

Коллоидный журнал, 2019, T. 81, № 5, стр. 547-556
Молекулярно-динамическое моделирование релаксационных процессов на границе жидкость–газ в одно- и двухкомпонентных леннард-джонсовских системах
В. Г. Байдаков 1, *, С. П. Проценко 1
1 Институт теплофизики, Уральское отделение Российской академии наук
620016 Екатеринбург, ул. Амундсена, 107а, Россия
* E-mail: baidakov@itp.uran.ru
Поступила в редакцию 14.02.2019
После доработки 19.02.2019
Принята к публикации 21.02.2019
Аннотация
При молекулярно-динамическом моделировании воспроизведен процесс формирования равновесной межфазной границы жидкость–газ в одно- и двухкомпонентных леннард-джонсовских системах. В двухкомпонентной системе второй компонент являлся летучей примесью. Начальное состояние создавалось приведением в контакт однородных жидкой и газовой фаз, имеющих равные температуры, давления и химические потенциалы. В процессе моделирования оценивались времена установления равновесных значений давления, состава, формы и толщины межфазной границы, относительной адсорбции, поверхностного натяжения. Расчеты проведены при температуре, близкой к температуре тройной точки растворителя. Установлено, что в процессе релаксации максимальная величина динамического поверхностного натяжения в 1.2–1.6 раз превышает равновесное значение, а время релаксации возрастает от 10 до 100 нс с увеличением концентрации летучего компонента в растворе до 0.25. В двухкомпонентной системе с ограниченным объемом газовой фазы равновесный межфазный слой формируется в два этапа. На первом этапе летучий компонент переносится в межфазный слой из приповерхностных областей жидкой и газовой фаз. При достижении равновесной парциальной плотности летучего компонента в газовой фазе происходит переход ко второму этапу, когда поверхностный слой пополняется в основном частицами жидкой фазы, что приводит к существенному возрастанию времен релаксации относительной адсорбции и поверхностного натяжения. Обсуждается проявление динамического поверхностного натяжения в процессах нуклеации.
1. ВВЕДЕНИЕ
Поверхностное натяжение играет важную роль в природных и технологических процессах. Оно отвечает за сферическую форму капли [1], зародышеобразование [2, 3], формирование и распад струй [4, 5], коллапс пузырьков [6, 7] и др.
Многие процессы, связанные с пенообразованием, диспергированием, смачиванием и т.п., протекают в условиях, когда равновесие между поверхностным слоем и объемными фазами не достигается. Поверхностное натяжение свежеобразованной границы фаз известно как динамическое поверхностное натяжение.
Наиболее подробная информация о динамическом поверхностном натяжении получена для систем, содержащих поверхностно-активные вещества (см. [8–10] и цитируемую в этих работах литературу). Ввиду малых времен релаксации, сведения о динамическом поверхностном натяжении однокомпонентных систем ограничены [11–14]. Для воды, по данным работ [11, 12], время формирования равновесной структуры поверхностного слоя оценивается в 10–4–10–3 с. Проследить эволюцию поверхностного натяжения на столь коротком интервале времени затруднительно. Тем не менее, в последних экспериментах по отрыву капель воды [15] было получено значение поверхностного натяжения, на 25% превышающее его равновесное значение.
Новые возможности в изучении динамического поверхностного натяжения на интервалах менее миллисекунды открывают методы молекулярно-динамического моделирования. Моделирование позволяет исследовать динамику формирования равновесного межфазного слоя и тем самым установить характерные времена релаксационных процессов.
В работе [16] методом молекулярной динамики в одно- и двухкомпонентных леннард-джонсовских системах исследовалось восстановление поверхностного слоя жидких капель после его удаления. Для однокомпонентной системы характерное время релаксации поверхностного натяжения составило 4 × 10–12 с. В случае бинарной системы выделены два характерных времени. Первое, короткое, связано с диффузией растворителя (первого компонента) на расстояниях, сопоставимых с толщиной межфазного слоя. Второе, более продолжительное время релаксации, обусловлено перераспределением компонентов смеси внутри переходного слоя.
В данной работе исследуются релаксационные процессы на плоской межфазной границе жидкость–газ в одно- и двухкомпонентных леннард-джонсовских системах. В бинарной смеси второй компонент является легколетучей добавкой. Примерами таких смесей, для которых характерно большое различие в энергиях взаимодействия частиц компонентов, являются растворы метан–гелий, метан–водород, аргон–неон. Отношение величин минимальной энергии взаимодействия частиц растворенного вещества и растворителя в этих системах составляет 0.024–0.29 [17]. При двухфазном равновесии компонент с более слабым межчастичным взаимодействием преобладает в газовой фазе. Жидкая фаза состоит в основном из частиц растворителя. Содержание летучего компонента в ней составляет доли процента. Кроме того, летучая примесь обычно адсорбируются в переходном слое, что приводит к понижению поверхностного натяжения раствора относительно поверхностного натяжения чистого растворителя [18–20].
При молекулярно-динамическом моделировании начальное неравновесное состояние в момент времени t = 0 создавалось удалением из равновесной двухфазной системы переходного слоя и приведением в контакт однородных жидкой и газовой фаз, которые имели равные температуры, давления и химические потенциалы. В процессе моделирования оценивались характерные времена установления механического равновесия между фазами, достижения равновесных составов, формы и толщины переходного слоя, исследовалась эволюция во времени поверхностного натяжения.
2. МОДЕЛЬ И ДЕТАЛИ МОДЕЛИРОВАНИЯ
Двухфазная система представляла собой слой жидкости, расположенный в центре ячейки перпендикулярно оси z, с двух сторон окруженный паровой фазой. На границы ячейки накладывались периодические граничные условия. Число частиц в ячейке N варьировалось от 24 100 до 70 800. Взаимодействие между частицами описывалось потенциалом Леннард-Джонса
(1)
${{\phi }_{{\alpha \beta }}}({{r}_{{ij}}}) = \left\{ \begin{gathered} 4{{\varepsilon }_{{\alpha \beta }}}\left[ {{{{\left( {\frac{{{{\sigma }_{{\alpha \beta }}}}}{{{{r}_{{ij}}}}}} \right)}}^{{12}}} - {{{\left( {\frac{{{{\sigma }_{{\alpha \beta }}}}}{{{{r}_{{ij}}}}}} \right)}}^{6}}} \right],\,\,\,\,{{r}_{{ij}}} \leqslant {{r}_{{\text{c}}}} \hfill \\ 0,{\text{ }}{{r}_{{ij}}} > {{r}_{{\text{c}}}}, \hfill \\ \end{gathered} \right.$Параметры потенциала ε11, σ11 и масса частицы первого компонента (растворителя) m1 использовались в качестве единиц приведения рассчитываемых величин к безразмерному виду. Единица температуры – ${{{{\varepsilon }_{{11}}}} \mathord{\left/ {\vphantom {{{{\varepsilon }_{{11}}}} {{{k}_{{\text{B}}}}}}} \right. \kern-0em} {{{k}_{{\text{B}}}}}}$, давления ${{{{\varepsilon }_{{11}}}} \mathord{\left/ {\vphantom {{{{\varepsilon }_{{11}}}} {\sigma _{{11}}^{3}}}} \right. \kern-0em} {\sigma _{{11}}^{3}}},$ числовой плотности ${1 \mathord{\left/ {\vphantom {1 {\sigma _{{11}}^{3}}}} \right. \kern-0em} {\sigma _{{11}}^{3}}},$ поверхностного натяжения ${{{{\varepsilon }_{{11}}}} \mathord{\left/ {\vphantom {{{{\varepsilon }_{{11}}}} {\sigma _{{11}}^{2}}}} \right. \kern-0em} {\sigma _{{11}}^{2}}},$ адсорбции ${1 \mathord{\left/ {\vphantom {1 {\sigma _{{11}}^{2}}}} \right. \kern-0em} {\sigma _{{11}}^{2}}}.$ Единица времени – ${{\sigma }_{{11}}}{{({{{{m}_{1}}} \mathord{\left/ {\vphantom {{{{m}_{1}}} {{{\varepsilon }_{{11}}}}}} \right. \kern-0em} {{{\varepsilon }_{{11}}}}})}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}.$ Далее все величины представляются в приведенном виде. Параметры потенциалов взаимодействия компонентов соотносились как ${{{{\varepsilon }_{{22}}}} \mathord{\left/ {\vphantom {{{{\varepsilon }_{{22}}}} {{{\varepsilon }_{{11}}}}}} \right. \kern-0em} {{{\varepsilon }_{{11}}}}}$ = 0.2926, ${{{{\sigma }_{{22}}}} \mathord{\left/ {\vphantom {{{{\sigma }_{{22}}}} {{{\sigma }_{{11}}}}}} \right. \kern-0em} {{{\sigma }_{{11}}}}}$ = 0.8076, массы частиц как ${{{{m}_{2}}} \mathord{\left/ {\vphantom {{{{m}_{2}}} {{{m}_{1}}}}} \right. \kern-0em} {{{m}_{1}}}}$ = 0.505. Такие отношения характерны для системы аргон–неон [17]. Параметры взаимодействия между частицами разного сорта рассчитывались по правилам Бертло, ${{\varepsilon }_{{12}}}$ = ${{({{\varepsilon }_{{11}}}{{\varepsilon }_{{22}}})}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}},$ и Лоренца, ${{\sigma }_{{12}}}$ = ${{({{\sigma }_{{11}}} + {{\sigma }_{{22}}})} \mathord{\left/ {\vphantom {{({{\sigma }_{{11}}} + {{\sigma }_{{22}}})} 2}} \right. \kern-0em} 2}.$
Длина ребер базовой ячейки, которая имела форму прямоугольного параллелепипеда, вдоль осей x и y составляла Lx = Ly = 30, а в направлении z варьировалось от 60 до 160. Расчеты проводились в NVT-ансамбле при температуре $T = 0.7$ с использованием пакета молекулярно-динамического моделирования LAMMPS [21]. Постоянство температуры обеспечивалось термостатом Nose–Hoover [22]. Шаг интегрирования уравнений движения по времени $\Delta {\kern 1pt} t$ = 0.002318.
В процессе моделирования определялись профили локальной числовой плотности ρ(z), температуры T(z), нормальной PN(z) и тангенциальной PT(z) компонент тензора давления. Для этого ячейка разбивалась вдоль оси z на слои толщиной $\Delta {\kern 1pt} z$ = 0.05. Процедура расчета указанных величин подробно описана в работе [23]. По распределениям $\rho (z),$ PN(z) и PT(z) определены ортобарические плотности жидкости ${{\rho }_{{\text{l}}}},$ газа ${{\rho }_{{\text{g}}}}$ и давление фазового равновесия ps.
Вследствие неравномерности испарения частиц с межфазных границ, жидкая пленка в процессе эволюции системы могла смещаться. Начало отсчета рассчитываемых распределений привязывалось к оси симметрии жидкой фазы (координате z0 ее центра масс), положение которой контролировалось через каждые 200 шагов интегрирования уравнений движения частиц.
Поверхностное натяжение находилось согласно его механическому определению
(2)
$\gamma = \frac{1}{2}\int\limits_{ - \infty }^{ + \infty } {\left( {{{P}_{{\text{N}}}}(z) - {{P}_{{\text{T}}}}(z)} \right)d{\kern 1pt} z} ,$Относительная адсорбция второго компонента на эквимолекулярной разделяющей поверхности первого компонента ${{z}_{{1,{\text{e}}}}}$ есть
(3)
${{\Gamma }_{{2(1)}}} = \int\limits_{{{{{L}_{z}}} \mathord{\left/ {\vphantom {{{{L}_{z}}} 2}} \right. \kern-0em} 2}}^{{{z}_{{1,{\text{e}}}}}} {\left( {{{\rho }_{2}}(z) - {{\rho }_{{2{\text{,l}}}}}} \right)dz} + \int\limits_{{{z}_{{1,{\text{e}}}}}}^{{{{{L}_{z}}} \mathord{\left/ {\vphantom {{{{L}_{z}}} 2}} \right. \kern-0em} 2}} {\left( {{{\rho }_{2}}(z) - {{\rho }_{{2,{\text{g}}}}}} \right)dz} .$Эффективная толщина межфазной границы раздела $\Delta $ определялась как расстояние, на котором плотность изменяется от значения ${{\rho }_{{10}}}$ = ${{\rho }_{{{\text{g }}}}} + 0.1\Delta \rho $ до ${{\rho }_{{90}}}$ = ${{\rho }_{{{\text{g }}}}} + 0.9\Delta \rho ,$ где $\Delta \rho $ = ${{\rho }_{{\text{l}}}} - {{\rho }_{{\text{g}}}}$ [24].
3. РЕЗУЛЬТАТЫ МОДЕЛИРОВАНИЯ
3.1. Равновесные состояния двухфазных систем
На первом этапе молекулярно-динамического моделирования при температуре T = 0.7, которая близка к температуре тройной точки первого компонента ${{T}_{{\text{t}}}}$ = 0.692 [23], получены равновесные двухфазные состояния в системах разного состава. Расчеты проводились в ячейках двух размеров, различающихся величиной Lz: 80 и 160. Протяженность жидкой фазы Lz, l в ячейке с Lz = 80 составляла примерно 40 единиц, а с Lz = 160 изменялась от 95 до 120. Распределения $\rho (z),$ ${{P}_{{\text{N}}}}(z),$ ${{P}_{{\text{T}}}}(z)$ получены как средние по интервалам в 1200–2400 единиц времени. Полное время выхода двухфазной системы на равновесие составляло не менее 106 шагов интегрирования уравнений движения частиц. Параметры равновесия, толщина межфазного слоя и поверхностное натяжение представлены в табл. 1.
Таблица 1.
Параметры модели при приведенной температуре T = 0.7: длина ячейки Lz, радиус обрезания потенциала взаимодействия rc, равновесное значение концентрации второго компонента в жидкой фазе ${{с }_{{\text{l}}}},$ плотности жидкой ${{\rho }_{{\text{l}}}}$ и газовой ${{\rho }_{{\text{g}}}}$ фаз, давление насыщения ${{p}_{{\text{s}}}},$ толщина межфазной границы ${{\Delta }_{{{\text{eq}}}}},$ поверхностное натяжение ${{\gamma }_{{{\text{eq}}}}},$ относительная адсорбция второго компонента $\Gamma _{{2(1)}}^{{{\text{eq}}}}$
| Lz | ${{r}_{{\text{c}}}}$ | ${{с }_{{\text{l}}}}$ | ${{\rho }_{{\text{l}}}}$ | ${{\rho }_{{\text{g}}}}$ | ${{p}_{{\text{s}}}}$ | ${{\Delta }_{{{\text{eq}}}}}$ | ${{\gamma }_{{{\text{eq}}}}}$ | $\Gamma _{{2(1)}}^{{{\text{eq}}}}$ |
|---|---|---|---|---|---|---|---|---|
| 80 | 2.5 | 0 | 0.787 | 0.0076 | 0.006 | 2.326 | 0.580 | – |
| 80 | 6.78 | 0 | 0.840 | 0.0021 | 0.002 | 1.975 | 1.089 | – |
| 80 | 6.78 | 0.032 | 0.849 | 0.0420 | 0.028 | 2.251 | 0.960 | 0.185 |
| 80 | 6.78 | 0.092 | 0.865 | 0.1232 | 0.081 | 2.705 | 0.749 | 0.416 |
| 80 | 6.78 | 0.174 | 0.887 | 0.2428 | 0.153 | 3.225 | 0.520 | 0.599 |
| 160 | 6.78 | 0 | 0.840 | 0.0022 | 0.003 | 1.975 | 1.085 | – |
| 160 | 6.78 | 0.032 | 0.848 | 0.0358 | 0.025 | 2.206 | 0.989 | 0.144 |
| 160 | 6.78 | 0.092 | 0.865 | 0.1231 | 0.081 | 2.705 | 0.747 | 0.417 |
| 160 | 6.78 | 0.168 | 0.886 | 0.2290 | 0.147 | 3.221 | 0.552 | 0.564 |
| 160 | 6.78 | 0.247 | 0.907 | 0.3630 | 0.225 | 3.950 | 0.366 | 0.634 |
Хорошо известна достаточно сильная зависимость плотностей сосуществующих фаз, давления насыщения и поверхностного натяжения от радиуса обрезания потенциала взаимодействия rc [25–27]. Как отмечено в работе [26], вкладом в поверхностное натяжение от взаимодействия на расстояниях, превышающих rc, можно пренебречь, если rc > 5.5. В нашей работе основной массив данных получен при rc = 6.78. Расчеты с rc = 2.5 проведены для иллюстрации влияния дальнодействующих взаимодействий на релаксационные процессы в межфазном слое (см. табл. 1).
После установления двухфазного равновесия через каждые 10 000 шагов интегрирования уравнений движения запоминались координаты и скорости частиц, которые в дальнейшем использовались для запуска независимых процессов восстановления межфазной границы по методике, изложенной ниже. По равновесным распределениям плотности определялись границы межфазных слоев (показаны серым цветом на рис. 1). Для создания исходной ступенчатой разделяющей границы между однородными фазами, с которой начинался процесс восстановления поверхностного слоя, удалялись все частицы межфазных слоев, а частицы газовой фазы и стенки ячейки, перпендикулярные оси z, смещались к слою жидкости. Возможные “избыточные” отталкивательные взаимодействия, после совмещения границ жидкости и газа, устранялись удалением из зон контакта частиц, расстояния между которыми вдоль оси z были меньше 0.75. Таким образом, в момент времени t = 0 объемные фазы, имеющие равные значения химического потенциала, температуры и давления, оказывались в контакте, и начинался процесс восстановления переходного слоя между ними.
Рис. 1.
Распределение числовой плотности $\rho (z)$ (а), нормальной ${{P}_{{\text{N}}}}(z)$ и тангенциальной ${{P}_{{\text{T}}}}(z)$ компонент тензора давления (б) по высоте ячейки в состоянии двухфазного равновесия при T = 0.7 и ${{с }_{{\text{l}}}} = $ 0.091: профиль плотности смеси (штриховая линия), профиль парциальной плотности первого компонента и ${{P}_{{\text{N}}}}(z)$ (сплошная линия), второго компонента и ${{P}_{{\text{T}}}}(z)$ (пунктирная линия).
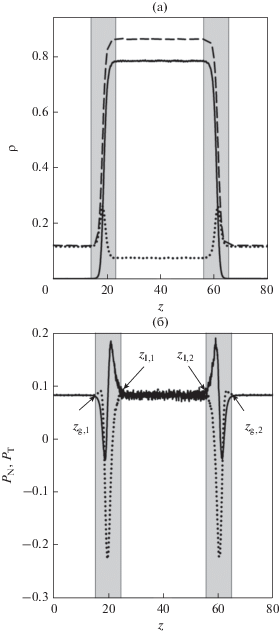
Для однокомпонентной системы плотности однородных участков жидкости и газа в начале и по завершении релаксации должны иметь близкие значения. В бинарной системе летучий компонент адсорбируется в межфазном слое (рис. 1), поэтому, после его удаления, средняя концентрация второго компонента в системе понижается. По окончании релаксации это приводит к изменению параметров равновесия.
3.2. Релаксационные процессы в однокомпонентной двухфазной системе
При фиксированном термодинамическом состоянии (p, T, c = const) из различных конфигураций частиц в ячейке, содержащей приведенные в контакт жидкую и газовую фазы, запускалось 200 независимых процессов восстановления межфазной границы. В каждом таком процессе на сотом, тысячном или десятитысячном шаге интегрирования уравнений движения частиц (в зависимости от стадии релаксации) рассчитывались распределения плотности $\rho (z)$ и компонент тензора давления ${{P}_{{\text{N}}}}(z)$ и ${{P}_{{\text{T}}}}(z).$ Полученные распределения, отвечающие одинаковым моментам времени от начала релаксации, затем усреднялись по числу наблюдаемых процессов восстановления межфазного слоя.
В течение первых 100 шагов (t = 0.116) распределение $\rho (z)$ сохраняло ступенчатую форму. Сразу после удаления межфазного слоя частицы, находящиеся вблизи искусственно созданной границы фаз, испытывали некомпенсированное взаимодействие со стороны частиц жидкой фазы. Это приводило к росту давления у границы фаз, которое примерно в 8 раз превышало его равновесное значение. Формирование межфазного слоя сопровождалось появлением двух волн сжатия, распространяющихся от межфазных границ внутрь жидкого слоя. В момент времени t ≈ 2.2 волны встречались в центре жидкого слоя, что приводило к генерации волн отражения, двигающихся к межфазным границам. В течение всего начального этапа релаксации (0 < t < 4.5) давление в жидкости отличалось от давления в газе, а в жидкой фазе значения ${{P}_{{\text{N}}}}(z)$ и ${{P}_{{\text{T}}}}(z)$ не совпадали. Отсутствие механического равновесия не позволяло использовать для определения поверхностного натяжения уравнение (2). Поэтому на стадии релаксации для оценки величины γ использовалась следующая модификация уравнения (2):
(4)
$\gamma = \frac{1}{2}\left[ {\int\limits_{{{z}_{{{\text{g}},1}}}}^{{{z}_{{{\text{l}},1}}}} {\left( {{{P}_{{\text{N}}}}(z) - {{P}_{{\text{T}}}}(z)} \right){\kern 1pt} dz} + } \right.\left. {\int\limits_{{{z}_{{{\text{l}},2}}}}^{{{z}_{{{\text{g}},2}}}} {\left( {{{P}_{{\text{N}}}}(z) - {{P}_{{\text{T}}}}(z)} \right){\kern 1pt} dz} } \right],$Равенство нормальной и тангенциальной компонент тензора давления в жидком слое достигалось при t ≈ 4.5. Дальнейшая релаксация переходного слоя проходила при установившемся механическом равновесии. По завершении релаксации максимальное значение разности ${{P}_{{\text{N}}}}(z) - {{P}_{{\text{T}}}}(z)$ снижалось примерно на 20% по отношению к ее значению при t ≈ 4.5.
Функции $\rho (z)$ и ${{P}_{{\text{N}}}}(z) - {{P}_{{\text{T}}}}(z)$, полученные в разные моменты времени, позволяют проследить эволюцию плотности в объемных фазах (рис. 2), толщины межфазного слоя (рис. 3) и поверхностного натяжения (рис. 4).
Рис. 2.
Эволюция плотности жидкой (1–3) и газовой (4, 5) фаз в процессе восстановлении межфазного слоя в моделях с радиусами обрезания потенциала Леннард-Джонса ${{r}_{{\text{c}}}}$ = 2.5 (точки 3, 4, толщина жидкого слоя Lz,l = 40) и ${{r}_{{\text{c}}}}$ = 6.78 (точки 1, 5, Lz,l = 40; точки 2, Lz,l = 94). Штриховые линии – равновесные значения плотностей ${{\rho }_{{\text{l}}}}$ и ${{\rho }_{{\text{g}}}}.$
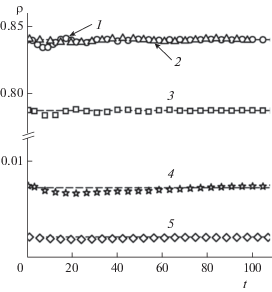
Рис. 3.
Толщина межфазного слоя как функция времени в линейном (a) и логарифмическом (б) масштабах в моделях с разным радиусом обрезания потенциала Леннард-Джонса: 1 – ${{r}_{{\text{c}}}} = $ 2.5, 2 – 6.78. Штриховые линии – равновесные значения толщины ${{\Delta }_{{{\text{eq}}}}}.$ Числа в рамках – показатель степени b в уравнении (5).
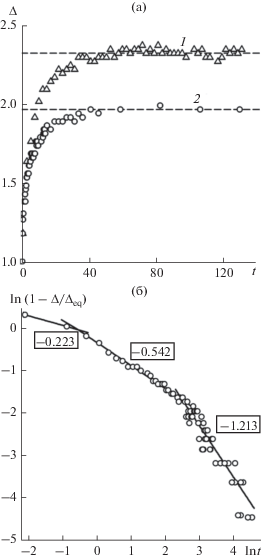
Рис. 4.
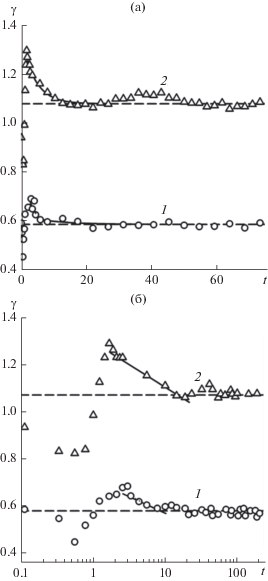
Динамическое поверхностное натяжение как функция времени в линейном (а) и логарифмическом (б) масштабах в моделях с разным радиусом обрезания потенциала Леннард-Джонса: 1 – ${{r}_{{\text{c}}}} = $ 2.5, 2 – 6.78. Штриховые линии – равновесные значения поверхностного натяжения ${{\gamma }_{{{\text{eq}}}}}$. Сплошные линии – аппроксимация уравнением (6).
Зависимость ${{\rho }_{{\text{g}}}}(t)$ имеет небольшой минимум, величина которого, как и равновесное значение ${{\rho }_{{\text{g}}}},$ определяется радиусом обрезания потенциала межчастичного взаимодействия. Уменьшение rc с 6.78 до 2.5 приводит к возрастанию времени выхода плотности на равновесное значение примерно в 1.5 раза.
При малой толщине слоя жидкости (Lz,l = 40) равновесное значение плотности ${{\rho }_{{\text{l}}}}$ устанавливается в процессе затухающих осцилляций функции ${{\rho }_{{\text{l}}}}(t)$. В ячейке с начальной длиной ребра Lz,l = 160 и Lz,l ≈ 95 волна сжатия затухает при однократном прохождении слоя жидкости, вследствие чего зависимость ${{\rho }_{{\text{l}}}}(t)$ очень слабая. Таким образом, рост толщины жидкого слоя снижает влияние волны сжатия на свойства объемных фаз.
В процессе релаксации толщина межфазного слоя ∆ возрастает и при t ≈ 60 отличается от равновесного значения ${{\Delta }_{{{\text{eq}}}}}$ не более чем на 3% (рис. 3). Когда rc = 6.78, формируется примерно на 25% более тонкий межфазный слой, чем при rc = 2.5. Относительная толщина межфазного слоя ${{\left[ {\Delta (t) - {{\Delta }_{{{\text{eq}}}}}} \right]} \mathord{\left/ {\vphantom {{\left[ {\Delta (t) - {{\Delta }_{{{\text{eq}}}}}} \right]} {{{\Delta }_{{{\text{eq}}}}}}}} \right. \kern-0em} {{{\Delta }_{{{\text{eq}}}}}}}$ изменяется по степенному закону
где a и b – индивидуальные постоянные.На зависимости $\Delta (t)$ можно выделить несколько этапов релаксации (рис. 3б). На начальном этапе увеличение толщины межфазной границы происходит в основном за счет частиц жидкой фазы. Это проявляется в образовании со стороны жидкого слоя “зоны обеднения”. В течение второго этапа межфазный слой расширяется примерно с одинаковой скоростью как в сторону жидкой, так и газовой фазы. Давления в жидкости и газе выравниваются, а толщина переходного слоя достигает 85% от величины ${{\Delta }_{{{\text{eq}}}}}.$ Третий этап протекает в условиях механического равновесия между объемными фазами. При достижении равновесной толщины ${{\Delta }_{{{\text{eq}}}}}$ устанавливаются равновесные значения нормальной и тангенциальной компонент тензора давления. Авторы работы [16] связывают этот этап с возбуждением на границе фаз капиллярных волн.
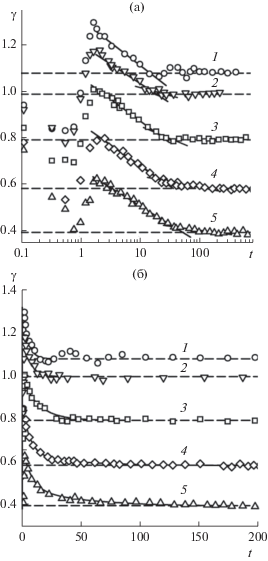
После удаления межфазного слоя на начальной стадии релаксации механическое равновесие нарушено. Используя уравнение (4) и проведя интегрирование только в пределах межфазного слоя, мы оценили величину поверхностного натяжения в процессе восстановления механического равновесия. Начальный этап релаксации характеризуется понижением поверхностного натяжения. Его минимальная величина составляет примерно 80% от равновесного значения ${{\gamma }_{{{\text{eq}}}}},$ после чего возрастает до максимума ${{\gamma }_{{\max }}},$ который на 20% больше ${{\gamma }_{{{\text{eq}}}}}.$ Последующее снижение γ до ${{\gamma }_{{{\text{eq}}}}},$ когда в объемных фазах устанавливается одинаковое давление, происходит по закону
(6)
$\gamma (t) = {{\gamma }_{{{\text{eq}}}}} + A\exp ({{ - t} \mathord{\left/ {\vphantom {{ - t} {{{\tau }_{\gamma }}}}} \right. \kern-0em} {{{\tau }_{\gamma }}}}).$4. Динамика формирования межфазной границы в двухкомпонентной системе
Интенсивность волны сжатия, возникающей в момент контакта двух однородных фаз, зависит от разности их плотностей. В свою очередь плотность фаз в существенной мере определяется их составом. Если в однокомпонентной системе (T = 0.7) плотности жидкости и газа различаются примерно в 380 раз, то в смеси это различие существенно меньше. При cl = 0.031 отношение ${{{{\rho }_{{\text{l}}}}} \mathord{\left/ {\vphantom {{{{\rho }_{{\text{l}}}}} {{{\rho }_{{\text{g}}}}}}} \right. \kern-0em} {{{\rho }_{{\text{g}}}}}} \approx $ 24, а при сl = 0.246 отношение ${{{{\rho }_{{\text{l}}}}} \mathord{\left/ {\vphantom {{{{\rho }_{{\text{l}}}}} {{{\rho }_{{\text{g}}}}}}} \right. \kern-0em} {{{\rho }_{{\text{g}}}}}} \approx $ 2.5. В двухфазной двухкомпонентной системе на границе фаз частицы испытывают притяжение не только со стороны жидкой, но и со стороны плотной газовой фазы. Некомпенсированность взаимодействий снижается по мере уменьшения отношения ${{{{\rho }_{{\text{l}}}}} \mathord{\left/ {\vphantom {{{{\rho }_{{\text{l}}}}} {{{\rho }_{{\text{g}}}},}}} \right. \kern-0em} {{{\rho }_{{\text{g}}}},}}$ поэтому на границе фаз в смеси генерируется более слабая волна сжатия, которая быстро затухает. Во всем исследованном диапазоне концентрации (cl = 0–0.25) рост давления в объемных фазах не превышал 15–20% от равновесного значения.
Относительно слабое возмущение волной сжатия объемных фаз проявляется в том, что в процессе восстановления межфазного слоя полная и парциальная плотности жидкой фазы, а также плотность первого компонента в газовой фазе, практически не изменяются со временем (рис. 5). Небольшое снижение парциальной плотности второго компонента в газовой фазе связано с установлением адсорбционного равновесия в межфазном слое, когда происходит его обогащение более летучим компонентом.
Рис. 5.
Эволюция плотности жидкой (1, 2, 5) и газовой (3, 4, 6) фазаx в процессе восстановлении межфазного слоя в системе с начальной концентрацией второго компонента ${{с }_{{\text{l}}}} = $ 0.168: 1, 3 – плотность смеси, 2, 6 – парциальная плотность первого компонента, 4, 5 – парциальная плотность второго компонента. Штриховые линии – равновесные значения плотности жидкой и газовой фаз смеси.
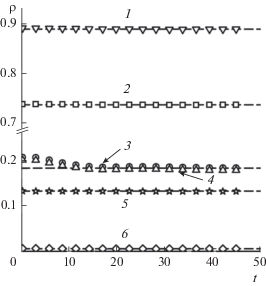
Рисунок 6 иллюстрирует эволюцию толщины межфазного слоя $\Delta (t)$ в процессе его восстановления. Равновесная толщина ${{\Delta }_{{{\text{eq}}}}}$ – линейная функция концентрации летучего компонента в жидкой фазе (вставка на рис. 6а). Как и в случае однокомпонентной системы, зависимость $\ln \left[ {1 - {{\Delta (t)} \mathord{\left/ {\vphantom {{\Delta (t)} {{{\Delta }_{{{\text{eq}}}}}}}} \right. \kern-0em} {{{\Delta }_{{{\text{eq}}}}}}}} \right]$ от ln t имеет характерные линейные участки (рис. 6б). На первом участке, в отсутствие механического равновесия, и втором участке, когда механическое равновесие восстанавливается, дополнительным фактором увеличения толщины межфазного слоя выступает диффузия в него летучего компонента как из газовой, так и из жидкой фазы. С началом третьего участка (t ≈ 6.6) диффузионный подвод происходит только из жидкой фазы. Зависимость $\Delta (t)$ на выделенных участках аппроксимирована функцией (5). На рис. 6б представлены значения показателя b этой функции.
Рис. 6.
Толщина межфазного слоя как функция времени в линейном масштабе (а) в системах с начальной концентрацией второго компонента ${{с }_{{\text{l}}}} = $ 0 (1), 0.031 (2), 0.09 (3), 0.168 (4), 0.246 (5) и в логарифмическом масштабе (б) при ${{с }_{{\text{l}}}}$ = 0.246. Штриховые линии – равновесные значения толщины ${{\Delta }_{{{\text{eq}}}}},$ сплошные линии – расчет по уравнению (5). Числа в рамках – показатель степени b в уравнении (5).
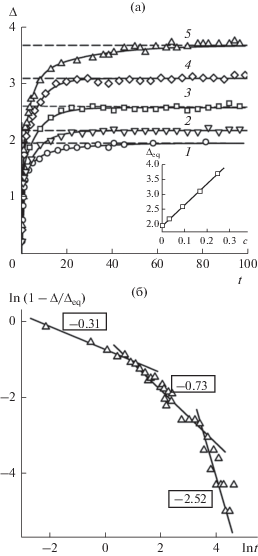
Установление равновесного состава в межфазном слое демонстрирует зависимость относительной адсорбции второго компонента ${{\Gamma }_{{{\text{2(1)}}}}}$ от времени (рис. 7). До t ≈ 4 величина ${{\Gamma }_{{{\text{2(1)}}}}}(t)$ отрицательна, что связано с появлением зон обеднения вторым компонентом жидкой и газовой фаз вблизи границ их контакта. При t > 4 происходит быстрый рост величины ${{\Gamma }_{{{\text{2(1)}}}}}(t).$ Здесь избыток второго компонента в межфазном слое образуется за счет поступления его из обеих фаз. Как следует из рис. 7, на зависимости ${{\Gamma }_{{{\text{2(1)}}}}}(t)$ имеются участки, на которых рост относительной адсорбции временно сменяется ее уменьшением. С приближением к равновесию рост ${{\Gamma }_{{{\text{2(1)}}}}}$ существенно замедляется. “Переключение” темпа роста ${{\Gamma }_{{{\text{2(1)}}}}}$ с быстрого на медленный происходит, когда прекращается пополнение межфазного слоя летучим компонентом из газовой фазы.
Рис. 7.
Эволюция относительной адсорбции второго компонента в процессе восстановления межфазного слоя в двухкомпонентных системах: 1 – ${{с }_{{\text{l}}}} = $ 0.031, 2 – 0.09, 3 – 0.168, 4 – 0.246. Штриховые линии – равновесные значения $\Gamma _{{2(1)}}^{{{\text{eq}}}},$ сплошные линии – аппроксимация экспоненциальными функциями.
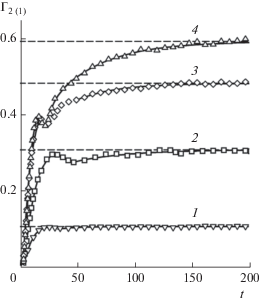
На начальной стадии релаксации характер изменения поверхностного натяжения в бинарной системе качественно такой же, как и в однокомпонентной (рис. 7). В течение первых 0.6–0.8 единиц времени поверхностное натяжение достигает наименьшего значения ${{\gamma }_{{{\text{min}}}}}$. Рост концентрации летучего компонента в жидкой фазе приводит к снижению относительной глубины минимума ${{{{\gamma }_{{{\text{min}}}}}} \mathord{\left/ {\vphantom {{{{\gamma }_{{{\text{min}}}}}} {{{\gamma }_{{{\text{eq}}}}}}}} \right. \kern-0em} {{{\gamma }_{{{\text{eq}}}}}}}$. Независимо от состава смеси последующий рост поверхностного натяжения происходит до t ≈ 2, когда достигается значение $\gamma = {{\gamma }_{{{\text{max}}}}}$. В момент t ≈ 2 равенство давления в объемных фазах еще не достигнуто. В исследованном интервале концентраций оно наступает по истечению 3–4.5 единиц времени с момента начала релаксации.
В отличие от однокомпонентной системы, после прохождения ${{\gamma }_{{{\text{max}}}}}$ на зависимости $\gamma (t)$ двухкомпонентной системы можно выделить два участка экспоненциального затухания (рис. 8а). Изменение наклона зависимости $\gamma (t)$ происходит примерно в то же время, когда наблюдается переход от “быстрого” темпа роста ${{\Gamma }_{{{\text{2(1)}}}}}$ к “медленному” (рис. 7). Участку “быстрого” уменьшения поверхностного натяжения отвечает время релаксации $\tau _{\gamma }^{{(1)}},$ “медленному” – $\tau _{\gamma }^{{(2)}}.$ Время $\tau _{\gamma }^{{(1)}}$ определяется поступлением в межфазный слой частиц второго компонента из обеих фаз. При неограниченных размерах контактирующих фаз релаксация определялась только этим процессом. Наибольшая величина $\tau _{\gamma }^{{(1)}}$ = (40 ± 2) × 10–12 с достигается при сl = 0.246, что в 4 раза больше времени релаксации γ в однокомпонентной системе.
Рис. 8.
Динамическое поверхностное натяжение в системах с разной концентрацией второго компонента в жидкой фазе: 1 – c1 = 0, 2 – 0.031, 3 – 0.09, 4 – 0.168, 5 – 0.246 в логарифмическом (а) и линейном (б) масштабах времени. Штриховые линии – равновесные значения ${{\gamma }_{{{\text{eq}}}}},$ сплошные линии – аппроксимация уравнением (6).
В исследуемых моделях газовая фаза имела толщину (размер в направлении оси z) примерно в 2.5 раз меньшую, чем контактирующая с ней жидкая фаза. При таком соотношении размеров поступление частиц легколетучего компонента из газовой фазы в межфазный слой прекращалось раньше, чем из жидкости. Пополнение переходного слоя частицами второго компонента только из жидкой фазы приводило к увеличению времени релаксации поверхностного натяжения. При концентрации сl = 0.246 время $\tau _{\gamma }^{{(2)}}$ = (1.1 ± 0.2) × × 10–10 с, что примерно в 2.5 раз больше $\tau _{\gamma }^{{(1)}}$ и в 10 раз превышает время релаксации в чистом растворителе.
4. ЗАКЛЮЧЕНИЕ
В молекулярно-динамических экспериментах исследован процесс формирования межфазной границы в двухфазной леннард-джонсовской системе при приведении в контакт однородных жидкости и пара, имеющих равные значения температуры, давления и химического потенциала. Расчеты проведены для одно- и двухкомпонентных систем при температуре, близкой к температуре тройной точки первого компонента – растворителя. Второй компонент в смеси являлся летучей примесью. Исследовалась релаксация плотности, давления, относительной адсорбции летучего компонента, толщины формируемого межфазного слоя, поверхностного натяжения.
В результате моделирования установлено следующее.
1. В системе с ограниченным размером жидкой фазы установлению механического равновесия предшествует появление волны сжатия, распространяющейся от межфазной границы сначала внутрь жидкости, а затем в обратном направлении.
2. Как и параметры фазового равновесия, время релаксации поверхностного натяжения зависит от радиуса обрезания потенциала межчастичных взаимодействий rc.
3. В однокомпонентной системе максимальная величина динамического поверхностного натяжения γmax достигается в конце этапа установления механического равновесия и в 1.2 раза превышает равновесное значение γeq. В модели с rc = 6.78 время релаксации поверхностного натяжения от γmax до γeq составляет около 10 пс.
4. В двухкомпонентной системе рост концентрации летучего компонента (от 0 до 0.25) приводит к увеличению времени релаксации γ до 40 пс. Максимальное значение γmax превосходит γeq в 1.4–1.6 раз.
5. Уменьшение протяженности газовой фазы (до 25 молекулярных диаметров) сопровождается увеличением времени релаксации поверхностного натяжения с 10 до 100 пс при возрастании содержания летучего компонента в жидкой фазе до 0.25.
Полученные результаты необходимо принимать во внимание при интерпретации данных по предельному перегреву растворов. Экспериментальные исследования [2, 28] и компьютерное моделирование [29, 30] зародышеобразования в бинарных растворах перегретых жидкостей свидетельствуют о превышении достигнутых перегревов над их теоретическими значениями, если в теории нуклеации игнорируется размерная зависимость поверхностного натяжения критических пузырьков [31]. Учет такой зависимости в рамках теории капиллярности Ван-дер-Ваальса приводит к сближению результатов теории нуклеации и данных натурных и компьютерных экспериментов [2, 32]. Как показано в работах [2, 28], при составе, близком к эквимолярному, поверхностное натяжение критических пузырьков в растворе выше, чем на плоской межфазной границе.
В отличие от однокомпонентной системы, где характерное время релаксации поверхностного натяжения близко к времени роста пузырька до критического размера, что оправдывает использование значения γeq для расчета ${{W}_{*}}$, в растворе ситуация иная. Так, согласно [29], в молекулярно-динамической модели раствора аргон–неон с концентрацией неона, равной 0.16, пузырек достигал критического размера за время ≈5 × 10–11 с, в то время как на плоской межфазной границе снижение поверхностного натяжения от γmax до γeq продолжалось в течение ≈3 × 10–10 с. Таким образом, время установления адсорбционного равновесия на плоской границе примерно на порядок выше времени роста зародыша до критического размера. В момент достижения пузырьком критического размера отношение ${\gamma \mathord{\left/ {\vphantom {\gamma {{{\gamma }_{{{\text{eq}}}}}}}} \right. \kern-0em} {{{\gamma }_{{{\text{eq}}}}}}}$ = 1.07, а ${{{{W}_{*}}\left( \gamma \right)} \mathord{\left/ {\vphantom {{{{W}_{*}}\left( \gamma \right)} {W\left( {{{\gamma }_{{{\text{eq}}}}}} \right)}}} \right. \kern-0em} {W\left( {{{\gamma }_{{{\text{eq}}}}}} \right)}}$ = 1.24. Этот факт, наряду с зависимостью γ от кривизны зародыша новой фазы, должен также учитываться при интерпретации как экспериментальных данных, так и результатов компьютерного моделирования нуклеации в перегретых жидких растворах.
БЛАГОДАРНОСТИ
Авторы выражают благодарность Суперкомпьютерному центру Института математики и механики Уральского отделения РАН за вычислительные ресурсы, предоставленные для выполнения данной работы.
Список литературы
Русанов А.И. Фазовые равновесия и поверхностные явления. Л.: Химия, 1967.
Baidakov V.G. Explosive Boiling of Superheated Cryogenic Liquids. Weinheim: Wiley, 2007.
Байдаков В.Г., Проценко С.П. // Докл. АН. 2004. Т. 49. С. 69.
Eggers J. // Rev. Mod. Phys. 1997. V. 69. P. 865.
Basaran O.A. // AIChE J. 2002. V. 48. P. 1842.
Brennen C.E. // J. Fluid Mech. 2002. V. 472. P. 153.
Ueno I., Hattori Y., Hosoya R. // Microgravity Sci. Technol. 2011. V. 23. P. 73.
Van den Tempel M., Lucassen-Reynders E.H. // Adv. Colloid Interface Sci. 1983. V. 18. P. 281.
Miller R., Joos P., Fainerman V.B. // Adv. Colloid Interface Sci. 1994. V. 49. P. 249.
Saint Vincent M.R.D., Petit J., Aytouna M., Delville J.P., Bonn D., Kellay H. // J. Fluid Mech. 2012. V. 692. P. 499 510.
Vandegrift A.E. // J. Colloid Interface Sci. 1967. V. 23. P. 43.
Owens D.K. // J. Colloid Interface Sci. 1969. V. 29. P. 496.
Kochurova N.N., Rusanov A.I. // J. Colloid Interface Sci. 1981. V. 81. P. 297.
Буланов Н.В., Скрипов В.П. // Коллоид. журн. 2003. Т. 65. С. 581.
Hauner I.M., Deblais A., Beattie J.K, Kellay H., Bonn D. // Phys. Chem. Lett. 2017. V. 8. P. 1599.
Lukyanov A.V., Likhtman A.E. // J. Chem. Phys. 2013. V. 138. P. 034712.
Hirschfelder J.O., Curtiss C.F., Bird R.B. Molecular Theory of Gases and Liquids. N.Y.: Wiley, 1954.
Baidakov V.G., Sulla I.I. // Int. J. Thermophys. 1995. V. 16. P. 909.
Каверин А.М., Андбаева В.Н., Байдаков В.Г. // Журн. физ. химии. 2006. Т. 80. С. 495.
Baidakov V.G., Grishina K.A. // Fluid Phase Equilib. 2013. V. 354. P. 245.
Plimpton S.J. // J. Comp. Phys. 1995. V. 17. P. 1.
Hoover W.G. // Phys. Rev. A. 1985. V. 31. P. 1695.
Baidakov V.G., Protsenko S.P., Kozlova Z.R., Chernykh G.G. // J. Chem. Phys. 2007. V. 126. 214505.
Lekner J., Henderson J.R. // Physica A. 1978. V. 94. P. 545.
Trokhymchuk A., Alejandre J. // J. Chem. Phys. 1999. V. 111. P. 8510.
Baidakov V.G., Chernykh G.G., Protsenko S.P. // Chem. Phys. Lett. 2000. V. 321. P. 315.
Dunikov D.O., Malyshenko S.P., Zhakhovskii V.V. // J. Chem. Phys. 2001. V. 115. P. 6623.
Baidakov V.G., Pankov A.S. // Int. J. Heat Mass Transfer. 2015. V. 86. P. 930.
Protsenko S.P., Baidakov V.G., Teterin A.S., Zhdanov E.R. // J. Chem. Phys. 2007. V. 126. P. 094502.
Baidakov V.G., Protsenko S.P., Bryukhanov V.M. // Chem. Phys. Lett. 2016. V. 663. P. 57.
Baidakov V.G. // J. Chem. Phys. 1999. V. 110. P. 3955.
Baidakov V.G., Boltachev G.Sh. // J. Chem. Phys. 2004. V. 121. P. 8594.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал