированной водой и этиленгликолем (ГОСТ 19 710-83) до и после оксифторирования использовали метод сидячей капли. Измерения проводили на лабораторном стенде для скоростной микросъемки. Для обеспечения одинаковых условий эксперимента с учетом рекомендаций [39] капли жидкости объемом 6.1 мкл наносили на поверхность полимерной пленки при температуре 20°C микрометрическим шприцeм, расположенным на фиксированной высоте от подложки. Среднее значение угла смачивания оценивали по результатам пяти независимых измерений. Относительная ошибка измерений составила ± 8%. Расчеты величины поверхностной энергии γ, ее полярной γP и дисперсионной γD составляющих проводили по уравнениям, приведенным в монографии [40].
ИК-спектры были измерены с помощью фурье-спектрометра Инфралюм ФТ801 (Люмэкс-Сибирь, РФ) в режиме нарушенного полного внутреннего отражения с использованием кристалла ZnSe.
Атомный состав поверхностных слоев пленок определяли методом рентгеновской фотоэлектронной спектроскопии (РФЭС) с помощью спектрометра JPS-9200 (JEOL, Япония) с полусферическим анализатором и двуханодной рентгеновской пушкой. Разрешение спектрометра по уровню Ag3d – 0.9 эВ (рентгеновское излучение MgKα). Глубина анализа составляет примерно 2 нм.
Изображения образцов были получены в автоэмиссионном сканирующем электронном микроскопе (СЭМ) JSM 7500 (JEOL, Япония) в режиме детектирования вторичных электронов при ускоряющем напряжении 10 кВ; предварительно на поверхность образцов напыляли слой платины толщиной 10−12 нм в установке магнетронного типа JFC-1600 Auto Fine Coater (JEOL, Япония).
Шероховатость поверхности полимерных пленок оценивали с помощью атомно-силового микроскопа (АСМ) СММ-2000 (ЗАО “Завод Протон-МИЭТ”, Россия).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Экспериментально установлено, что при всех условиях модификации наблюдается увеличение массы полимерных пленок. Это свидетельствует о протекании химических реакций полимера с компонентами газовой смеси. По экспериментальным данным построены кинетические зависимости степени модификации пленок ПЭНП и ПЭТФ от состава модифицирующей газовой смеси (рис. 1). На начальном участке кривых (примерно до 1 мин) увеличение массы (рост степени модификации $C_{{\text{A}}}^{{\text{M}}}$) пленок определяется, очевидно, скоростью химических реакций (Vr), а на конечном (после 50 мин оксифторирования) – скоростью диффузии компонентов газовой смеси вглубь полимера (Vr). На среднем участке кинетических зависимостей (от 1 до 50 мин) процесс в большей степени лимитируется скоростью поступления реагентов к более глубоким слоям исходного полимера, т.е. их диффузией через уже модифицированные слои полимера. Для количественного сравнения скоростей оксифторирования на начальном и конечном участках кинетических зависимостей были рассчитаны значения Vr и Vd по касательным, проведенным к этим участкам графиков соответствующих зависимостей.
Рис. 1.
Кинетические зависимости степени оксифторирования $(C_{{\text{A}}}^{{\text{M}}})$ ПЭНП (а) и ПЭТФ (б) при различном составе газовой смеси (об. %): 1 – 7.5 F2–0.25 O2, 2 – 7.5 F2–6 O2, 3 – 7.5 F2–10 O2.
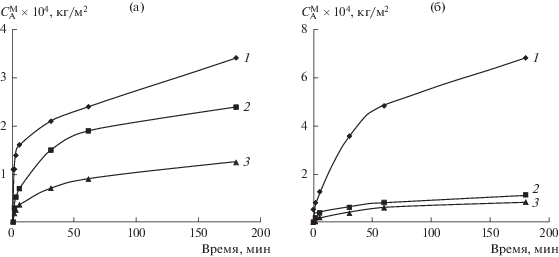
Полученные значения Vф и Vд представлены в табл. 1. Из приведенных в табл. 1 данных следует, что кинетика оксифторирования ПЭНП и ПЭТФ имеет некоторые общие закономерности. Сравнение значений Vr и Vd показывает, что при переходе к диффузионно-контролируемой стадии модификации скорость процесса снижается на два порядка. Повышение в газовой смеси концентрации кислорода с 0.25 до 6−10% уменьшает скорость модификации на обеих стадиях процесса. Однако если для ПЭНП и ПЭТФ Vr снижается примерно одинаково, в 2.5−6 раз, то на второй стадии Vd уменьшается для ПЭТФ в 5−10 раз, а для ПЭНП лишь в 2.5 раза. Это различие связано с тем, что оксифторирование поверхностного слоя ПЭТФ, по-видимому, сопровождается более глубокой его перестройкой с образованием плотного модифицированного слоя с меньшим свободным объемом, что снижает скорость диффузии реагентов вглубь полимера.
Таблица 1.
Расчетные значения скорости модификации полимеров на различных участках кинетических кривых
| Состав газовой смеси, F2/O2, об. % | Скорость процесса модификации | |||
|---|---|---|---|---|
| ПЭТФ | ПЭНП | |||
| скорость модификации
на начальном
участке, Vr × 107, кг/(м2 мин) |
скорость модификации
на диффузионном участке, Vd × 107, кг/(м2 мин) |
скорость модификации
на начальном участке, Vr × 107, кг/(м2 мин) |
скорость модификации
на диффузионном участке, Vd × 107, кг/(м2 мин) |
|
| 7.5/0.25 | 400 | 13 | 700 | 8 |
| 7.5/6.0 | 150 | 2.5 | 260 | 4 |
| 7.5/10.0 | 100 | 1.6 | 120 | 3 |
Оксифторирование обоих полимеров приводит к образованию в полимерной цепи как фторсодержащих фрагментов (CHF, СF2, СF3), так и карбонил- и карбоксилсодержащих групп, что подтверждается данными ИК-спектроскопии (рис. 2 и 3).
Рис. 2.
ИК-спектры исходной (1) и оксифторированных смесью состава 7.5% F2–10% O2 в течение 5 (2) и 180 мин (3) пленок ПЭНП.
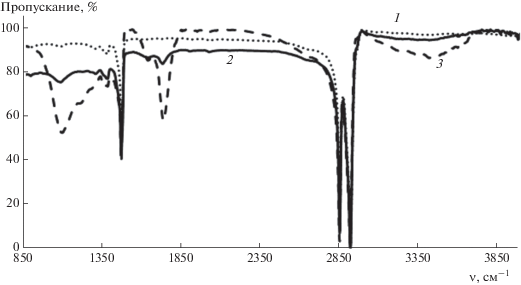
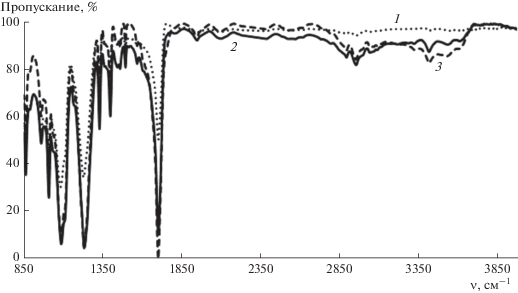
Рис. 3.
ИК-спектры исходной (1) и оксифторированных смесью состава 7.5% F2–10% O2 в течение 5 (2) и 180 мин (3) пленок ПЭТФ.
В спектре ПЭНП (рис. 2) появляются широкая диффузная полоса поглощения в области 1000−1300 см−1 с максимумом при 1200 см−1, обусловленная колебаниями связей С–F, а также уширенные полосы с максимумами при 1662 см−1 (слабая) и 1743 см−1 (средняя), соответствующие карбонильной группе фрагментов ‒СНF(C=O)СНF– и –СНF(C=O)СН2– либо –СНF(C=O)О–. Широкая полоса в области 3250−3500 см−1, обусловленная валентными колебаниями гидроксила, свидетельствует о наличии в модифицированном слое полимера групп –С(О)ОН, которые образуются в результате гидролиза фторангидридных групп атмосферной влагой. При оксифторировании ПЭНП смесями с меньшим содержанием кислорода наблюдаются аналогичные изменения в ИК-спектре. В то же время эксперимент показал, что с увеличением в газовой смеси доли кислорода интенсивность поглощения в области колебаний связей С–F уменьшается, а в области колебаний связей С=О возрастает. Аналогичные результаты были получены и для пленок ПП, что свидетельствует об общности этих изменений для полиолефинов.
При оксифторировании ПЭТФ также наблюдается образование фторированных и новых окисленных фрагментов в макромолекулах полимера, что подтверждают ИК-спектры образцов, модифицированных смесью состава 7.5% F2 + + 10% О2 (рис. 3). К поглощению CF-группами можно отнести уширенную полосу в районе 1030 и в области 1150−1230 см−1. Полоса колебаний новых групп C=O накладывается на сигнал аналогичных групп исходного полимера, более слабое поглощение в области 1950−1825 см−1 может быть отнесено к фрагментам −C(O)F, −C(O)OH или −CHFC(O)O−. Наличие карбоксильных групп подтверждается широкой полосой поглощения в области 3250−3500 см−1.
Таким образом, воздействие на поверхность полимера смесями фтора и кислорода приводит наряду с замещением атомов водорода в полимерной цепи на атомы фтора к образованию значительного числа кислородсодержащих фрагментов, количество которых можно регулировать, изменяя условия реакции и соотношение реагентов. Ориентировочная оценка толщины модифицированного слоя, выполненная исходя из степени модификации полимеров и зависимости “степень модификации–толщина модифицированного слоя” [9], показывает, что эта величина варьируется, как правило, от 0.1 до 4.0 мкм в зависимости от условий обработки и природы полимера. В то же время отметим, что с точки зрения смачиваемости толщина модифицированного слоя принципиального значения не имеет, т.к. она определяется химическим составом нескольких верхних молекулярных слоев (и шероховатостью поверхности).
Образование в модифицированном слое карбонильных и, особенно, карбоксильных групп закономерно приводит к улучшению смачиваемости поверхности полимера. На рис. 4 представлены зависимости ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ от времени модификации t (при различном содержании O2 в оксифторирующей смеси).
Рис. 4.
Кинетические зависимости краевого угла воды для пленок ПЭНП (а) и ПЭТФ (б), оксифторированных смесями состава (об. %): 7.5 F2–0.25 O2 (1), 7.5 F2–6 O2 (2) и 7.5 F2–10 O2 (3).
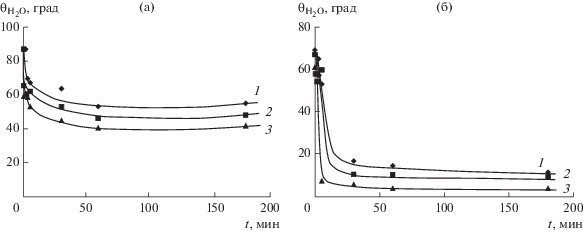
Из кинетических зависимостей (рис. 4а) следует, что оксифторирование весьма существенно уменьшает ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ как для ПЭНП, так и для ПЭТФ. Это уменьшение максимально при обработке полимеров газовой смесью состава 7.5% F2 + 10% O2 и для ПЭНП двукратно (с 87° до 42°). Аналогичное уменьшение ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ наблюдается при модификации ПП (с 87° до 53°) и ПП‑ЖЭ (с 83° до 59°). Еще более существенное уменьшение ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ имеет место в случае ПЭТФ – с 67° до 4°, что соответствует практически полному растеканию воды по поверхности полимера. Из кинетических зависимостей также следует, что наиболее интенсивно ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ уменьшается при малых временах модификации поверхности полимера, примерно до t = 30 мин. В дальнейшем он изменяется незначительно, что обусловлено высокой скоростью реакций на начальных стадиях оксифторирования (рис. 1 и табл. 1), в результате которых происходит существенное изменение химической природы поверхности полимера. Об этом свидетельствуют также данные РФЭС, приведенные в табл. 2.
Таблица 2.
Зависимость содержания элементов в поверхностном слое полимера от времени оксифторирования газовой смесью состава 7.5% F2–0.25% O2
| Время оксифторирования, мин |
Полимер | |||||
|---|---|---|---|---|---|---|
| ПЭНП | ПЭТФ | |||||
| содержание элемента, ат. % | содержание элемента, ат. % | |||||
| С 1s | F 1s | O 1s | С 1s | F 1s | O 1s | |
| 0 | 100* | – | – | 72.3 | – | 27.8 |
| 2 | 51.4 | 43.5 | 5.1 | 50.4 | 35.0 | 14.6 |
| 5 | 48.7 | 46.5 | 4.8 | 47.4 | 34.8 | 17.8 |
| 180 | 47.4 | 47.3 | 5.4 | 39.8 | 37.0 | 23.2 |
Согласно данным табл. 2, на начальном этапе оксифторирования ПЭНП (в течение первых 2 мин) реакция протекает с большой скоростью и достигается соотношение С : F, равное примерно 5 : 4. Бóльшая часть образовавшихся связей С−F приходится на фрагменты СFН. При дальнейшем оксифторировании количество введенного в полимер фтора незначительно увеличивается, и соотношение С : F при t = 180 мин достигает 1 : 1 за счет возрастания доли фрагментов СF2. В составе поверхностного слоя с началом оксифторирования обнаруживаются кислородсодержащие фрагменты, образование которых обусловлено присутствием кислорода во фторирующей газовой смеси. За счет увеличения количества СF2-групп при продолжительной модификации гидрофильность поверхности полимера снижается и ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ несколько возрастает. Таким образом, при оксифторировании ПЭНП с целью уменьшения его смачиваемости водой следует разумно ограничивать время модификации.
Содержание фтора в поверхностном слое ПЭТФ за 2 мин достигает 35% и с увеличением времени оксифторирования практически не изменяется. В этой связи снижение содержания углерода и рост содержания кислорода при дальнейшей модификации можно отнести на счет реакций деструкции, например разрыва связей С−О сложноэфирных групп под действием выделяющегося фтористого водорода.
Кроме химического состава на смачиваемость и поверхностную энергию полимера определенное влияние может оказывать морфология поверхности [41–45]. На шероховатых поверхностях реализуется один из двух типов смачивания, которые описываются уравнениями Венцеля или Касси [41–43]. Первый из них – “гомогенное” смачивание, при котором жидкость контактирует со всей поверхностью твердого тела, полностью заполняя имеющиеся на ней впадины, второй – “гетерогенное” смачивание, при котором воздух “защемляется” во впадинах.
При малых значениях шероховатости поверхности применима модель Венцеля (гомогенное смачивание), согласно которой с возрастанием шероховатости краевой угол увеличивается в случае гидрофобных поверхностей (θ > 90°) и уменьшается на гидрофильных поверхностях (θ < 90°). Изменение угла смачивания с учетом шероховатости поверхности описывается уравнением Венцеля–Дерягина [43–45]:
где K – коэффициент шероховатости, равный отношению площади реальной поверхности к площади ее проекции на плоскость, т.е. к площади идеально гладкой поверхности, а θr и θ0 – углы смачивания шероховатой и гладкой поверхностей соответственно.В соответствии с [41, 44] применимость формул Венцеля−Дерягина и Касси−Бакстера обусловлена тем, что характерные размеры неоднородностей микрорельефа малы по сравнению с капиллярной постоянной
где ${{\sigma }_{{{\text{lv}}}}}$ – свободная поверхностная энергия на границе жидкость−пар, $\Delta \rho $ – разность плотностей жидкости и пара, $g$ – ускорение свободного падения. (Капиллярная постоянная для воды при нормальных условиях ≈3.8 мм; характерные размеры наблюдаемых неоднородностей не превышают 1 мкм.)Формула Венцеля−Дерягина носит универсальный характер; вносимые в нее изменения и дополнения лишь уточняют способ расчета коэффициента шероховатости K (см., например, [42, 45]).
Для оценки вклада шероховатости поверхности полимера в его смачиваемость до и после модификации изучено влияние оксифторирования на морфологию полимерных пленок. На рис. 5 представлены СЭМ-изображения поверхностей исходного и оксифторированного в течение 180 мин газовой смесью состава 7.5% F2 + 10% O2 образцов ПЭНП. Видно, что в результате модификации поверхность становится более шероховатой и на ней формируются протяженные волнообразные выступы размером 0.5 мкм × 1.0 мкм и более. Такое же изменение морфологии наблюдается и при оксифторировании ПП.
Рис. 5.
СЭМ-изображения поверхности пленок ПЭНП: исходной (а) и оксифторированной смесью состава 7.5% F2–10% O2 в течение 180 мин (б).

Изменение морфологии поверхности в результате оксифторирования ПЭТФ выражено значительно слабее (рис. 6). Это, вероятно, связаны с наличием в макромолекулах этого полимера шарнирных мостиков C−O−C, что обеспечивает релаксацию напряжений, возникающих в результате полимераналогичных превращений, за счет гибкости полимерной цепи.
Рис. 6.
СЭМ-изображения поверхности пленок ПЭТФ: исходной (а) и оксифторированной смесью состава 7.5% F2–10% O2 в течение 180 мин (б).

За ориентировочное значение коэффициента шероховатости K для поверхности полимера до и после оксифторирования принимали значение L0 (отношение длины линии профиля к длине ее проекции на среднюю линию), которое было получено нами с помощью АСМ. Эта величина для исходной пленки ПЭНП равна 1.024, а для оксифторированной в течение 180 мин смесью 7.5% F2 + 0.25% O2 – 1.097. Для пленок ПЭТФ значения L0 составили 1.001 и 1.003 соответственно. Сравнивая эти пары значений для пленок каждого полимера, можно заключить, что в случае ПЭНП уменьшение ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ за счет увеличения шероховатости поверхности не должно превысить 7%, а для ПЭТФ – 0.1%. Из этого следует, что, в отличие от известного случая [41, 44], вклад микрошероховатости поверхности оксифторированных пленок ПЭНП и ПЭТФ в значения ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ незначителен. По всей видимости, это связано с соотношением характерных размеров неоднородностей, формирующихся в процессе газофазной модификации (~100 нм) [46, 47], и в процессах, описанных в [41, 44] (более 1 мкм).
К аналогичному выводу можно прийти, сравнивая экспериментальную кинетическую зависимость угла смачивания водой оксифторированного ПЭНП с расчетными, полученными в двух предположениях: 1) об определяющем вкладе микрорельефа в изменение смачиваемости поверхности образца (рис. 7а) и 2) об отсутствии влияния микрорельефа на смачиваемость образца (рис. 7б). Необходимые для этого значения “истинной” площади поверхности полимера рассчитывали по данным СЭМ на основании анализа яркостей пикселей [46, 47] и с учетом калибровки масштаба яркости пикселей по данным АСМ.
Рис. 7.
Экспериментальные (1) и расчетные (2) зависимости ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ от времени оксифторирования пленки ПЭНП смесью 7.5% F2–10% O2, полученные в предположениях об определяющей роли наблюдаемого микрорельефа поверхности в смачиваемости пленки (а) и об отсутствии такого влияния (б).
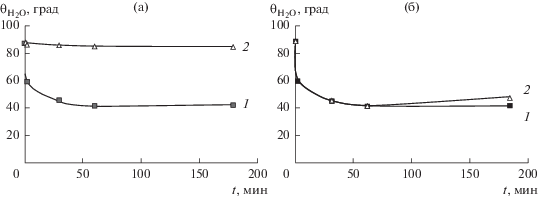
На рис. 7а видно, что расчетная зависимость не совпадает с экспериментальной, а на рис. 7б, напротив, наблюдается хорошее совпадение экспериментальных и рассчитанных значений ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}.$ Таким образом, очевидно доминирующее влияние изменений химического состава в результате модификации пленок ПЭНП на изменение их смачиваемости.
Вклад формирующегося в процессе модификации микрорельефа в изменение угла смачивания становится заметным (рис. 7б) только при продолжительной (t = 180 мин) обработке образца и в рассматриваемых условиях составляет не более 5°, т.е. если бы влияния рельефа “не было”, то ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ был бы больше, чем фактически наблюдаемый, примерно на 5°.
На основании экспериментальных данных были рассчитаны значения поверхностной энергии и ее составляющих для ряда образцов в зависимости от условий их оксифторирования (табл. 3). Анализ полученных результатов показывает, что обработка пленок полимеров смесями фтора с кислородом является эффективным методом регулируемого изменения полярной и дисперсионной составляющих свободной поверхностной энергии, а значит, и ее в целом. Изменение этих параметров происходит в широком диапазоне значений и зависит как от продолжительности модификации, так и от соотношения компонентов газовой смеси. В проведенной серии экспериментов максимальное увеличение свободной поверхностной энергии было достигнуто при использовании газовой смеси состава 7.5% F2 + 10% O2, которое составило, в частности, для ПЭНП 2 раза (с 23.5 до 52.6 мН/м). Такой эффект достигается тем, что одновременное использование фтора и кислорода позволяет формировать в поверхностном слое полимера не только фторсодержащие фрагменты, но и карбонильные и карбоксильные группы. Наличие последних подтверждается не только ИК-спектрами оксифторированных образцов, но и тем, что в результате модификации наблюдается существенное возрастание полярной составляющей свободной поверхностной энергии.
Таблица 3.
Значения поверхностной энергии (γ), ее полярной (γP) и дисперсионной (γD) составляющих для исследованных полимеров в зависимости от условий их оксифтрорирования
| Время оксифторирования, мин | Состав газовой смеси, об. % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 7.5 F2 + 0.25 O2 | 7.5 F2 + 6 O2 | 7.5 F2 + 10 O2 | |||||||
| ПЭНП | |||||||||
| γD, мН/м | γP, мН/м | γ, мН/м | γD, мН/м | γP, мН/м | γ, мН/м | γD, мН/м | γP, мН/м | γ, мН/м | |
| 0 | 10.0 | 16.8 | 26.8 | 10.0 | 16.8 | 26.8 | 10.0 | 16.8 | 26.8 |
| 1 | 17.6 | 16.8 | 34.4 | 17.6 | 20.1 | 37.7 | 21.4 | 21.3 | 42.7 |
| 2 | 18.5 | 16.2 | 34.7 | 15.9 | 25.0 | 40.9 | 22.2 | 21.4 | 43.6 |
| 5 | 18.8 | 17.9 | 36.7 | 21.0 | 20.1 | 41.2 | 18.1 | 28.5 | 46.6 |
| 30 | 17.1 | 21.6 | 38.7 | 18.1 | 28.7 | 46.8 | 12.4 | 40.6 | 53.0 |
| 60 | 18.0 | 29.1 | 47.1 | 11.6 | 40.8 | 52.4 | 9.5 | 47.8 | 57.3 |
| 180 | 20.9 | 25.0 | 45.9 | 9.6 | 42.1 | 51.7 | 12.1 | 43.5 | 55.6 |
| ПЭТФ | |||||||||
| γD, мН/м | γP, мН/м | γ, мН/м | γD, мН/м | γP, мН/м | γ, мН/м | γD, мН/м | γP, мН/м | γ, мН/м | |
| 0 | 19.4 | 17.5 | 36.9 | 19.4 | 17.5 | 36.9 | 19.4 | 17.5 | 36.9 |
| 1 | 24.5 | 13.9 | 38.4 | 27.5 | 16.9 | 44.4 | 24.6 | 19.6 | 44.2 |
| 2 | 25.0 | 15.6 | 40.6 | 21.3 | 23.3 | 44.6 | 25.2 | 19.9 | 45.1 |
| 5 | 10.2 | 36.8 | 47.0 | 26.5 | 18.1 | 44.6 | – | – | – |
| 30 | – | – | – | – | – | – | – | – | – |
| 60 | – | – | – | – | – | – | – | – | – |
| 180 | – | – | – | – | – | – | – | – | – |
| ПП-ЖЭ | |||||||||
| γD, мН/м | γP, мН/м | γ, мН/м | γD, мН/м | γP, мН/м | γ, мН/м | γD, мН/м | γP, мН/м | γ, мН/м | |
| 0 | 21.1 | 7.4 | 28.5 | 21.1 | 7.4 | 28.5 | 21.1 | 7.4 | 28.5 |
| 180 | 32.6 | 10.2 | 42.8 | – | – | – | 13.6 | 31.3 | 44.9 |
| ПП | |||||||||
| 0 | 16.1 | 7.4 | 23.5 | 16.1 | 7.4 | 23.5 | 16.1 | 7.4 | 23.5 |
| 180 | 32.6 | 10.2 | 42.8 | – | – | – | 19.4 | 27.8 | 47.2 |
Следует отметить результат, полученный для пленкок ПЭТФ, обработка которых газовой смесью состава 7.5% F2 + 10% O2 позволяет добиться практически полного смачивания их поверхности водой уже при t = 30 мин. (Это делает невозможным расчет значений γP и γD и, следовательно, γ полимера при t ≤ 30 мин.) Для достижения менее значимых изменений γ должны быть уменьшены время обработки и/или концентрация кислорода в смеси (например, путем снижения давления в реакторе, а тем самым и концентрации реагентов) [48, 49].
В последнее время большой интерес вызывает моделирование и создание систем с изменяющейся смачиваемостью поверхности, например, за счет ее особой текстуры [50–53]. Найденные нами условия максимального увеличения смачиваемости полимера позволяют варьировать смачиваемость полимерной пленки путем ее деформации растяжением. Так, установлено обратимое изменение ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ для оксифторированной пленки жесткоэластичного полипропилена (ПП-ЖЭ) при ее одноосной деформации растяжением. Этот эффект демонстрируют фотографии капли воды на поверхности такой пленки (верхний ряд изображений на рис. 8). Видно существенное изменение смачиваемости оксифторированной пленки ПП-ЖЭ при ее одноосном растяжении. На растянутой части пленки ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ увеличивается на 30% и достигает значения, измеряемого на немодифицированной пленке, а после снятия растягивающего усилия уменьшается практически до первоначального значения. Некоторое отличие угла смачивания после снятия растягивающего усилия от исходного значения, по-видимому, связано с незавершеностью релаксационных процессов в полимерной пленке после снятия напряжения к моменту проведения измерений.
Рис. 8.
Верхний ряд – фотографии капель воды на исходной и модифицированных (смесь 7.5% F2–10% O2, 3 ч) пленках ПП-ЖЭ: (а) исходная пленка, (б–г) оксифторированная пленка до растяжения, растянутая на 40% и после снятия нагрузки соответственно. Нижний ряд – СЭМ-изображения поверхности фторированной пленки ПЭНП до (а) и после (б) одноосного растяжении (остаточная деформация 100%), стрелкой показано направление растяжения [55].

Наблюдаемый эффект является примером обратимого изменения поверхностных свойств оксифторированной полимерной пленки. Его возникновение обусловлено тем, что полимерные пленки после фторирования представляют собой систему типа “твердое покрытие на податливом основании” [54, 55]. Деформация таких систем (как это показано на рис. 8 на примере пленки фторированного ПЭНП [55]) приводит к фрагментации “твердого” покрытия, т.е. фторированного слоя, с обнажением материала “податливого” основания, поверхность которого менее гидрофильна, чем поверхность оксифторированного полимера. Аналогичная фрагментация, очевидно, происходит и в случае одноосного растяжения оксифторированного образца ПП-ЖЭ, что приводит к возрастанию краевого угла воды. Не исключено также влияние на краевой угол изменения морфологии поверхности пленки при растяжении. После снятия растягивающего усилия восстанавливаются первоначальные размеры пленки ПП-ЖЭ, и, как следствие, ${{{\theta }}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ уменьшается почти до первоначального значения.
ЗАКЛЮЧЕНИЕ
Изучена газофазная модификация поверхности полимеров смесями фтора с кислородом при варьировании в широких диапазонах их объемного соотношения и времени модификации. Такой вариант поверхностной модификации приводит к росту гидрофильности поверхности полимеров, который тем больше, чем выше доля кислорода в смеси газов и чем продолжительней модификация (в определенном временнóм диапазоне). Этот эффект связан с формированием на поверхности полимера не только фторированных фрагментов макромолекул полимера, но и кислородсодержащих групп, в том числе карбоксильных. Варьируя условия модификации, можно в достаточно широком интервале управлять свойствами полимера: смачиваемостью, свободной поверхностной энергией и адгезией [1, 15, 30, 37]. Большое значение имеет возможность реализовать в определенных условиях практически полное растекание воды по поверхности полимера.
Рассмотрен вклад изменения в результате модификации морфологии поверхности полимера в краевой угол воды. Показано, что вклад формирующегося в процессе оксифторирования микрорельефа в изменение значения краевого угла становится заметным только при модификации образца в течение 180 мин и в рассматриваемых условиях составляет не более 5°.
Продемонстрировано, что увеличение смачиваемости поверхности полимера в результате оксифторирования можно использовать для получения полимерной пленки, у которой смачиваемость поверхности изменяется при ее деформации растяжением.
Авторы выражают благодарность А.М. Зуевой за предоставленные данные ИК-спектроскопии и И.В. Нагорновой за получение СЭМ-изображений.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (грант № 18-29-05037 мк).
Список литературы
Щукин Е.Д. Перцов А.В., Амелина Е.А. Коллоидная химия. 4-ое изд. испр. М.: Высш. школа, 2006.
Ye Y., Xu Y., Liu Y., Tu W., Liao J. // Polym. Mater. Sci. Technol. Eng. 2013. V. 29. № 11. P. 187.
Barish J.A., Goddard J.M. // J. Appl. Polym. Sci. 2011. V. 120. P. 2863.
Callies M., Quére D. // Soft Matter. 2005. V. 1. P. 55.
Feng X., Jiang L. // Adv. Mater. 2006. V. 18. P. 3063.
Gao L., McCarthy T.J. // Langmuir. 2007. V. 23. P. 3762.
Yi C., Li C.-W., Ji S., Yang M. // Anal. Chim. Acta. 2006. V. 560. № 1–2. P. 1.
Vinogradova O.I., Belyaev A.V. // J. Phys.: Condens. Matter. 2011. V. 23. 184104.
Назаров В.Г. Поверхностная модификация полимеров. М.: МГУП, 2008.
Logtenberg H., Lopez-Martinez M.J., Feringa B.L., Browne W.R., Verpoorte E. // Lab Chip. 2011. V. 11. P. 2030.
Сахно О.Н., Селиванов О.Г., Чухланов В.Ю. Биологическая устойчивость полимерных материалов / Под ред. Трифоновой Т.А. Владимир: Изд. ВлГУ, 2014.
Смирнов В.Ф., Мочалова А.Е., Смирнова О.Н., Захарова Е.А., Кряжев Д.В., Смирнова Л.А. // Поволжский экологический журнал. 2011. С. 537.
Калинина И.Г., Белов Г.П., Гумаргалиева К.З., Петронюк Ю.С., Семенов С.А. // Хим. физика. 2011. Т. 30. № 2. С. 70.
Nie Z., Kumacheva E. // Nat. Mater. 2008. V. 7. P. 277.
Назаров В.Г., Столяров В.П., Молчанов С.П., Юрасик Г.А., Артеменко М.Н. // Высокомолек. соед. А. 2013. Т. 55. С. 1343.
Кучма А.Е., Щекин А.К., Есипова Н.Е., Татьяненко Д.В., Ицков С.В., Савин А.В. // Коллоид. журн. 2017. Т. 79. С. 297.
Высоцкий В.В., Ролдугин В.И., Урюпина О.Я., Сенчихин И.Н., Зайцева А.В. // Коллоид. журн. 2017. Т. 79. С. 134.
Молчанов С.П., Ролдугин В.И., Чернова-Хараева И.А., Юрасик Г.А. // Коллоид. журн. 2017. Т. 79. С. 182.
Назаров В.Г., Столяров В.П., Евлампиева Л.А., Баранов В.А., Гагарин М.В. // Высокомолек. соед. А. 2009. Т. 51. С. 478.
Молчанов С.П., Ролдугин В.И., Чернова-Хараева И.А., Юрасик Г.А. // Коллоид. журн. 2016. Т. 78. С. 588.
Schofield W.C.E., Badyal J.P.S. // Polymer. 2011. V. 52. P. 5732.
Niu J., Liu D., Ding H., Wu Y. // Eur. Phys. J. Appl. Phys. 2012. V. 57. P. 10801.
Reznickova A., Kolsra Z., Hnatowicz V., Stopka P., Svorcik V. // Nucl. Instrum. Methods Phys. Res. B. 2011. V. 269. P. 83.
Oehr C. // Nucl. Instrum. Methods Meth. Phys. Res. B. 2003. V. 208. P. 40.
Назаров В.Г., Кондратов А.П., Столяров В.П., Евлампиева Л.А., Баранов В.А., Гагарин М.В. // Высокомолекуляр. соединения. А. 2006. Т. 48. С. 1976.
Валуев И.Л., Венчугова Л.В., Обыденнова И.В., Валуев Л.И. // Высокомолек. соед. Б. 2014. Т. 56. С. 188.
Kharitonov A.P. // J. Fluor. Chem. 2000. V. 103. P. 123.
Харитонов А.П., Логинов Б.А. // Рос. хим. журн. (Журн. Рос. хим. об-ва им. Д.И. Менделеева). 2008. Т. 52. № 3. С. 106.
Щербина А.А., Чалых А.Е. // Физикохимия поверхности и защита материалов. 2015. Т. 51. С. 231.
Назаров В.Г., Столяров В.П. // Коллоид. журн. 2016. Т. 78. С. 59.
Kharitonov A.P., Simbirtseva G.V., Tressaud A. Durand E., Labrugere C., Dubois M. // J. Fluor. Chem. 2014. V. 165. P. 49.
Le Roux J.D., Paul D.R., Arendt M.F., Yuan Y., Cabasso I. // J. Membr. Sci. 1994. V. 90. P. 37.
Toit F.J., Sanderson R.D., Engelbrecht W.J., Wagener J.B. // J. Fluor. Chem. 1995. V. 74. P. 43.
Taege R., Ferrier G. // Eur. Coatings J. 2006. № 5−6. P. 36.
Toit F.J., Sanderson R.D. // J. Fluor. Chem. 1999. V. 98. P. 107.
Харитонов А.П. Кинетика и механизм прямого фторирования полимеров. Дис. … докт. физ.-мат. наук. Черноголовка: Институт энергетических проблем химической физики РАН, 2005.
Перцов А.В., Соболева А.В., Проценко П.В., Назаров В.Г. // Коллоид. журн. 2008. Т. 70. С. 812.
Химическая энциклопедия / Под ред. Кнунянца И.Л., Зефирова Н.С., Кулова Н.Н. М.: Советская энциклопедия, 1990. Т. 2. С. 387.
Молчанов С.П., Ролдугин В.И., Чернова-Хараева И.Л. // Коллоид. журн. 2015. Т. 77. С. 755.
Ван Кревелен Д.В. Свойства и химическое строение полимеров. М.: Химия, 1976.
Бойнович Л.Б., Емельяненко А.М. // Успехи химии. 2008. Т. 77. С. 619.
Chung J.Y., Youngblood J.P., Stafford C.M. // Soft Matter. 2007. V. 3. P. 1163.
Lin Y.-H. // Opt. Express. 2008. V. 16. P. 17591.
Емельяненко А.М., Бойнович Л.Б. // Заводская лаборатория. Диагностика материалов. 2010. Т. 76. № 9. С. 27.
Далакова Н.В., Елекоева К.М., Кашежев А.З., Манукянц А.Р., Понежев М.Х., Прохоренко А.Д., Созаев В.А. // Известия Российской академии наук. Серия физическая. 2014. Т. 78. С. 512.
Drozdov S.A., Nazarov V.G., Nozdrachev S.A., Rudyak Yu.V., Rytikov G.O. // Nanosystems: Physics, Chemistry, Mathematics. 2017. 8. P. 137.
Копачев Е.С., Ноздрачев С.А., Петрушин В.Н., Рудяк Ю.В., Рытиков Г.О., Назаров В.Г. // Физическая мезомеханика. 2015. Т. 18. № 6. С. 98.
Prorokova N.P., Istratkin V.A., Kumeeva T.Yu., Vavilova S.Yu., Kharitonov A.P., Bouznik V.M. // RSC Adv. 2015. V. 5. P. 44545.
Истраткин В.А. Придание новых потребительских свойств синтетическим волокнистым материалам методом прямого газового фторирования. Дис. … канд. тех. наук. Иваново: Институт химии растворов им. Г.А. Крестова РАН, 2016.
Khongtong S.T., Ferguson G.S. // J. Am. Chem. Soc. 2002. V. 124. P. 7254.
Julthongpiput S.D., Fasolka M.J., Zhang W.H., Nguyen T., Amis E.J. // Nano Lett. 2005. V. 5. P. 1535.
Назаров В.Г., Столяров В.П., Гагарин М.В. // Высокомолек. соед. А. 2015. Т. 57. С. 274.
Лебедев-Степанов П.В, Кадушников Р.М., Мол-чанов С.П., Иванов А.А., Митрохин В.П., Вла-сов К.О., Рубин Н.И., Юрасик Г.А., Назаров В.Г., Алфимов М.В. // Российские нанотехнологии. 2013. Т. 8. № 3−4. С. 5.
Волынский А.Л., Бакеев Н.Ф. Структурная самоорганизация аморфных полимеров. М.: Физматлит, 2005.
Назаров В.Г., Волынский А.Л., Ярышева Л.М., Столяров В.П., Бакеев Н.Ф. // Высокомолек. соед. А. 2012. Т. 54. С. 1355.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал