

Коллоидный журнал, 2019, T. 81, № 4, стр. 532-540
Эмульсионный синтез и электрофоретическое концентрирование наночастиц золота в растворе бис(2-этилгексил)сульфосукцината натрия в н-декане
Н. О. Шапаренко 1, *, А. Т. Арымбаева 1, М. Г. Демидова 1, П. Е. Плюснин 1, 2, А. Н. Колодин 1, 2, Е. А. Максимовский 1, 2, И. В. Корольков 1, 2, А. И. Булавченко 1, 2
1 Институт неорганической химии им. А.В. Николаева СО РАН
630090 Новосибирск, пр. академика Лаврентьева, 3, Россия
2 Новосибирский государственный университет
630090 Новосибирск, ул. Пирогова, 1, Россия
* E-mail: nikshapar@mail.ru
Поступила в редакцию 25.12.2018
После доработки 31.01.2019
Принята к публикации 04.02.2019
Аннотация
Органозоли наночастиц золота получены восстановлением раствора золотохлористоводородной кислоты гидразином в динамической эмульсии воды в н-декане, стабилизированной поверхностно-активным веществом бис(2-этилгексил)сульфосукцинатом натрия. Наночастицы золота охарактеризованы методами фазового анализа рассеянного света, фотон-корреляционной спектроскопии, просвечивающей и сканирующей электронной микроскопии, рентгенофазового анализа, спектрофотометрии, термического анализа и элементного анализа. Определены гидродинамический диаметр (≈8 нм), электрофоретическая подвижность (3.9 × 10–10 м2/(В с)) и электрокинетический потенциал (30 мВ) синтезированных частиц. Неводным электрофорезом из органозоля выделен жидкофазный концентрат наночастиц, из которого термообработкой получены гидрофобные (краевой угол капли воды 115°) электропроводящие пленки (удельное сопротивление 0.41 Ом на квадрат).
ВВЕДЕНИЕ
Высококонцентрированные органозоли наночастиц металлов представляют существенный интерес для нанохимии и нанотехнологии [1–7]. Одной из наиболее перспективных областей их применения является использование в печатных технологиях в качестве металлсодержащих чернил-красок (metal based inkjet inks) для принтерной печати микросхем и плат. Чернила должны быть стабильны и иметь четко определенные вязкость, поверхностное натяжение, размеры, электрокинетический потенциал и концентрацию частиц металла [8–10].
Одним из самых распространенных и популярных способов синтеза органозолей и ультрадисперсных порошков является мицеллярный (микроэмульсионный) синтез [11–14]. Он позволяет получать стабильные органозоли с качественными наночастицами заданного размера с узкой функцией распределения [15]. Однако микроэмульсионный подход дает возможность синтезировать органозоли только с низкой концентрацией наночастиц, что обусловлено незначительной солюбилизационной емкостью мицелл по отношению к водным растворам вводимых реагентов. В наших работах [16–19] разработана методика электрофоретического концентрирования разнообразных наночастиц из мицеллярных растворов, позволяющая доводить концентрацию металла в органозоле до нескольких моль/л и формировать из них термической или водно-спиртовой обработкой электропроводящие покрытия [20]. Тем не менее, введение дополнительной стадии концентрирования не решило проблемы чрезвычайно низкой производительности процесса в целом: обычно из 100 мл микроэмульсии удается выделить не более 50 мкл жидкого концентрата с концентрацией порядка молярной. В работах [20, 21] сделана попытка увеличения производительности процесса посредством проведения восстановления азотнокислого серебра гидразином в динамической эмульсии типа вода/масло, стабилизированной бис(2-этилгексил)сульфосукцинатом натрия (Аэрозолем ОТ, АОТ). В итоге объем электрофоретического концентрата увеличился более чем в 30 раз, а размер и стабильность наночастиц остались прежними.
Цель данной работы заключалась в разработке эмульсионного синтеза наночастиц золота с последующим электрофоретическим концентрированием и определением характеристик полученных органозолей и твердофазных композитов Au–AOT.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
В качестве дисперсионной среды для синтеза наночастиц использовали н-декан (99%, эталонный). Исходный раствор золота(III) состава 0.25 моль/л HAuCl4 + 3 моль/л HCl готовили растворением золотой проволоки (содержание металла 99.9%) в царской водке с последующим получением хлоридного комплекса и разбавлением до соответствующей концентрации с помощью HCl. В качестве восстановителя использовали моногидрат гидразина (99%, Вектон). Стабилизатором эмульсии служило мицеллообразующее поверхностно-активное вещество – бис(2-этилгексил)сульфосукцинат натрия (АОТ) с содержанием основного вещества 97% (Sigma Aldrich). Концентрация АОТ в н-декане составляла 0.25 моль/л. В работе использовали соляную кислоту (“х. ч.”) и сульфат натрия (“ч. д. а.”).
Эмульсионный синтез наночастиц Au
Наночастицы золота в динамической эмульсии получали следующим образом. В 10 мл раствора АОТ в н-декане при перемешивании на магнитной мешалке (500 об./мин) сначала вводили 10 мл водного раствора HAuCl4 с концентрацией 0.02 моль/л, а затем такой же объем раствора гидразина с концентрацией 10 моль/л. Фоновый электролит содержал 3 моль/л HCl и 3.55 моль/л Na2SO4. Итоговое соотношение объемов водной и органической фаз составляло 2 : 1. Восстановление вели в течение 60 мин при интенсивном перемешивании при комнатной температуре. После стадии синтеза обратную эмульсию разрушали выдерживанием в термостате при 60°C в течение 35 мин до полного разделения фаз и затем центрифугировали 10 мин со скоростью 1500 об./мин. Отделенную органическую фазу перемешивали 2 ч на магнитной мешалке в открытом стакане до полного испарения воды. Аликвоту органозоля разбавляли и анализировали спектрофотометрически на содержание золота; выход составлял 60–70%. Далее органозоль подвергали электрофоретическому концентрированию.
Электрофоретическое концентрирование
Перед проведением электрофореза в обезвоженный органозоль вводили 1 об. % воды. Полученную микроэмульсию заливали в электрофоретическую ячейку конденсаторного типа с горизонтально ориентированными плоскопараллельными медными электродами площадью 16 см2 и межэлектродным зазором 1 см и подвергали электрофорезу в течение 30–60 мин при постоянном напряжении 300–600 В с целью дополнительного концентрирования и отделения наночастиц от побочных продуктов реакции. В процессе электрофореза жидкий концентрат наночастиц оседал на катоде с образованием четкой границы рафинат/концентрат. По окончании электрофореза концентрат отбирали микродозатором.
Методы определения характеристик органозолей и композита
Эффективный гидродинамический диаметр частиц определяли методом динамического рассеяния света (фотон-корреляционной спектроскопии – ФКС) на спектрометре NanoOmni (Brookhaven Instruments, США). Автокорреляционная функция обрабатывалась с использованием полимодального анализа при помощи алгоритма Non Negatively Constrained Least Squares (NNLS). Перед измерениями органозоли очищали от пыли пятикратным циклическим фильтрованием через мембранный PTFE-фильтр с диаметром пор 0.2 мкм (Sartorius, Германия) непосредственно в измерительную 1-см ячейку из стекла (Brookhaven Instruments, США). Мощность твердотельного лазера с длиной волны 640 нм составляла 35 мВт, рассеянные частицами фотоны детектировались под углом 90° к источнику излучения. Время накопления фотонов для одного измерения составляло 10–30 с, гидродинамический диаметр определялся как среднее из 20–30 измерений. Усредненный по интенсивности светорассеяния гидродинамический диаметр D рассчитывался для сферических частиц с использованием формулы Стокса–Эйнштейна
где kB – постоянная Больцмана, T – температура, d – коэффициент диффузии.Электрокинетический потенциал (ζ-потенциал) наночастиц измеряли на том же приборе методом лазерного электрофореза с использованием опции фазового анализа рассеянного света (Phase Analysis Light Scattering, PALS). Рассеянные частицами фотоны детектировались под углом 15°. Измерения проводили в специальной ячейке SRR2, устойчивой к действию органических растворителей, с плоскопараллельными палладиевыми электродами площадью примерно 45 мм2 с межэлектродным зазором 3.45 мм. Электроды также обеспыливали непрерывным пропусканием через них растворителя в течение 20 мин с помощью специализированной фильтрующей системы BI-SFS (Brookhaven Instruments, США); скорость потока составляла 7.8 мл/мин. Измерения скорости движения наночастиц проводили в ручном режиме в диапазоне напряжений от 75 до 200 В (рис. 1). Для каждого напряжения среднее значение скорости определялось из 10–20 измерений. Для всех систем зависимости скорости от напряженности поля были линейными, что свидетельствует о выполнимости критериев “истинного” (линейного) электрофореза [22]. Электрофоретическую подвижность (μе) определяли из тангенса угла наклона такой зависимости, а расчет ζ-потенциала производили по формуле Хюккеля–Онзагера
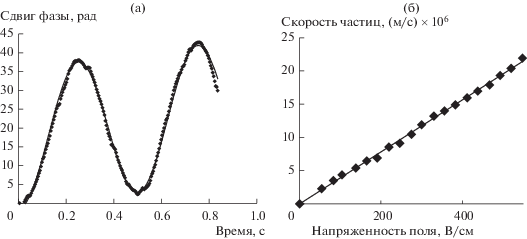
где ζ – дзета-потенциал, ε – диэлектрическая проницаемость, ε0 – электрическая постоянная, η – вязкость растворителя.Рис. 1.
Измерение электрокинетического потенциала наночастиц Au методом PALS. Зависимости сдвига фазы от времени при напряженности электрического поля 412 В/см (а) и скорости движения наночастиц от напряженности поля (б).
Динамическую вязкость н-декана определяли с помощью вискозиметра MicroVISC (ReoSense Inc., США). Измерения проводили в воздушном температурном контроллере MicroVISC TC при 20°C; точность поддержания температуры составляла ±0.02°C. Диэлектрическую проницаемость н-декана определяли, используя анализатор диэлектрической постоянной BI-870 (Brookhaven Instruments, США), с помощью зонда, состоящего из двух коаксиальных стальных цилиндров. Вязкость и диэлектрическая проницаемость н-декана составили 0.907 мПа с и 1.96 соответственно.
Плотность электрофоретических концентратов измеряли взвешиванием известных их объемов на аналитических весах ViBRA HTR-220CE (Sinko Denshi, Япония).
Электронные спектры поглощения наночастиц Au в микроэмульсиях регистрировали на спектрофотометре UV-1700 (Shimadzu, Япония) в кварцевых кюветах с длиной оптического пути 0.2 см.
Диаметр металлического ядра наночастиц (т.е. диаметр наночастиц без адсорбционного слоя) определяли методом просвечивающей электронной микроскопии (ПЭМ) с помощью микроскопа JEM-2010 с максимальным разрешением 0.2 нм. Электрофоретический концентрат наночастиц Au разбавляли в 500 раз н-гексаном, затем каплю раствора наносили на углеродную или безуглеродную подложку и высушивали при комнатной температуре. Функцию распределения наночастиц по размеру строили из 100–200 измерений при различных увеличениях. Для сопоставления данных ПЭМ и ФКС использовали диаметр, усредненный по числу частиц.
Синхронный термический анализ (термогравиметрия (ТГ) + дифференциальная ТГ (ДТГ) + + дифференциальная сканирующая калориметрия (ДСК)) твердофазного композита Au–AOT проводили на приборе STA 449F1 Jupiter (NETZSCH, Германия) в атмосфере синтетического воздуха (80 об. % Ar, 20 об. % O2) в температурном интервале 30–1100°C. Использовали закрытые тигли из Al2O3, скорость нагрева – 10 град/мин, скорость потока газов – 40 мл/мин аргона, 10 мл/мин кислорода.
Рентгенофазовый анализ поликристаллов проводили на дифрактометре Shimadzu XRD-7000 (CuKα-излучение, Ni-фильтр, диапазон 2θ = = 5°–70°, шаг – 0.03°, время накопления – 1 с). Образец для исследования готовили следующим образом: поликристаллы растирали в агатовой ступке в присутствии гептана и полученную суспензию наносили на полированную сторону стандартной кварцевой кюветы. После высыхания гептана образец представлял собой тонкий ровный слой (толщина ∼100 мкм). Индицирование дифрактограммы проводили по данным картотеки PDF [23]. Расчет областей когерентного рассеяния (ОКР) проводили по уравнению Шеррера с учетом полуширин эталона Si. Погрешность определения ОКР составляет порядка 10%.
Элементный анализ композита (использовали навески массой 3–6 мг) выполняли на анализаторе Vario Micro cube (Elementar Analysensysteme GmbH, Германия).
Методика получения пленок и определение их характеристик
Электропроводящие пленки получали следующим образом: 20 мкл концентрата наносили на стеклянную пластинку 2 × 2 см2 методом “Doctor Blade”. Высохшая пленка имела цвет от темно-красного до практически черного, электронный спектр поглощения наночастиц золота в пленке был идентичен спектрам органозолей. На следующий день пластинку выдерживали 2 ч при температуре 250°C в программируемой лабораторной печи L 03/12 (Чехия).
Сопротивление пленок измеряли с помощью мультиметра Keithley 2110 51/2.
Морфологию проводящих пленок исследовали в сканирующем электронном микроскопе (СЭМ) JSM 6700F (JEOL, Япония), а их состав – методом энергодисперсионного рентгеновского анализа (EDX), используя приставку к микроскопу Quantax 200 (Bruker, Германия).
Измерения смачиваемости золотых пленок проводили на приборе OCA 15 PRO (DataPhysics Instruments GmbH, Германия), оснащенном измерительной видеосистемой с USB-камерой, а также светосильным измерительным объективом с настраиваемым углом наблюдения. Все образцы находились в термостатированном боксе, оснащенном элементом Пельтье, при T = 298 ± 2 K и p = 1 бар. В качестве тестовой жидкости использовали дистиллированную воду. Объем капель был постоянным и составлял ≈2.5 мкл. Значения краевого угла измеряли в режиме лежащей капли по методу Юнга–Лапласа. Проводящие золотые пленки предварительно промывали в бидистиллированной воде, после чего высушивали в атмосфере чистого воздуха в течение нескольких часов. Конечное значение краевого угла вычисляли как среднее из трех измерений на различных участках подложки.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Эмульсионный синтез
АОТ является слабым стабилизатором обратных эмульсий, значение его гидрофильно-липофильного баланса равно 10.2 по сравнению, например, с 4.3 для популярного стабилизатора моноолеата сорбитана (Span 80) [24]. Поэтому для синтеза обратные эмульсии, стабилизированные АОТ, следует применять только в динамическом режиме. В эмульсии исходные реагенты находятся в водных микрокаплях, их столкновение может привести к коалесценции и неконтролируемому восстановлению золота гидразином с выделением “черных” порошков золота. Именно это мы и наблюдали при введении HAuCl4 в 3 моль/л раствор HCl. Золото в растворе соляной кислоты находится в виде комплексного аниона ${\text{AuCl}}_{{\text{4}}}^{--}$ и в мицеллы анионного АОТ не переходит. Поэтому восстановление происходит в эмульсионных микрокаплях при их столкновениях и коалесценции.
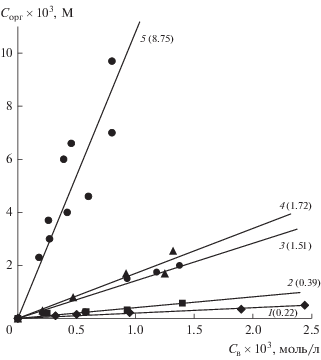
Однако насыщение солянокислого раствора золота сульфатом натрия (солью с сильными дегидратирующими свойствами) привело к заметному переходу HAuCl4 из микрокапель в мицеллы АОТ (рис. 2). В равновесных условиях при соотношении объемов водной и органической фаз, равном 1 : 1, степень извлечения составила 90%. По-видимому, переход происходит по механизму простого физического распределения [25] посредством высаливания HAuCl4 в ядро мицелл АОТ [26]. Таким образом, при эмульгировании раствора HAuCl4 + 3 моль/л HCl + Na2SO4 (нас.) Au(III) за несколько минут практически полностью переходит из микрокапель эмульсии в мицеллы АОТ.
Рис. 2.
Изотермы экстракции и коэффициенты распределения ${\text{AuCl}}_{4}^{ - }$ (приведены в скобках) при постоянной концентрации соляной кислоты (3 моль/л) и различном содержании сульфата натрия: 1 – 0, 2 – 0.5, 3 – 2, 4 –2.25, 5 – 3.55 моль/л.
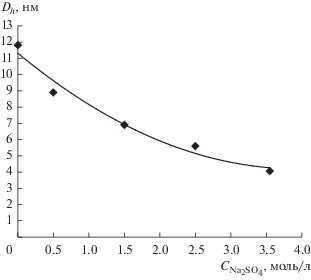
Иная ситуация возникает в случае второго реагента – гидразина. При эмульгировании раствора гидразина бóльшая часть восстановителя остается в микрокаплях эмульсии [20]. Однако за счет обменных взаимодействий осмотической природы между микрокаплями и мицеллами часть гидразина переходит в мицеллы АОТ. В результате восстановление золота(III) гидразином протекает в мицеллах за счет межмицеллярного обмена [20] и приводит к образованию стабильных наночастиц малого размера. Высокая концентрация сульфата натрия в эмульсионных микрокаплях способствует формированию мицелл малого размера (рис. 3) за счет осмотического эффекта. После синтеза эмульсия была разрушена нагреванием, а полученный органозоль обезвожен.
Характеристики полученных наночастиц в органозолях
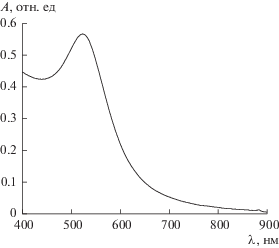
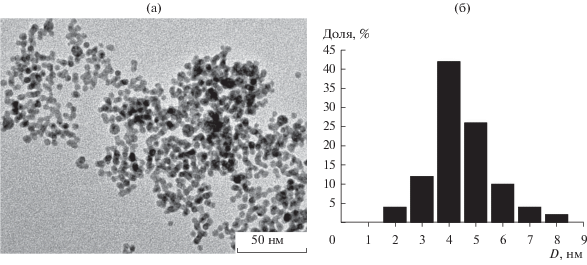
Спектр поверхностного плазмонного поглощения наночастиц золота приведен на рис. 4. Длина волны максимума поглощения наночастиц золота составляла 520 нм. Среднечисленный диаметр наночастиц золота равен 8 нм по данным ФКС. По данным ПЭМ среднечисленный диаметр наночастиц золота составил 4 нм (рис. 5). Разница диаметров частиц, полученных двумя независимыми методами, определяется толщиной адсорбционного слоя, не фиксируемого ПЭМ.
Рис. 4.
Спектр поверхностного плазмонного резонанса органозоля золота (8.6 × 10–4 моль/л); дисперсионная среда – 0.25 моль/л раствор АОТ в н-декане. Толщина кварцевой кюветы 0.2 см.
По данным PALS (рис. 1а) полученные наночастицы имели положительный поверхностный заряд. Зависимость скорости движения наночастиц золота от напряженности поля (напряжение 5–200 В) была линейной (рис. 1б). По полученной зависимости из угла наклона была рассчитана электрофоретическая подвижность и, соответственно, ζ-потенциал. Электрофоретическая подвижность составила 3.9 × 10–10 м2/(В с), что соответствует значению ζ-потенциала, равному 30 мВ.
Электрофоретическое концентрирование наночастиц
Наличие на наночастиц золота стабильного поверхностного заряда, компенсированного диффузным слоем противоионов, позволяет провести их электрофоретическое концентрирование. Электрофорез проводили в ячейке конденсаторного типа, в процессе электрофореза жидкий концентрат наночастиц оседал на электродах и стекал на дно ячейки, образуя тонкий легкоподвижный слой малого объема.
Чтобы оценить эффективность электрофоретического концентрирования, определяли степень извлечения и коэффициент концентрирования следующим образом: по окончанию электрофореза из межэлектродного пространства в верхней части ячейки осторожно отбирали 1–2 мл рафината, а со дна ячейки – 50–100 мкл концентрата. Затем по спектрам плазмонного поглощения определяли концентрацию наночастиц золота в рафинате и концентрате. По найденным значениям концентрации определяли степень извлечения и коэффициент концентрирования. По данным спектрофотометрии содержание золота в исходном растворе после синтеза составляло 1.6 × 10–2 моль/л, в рафинате 9.1 × 10–6 моль/л, в концентрате 2.6 моль/л. Таким образом, коэффициент концентрирования равен 160, а степень извлечения золота составляет 99.9%.
Получение твердофазных композитов и проводящих пленок
При полном испарении растворителя из электрофоретического концентрата образуется сухой твердофазный композит Au–АОТ. По данным элементного микроанализа содержание в нем AOT составило 17.8%, гидразина – 1.4% и, соответственно, золота – 80.8%. Найденное содержание золота хорошо совпадает с определенным методом ТГ – 79.3%. При хранении в открытом сосуде в комнатных условиях наночастицы золота в твердофазном композите также не подвержены коагуляции. Термообработка при 250°C концентрата и пленок на его основе приводит к заметному изменению содержанию компонентов-неметаллов. Согласно данным элементного микроанализа (табл. 1) концентрация азота и углерода падает в 10–20 раз в композите и до 30–100 раз в случае пленок. Менее существенно изменяется содержание водорода и серы. Таким образом, при термообработке происходит эффективная металлизация концентрата и особенно пленок и удаление ПАВ.
Согласно данным термогравиметрического анализа (рис. 6) первая стадия потери массы, протекающая в интервале температур 30–100°C, приводит к уменьшению массы на 1.8% и представляет собой удаление молекул растворителя. Безводный образец твердофазного композита Au–AOT в атмосфере синтетического воздуха устойчив до температуры ≈ 130°C. В температурном диапазоне 130–160°C имеют место незначительные потери массы (0.5%), сопровождающиеся экзоэффектом. Далее, при температуре 160–200°C следует еще одна стадия, которая также сопровождается экзоэффектом и потерей еще 1% массы. В интервале 200–340°C протекают несколько плохо разделенных стадий, характеризующихся эндоэффектами и представляющих собой процессы разложения органической матрицы композита. Последняя стадия разложения при 340–430°C сопровождается экзоэффектом и приводит к полному окислению (сгоранию) промежуточных углеродсодержащих продуктов. Конечный продукт представляет собой металлическое золото, что подтверждается эндоэффектом на ДСК-кривой при температуре 1060°C, который соответствует процессу плавления металла.
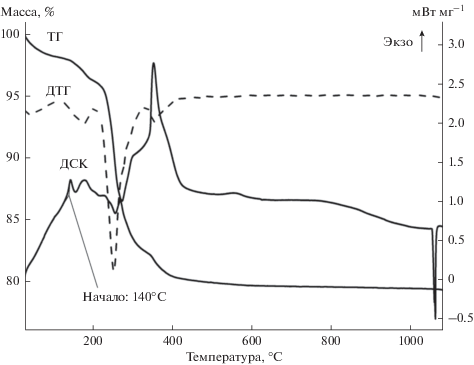
Первая стадия разложения композита представляет собой процесс укрупнения (спекания) наночастиц золота. Самопроизвольный процесс укрупнения наночастиц золота протекает с выделением теплоты, что приводит к частичному разложению органической матрицы, о чем свидетельствует незначительная потеря массы на ТГ-кривой. Конечная масса при температуре 1050°C, т.е. до плавления золота, составляет 79.32% от исходной, при температуре 1100°C (после плавления) – 78.96%.
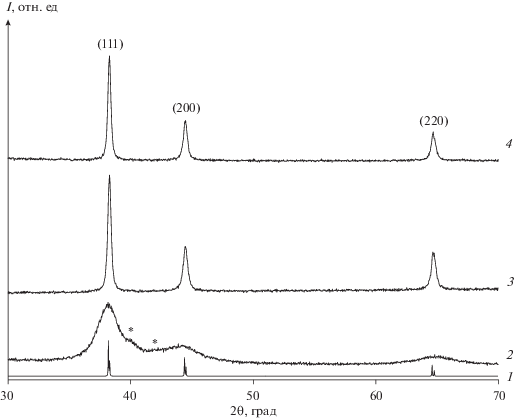
По результатам РФА образец композита Au–АОТ состоит из наноразмерных частиц золота и неизвестной фазы (рис. 7). В области 2θ = 39.4°–42.5° наблюдаются широкие дифракционные пики, не относящиеся к металлическому золоту. Поэтому возможна лишь грубая оценка ОКР Au по пику (111), которая дает значение 7.5 нм. После отжига при 250°C на дифрактограмме остаются лишь пики чистого Au. Дифрактограммы отожженного композита и пленки различаются только полуширинами пиков. Для пленки размеры ОКР, определенные по пикам (111), (200), (220), составляют 65, 43 и 40 нм соответственно; для композита они равны 54, 33 и 45 нм.
Рис. 7.
1 – Теоретическая дифрактограмма золота (PDF № 000-04-0784), 2–4 экспериментальные дифрактограммы (2 – композит, 3 и 4 – композит и пленка, отожженные при 250°С). Звездочками указаны неиндицированные уширения пиков.
На рис. 8а приведено СЭМ-изображение проводящей пленки. Она имеет пористую структуру и состоит из частиц размером 50–200 нм. Учитывая исходный размер наночастиц золота в концентрате, можно сделать вывод, что каждая частица сформировалась в результате сплавления (спекания) 10–40 первичных наночастиц. Толщина полученных пленок составляла от 500 нм до 4 мкм. В EDX-спектре (рис. 8б) идентифицированы золото, следы углерода и кремний (материал подложки). Проводимость пленок не превышала 0.41 Ом на квадрат (измерения проведены по четырехконтактной схеме).
Рис. 8.
(а) СЭМ-изображение проводящей пленки золота (на врезке – капля воды на ее поверхности); (б) EDX-спектр пленки.

Полученные пленки являются гидрофобными: наблюдаемый краевой угол воды Ө составляет 115 ± 3° (врезка на рис. 8а). Согласно литературным данным гладкая, химически однородная поверхность золота является гидрофильной. Авторами статьи [27] показано, что в случае гладкой и чистой поверхности золота краевой угол воды Өгл равен примерно 60°–65°. По данным СЭМ полученные образцы имеют шероховатую поверхность (рис. 8а), смачивание которой может проходить как в гомогенном [28], так и в гетерогенном [29] режиме. В условиях гомогенного смачивания для гидрофильных материалов (Өгл < 90°) наблюдаемый краевой угол должен уменьшаться с ростом шероховатости поверхности в соответствии с теорией Венцеля–Дерягина [28]. Таким образом, в условиях гомогенного смачивания шероховатых золотых пленок значение краевого угла должно быть меньше 60°–65°. В свою очередь, в режиме гетерогенного смачивания можно получить как гидрофильные, так и гидрофобные и супергидрофобные золотые пленки, а наблюдаемый краевой угол зависит от степени заполнения пор тестовой жидкостью согласно теории Касси–Бакстера [29]. Поскольку для полученных пленок измеренный краевой угол воды превышает значение угла для гладкой поверхности золота, их смачивание протекает в гетерогенном режиме. Вода полностью не затекает в имеющиеся поры: образуются воздушные “карманы” внутри пористой структуры пленок.
ЗАКЛЮЧЕНИЕ
Предложен эмульсионный вариант синтеза и концентрирования наночастиц золота. По сравнению с мицеллярным синтезом производительность выросла в 30 раз. Следует также отметить, что механизм эмульсионного синтеза еще до конца не установлен. На данном этапе исследований очевидно, что для получения наночастиц в качестве стабилизатора эмульсий следует использовать мицеллообразующие ПАВ. При этом в случае использования слабого эмульгатора типа АОТ один из реагентов должен быть предварительно (до введения второго реагента) полностью переведен в мицеллы. Микрокапли эмульсии играют роль огромных (по сравнению с мицеллами) резервуаров, выполняющих роль поставщика реагентов. В мицеллы реагенты переходят за счет обмена типа микрокапля–мицелла, а процессы образования зародышей, их рост и стабилизация происходят в мицеллах за счет межмицеллярного обмена. Получены и подробно исследованы органозоли наночастиц золота, а также твердофазные композиты Au–AOT и пленки на их основе.
Список литературы
Blosi M., Albonetti S., Gatti F., Baldi G., Dondi M. // Dyes Pigm. 2012. V. 94. P. 355.
Cardellini A., Fasano M., Bigdeli M.B., Chiavazzo E., Asinari P. // J. Phys.: Condens. Matter. 2016. V. 28. 483 003.
Chandrasekar M., Suresh S. // Heat Transfer Eng. 2009. V. 30. P. 1136.
Minkowycz W.J., Sparrow E.M., Abraham J.P. Advances in Numerical Heat Transfer. V. 4. Boca Raton: CRC Press, 2013. P. 137.
Burns A., Ow H., Wiesner U. // Chem. Soc. Rev. 2006. V. 35. P. 1028.
Zhang Q., Shu X.-Z., Lucas J.M., Toste F.D., Somorjai G.A., Alivisatos A.P. // Nano Lett. 2014. V. 14. P. 379.
Parent M.E., Yang J., Jeon Y., Toney M.F., Zhou Z.-L., Henze D. // Langmuir. 2011. V. 27. P. 11845.
Kamyshny A., Magdassi S. // Small. 2014. V. 10. P. 3515.
Kamyshny A., Steinke J., Magdassi S. // Open Appl. Phys. J. 2011. V. 4. P. 19.
Magdassi S. The Chemistry of Inkjet Inks. Singapore: World Scientific Publ., 2010.
Hollamby M.J., Hudson L. // Adv. Colloid Interface Sci. 2006. V. 188. P. 128.
Jankiewicz B.J., Jamiola D., Choma J., Jaroniec M. // Adv. Colloid Interface Sci. 2012. V. 170. P. 28.
Andersson M., Pedersen J.S., Palmqvist A.E.C. // Langmuir. 2005. V. 21. P. 11387.
Bagwe R.P., Khilar K.C. // Langmuir. 2000. V. 16. P. 905.
Kim D.W., Oh S.G., Lee J.D. // Langmuir. 1999. V. 15. P. 1599.
Bulavchenko A.I., Popovetskiy P.S. // Langmuir. 2010. V. 26. № 2. P. 736.
Булавченко А.И., Сапьяник А.А., Демидова М.Г. // Журн. физ. химии. 2014. Т. 88. С. 502.
Bulavchenko A.I., Shaparenko N.O., Demidova M.G. // Electrophoresis. 2017. V. 38. P. 1678.
Bulavchenko A.I., Pletnev D.N. // J. Phys. Chem. C. 2008. V. 112. P. 16365.
Bulavchenko A.I., Arymbaeva A.T., Demidova M.G., Popovetskiy P.S., Plyusnin P.E., Bulavchenko O.A. // Langmuir. 2018. V. 34. P. 2815.
Булавченко А.И., Арымбаева А.Т., Демидова М.Г. Патент РФ № 2644176 // Б.И. 2018. № 4. С. 12.
Kuo S., Osterle F. // J. Colloid Interface Sci. 1967. V. 25. P. 421.
Powder Diffraction File, release 2010, International Centre for Diffraction Data, Pennsylvania, USA.
Ghosh S., Naskar M.K. // RSC Adv. 2013. V. 3. P. 4207.
Гиндин Л.М. Экстракционные процессы и их применение. М.: Наука, 1984.
Bulavchenko A.I., Podlipskaya T.Y., Arymbaeva A.T. // Sep. Sci. Technol. 2010. V. 46. P. 54.
Erb R.A. // J. Phys. Chem. 1968. V. 72. P. 2412.
Wenzel R.N. // Ind. Eng. Chem. 1936. V. 28. P. 988.
Choi W., Tuteja A., Mabry J.M., Cohen R.E., McKinley G.H. // J. Colloid Interface Sci. 2009. V. 339. P. 208.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал