

Коллоидный журнал, 2020, T. 82, № 1, стр. 119-130
Тензиометрические и реологические характеристики фракций гуминовых и гиматомелановых кислот
С. Л. Хилько 1, *, М. И. Рогатко 1, Р. А. Макарова 1, Р. Г. Семенова 1
1 Институт физико-органической химии и углехимии им. Л.М. Литвиненко
283114 Донецк-114,
ул. Р. Люксембург, 70, Украина
* E-mail: sv-hilko@yandex.ru
Поступила в редакцию 09.04.2019
После доработки 12.06.2019
Принята к публикации 24.06.2019
Аннотация
Методами кольца Дю Нуи и формы висячей капли исследованы тензиометрические (динамическое и равновесное поверхностное натяжение) и поверхностные реологические (модуль вязкоупругости и фазовый угол) характеристики водных растворов фракций гуминовых и гиматомелановых кислот на границе с воздухом. Установлено, что фракция гиматомелановых кислот с низкой молекулярной массой обладает высокой поверхностной активностью, а ее поверхностные слои характеризуются высокими значениями модулей вязкоупругости и упругости. Экспериментальные зависимости равновесного поверхностного натяжения и модуля вязкоупругости от концентрации растворов солей гиматомелановых кислот хорошо описываются в рамках модели реальных двумерных растворов для полимолекулярной адсорбции полиэлектролитов.
ВВЕДЕНИЕ
Гуминовые и гиматомелановые кислоты считаются наиболее химически активными компонентами органогенных субстратов (угля, торфа, сапропеля, горючих сланцев) [1, 2], что делает их уникальными объектами для решения важных практических задач различного плана. Гиматомелановые кислоты (ГМК) – это спирторастворимые фракции гуминовых кислот (ГК), которые, в свою очередь, являются растворимыми в щелочах и нерастворимыми в кислотах фракциями гумусовых веществ. В настоящее время гуминовые вещества признаны одним из перспективных возобновляемых, экономически выгодных и экологически безопасных источников сырья для получения важных химических продуктов методами “зеленой” химии [3–5]. Уже имеется значительный опыт применения различных видов гуминовых удобрений и стимуляторов роста растений [6]. Природные полимеры на основе гуминовых веществ являются активными комплексообразующими сорбентами по отношению к тяжелым металлам, радионуклидам, ароматическим углеводородам, гербицидам и другим органическим экотоксикантам [7–15]. Гуминовые вещества проявляют выраженные свойства окислительно-восстановительных полимеров [16–19], ингибиторов коррозии металлов [20, 21], антиоксидантов [22–26], а также являются основой для получения лекарств и биологически активных добавок [27–30]. Кроме того, макромолекулы гуминовых веществ способны к образованию комплексов полимер–лекарство, что может быть перспективно для разработки новых носителей лекарственных препаратов [31, 32].
Интерес к гуминовым соединениям обусловлен их составом. Гуминовые вещества представляют собой супрамолекулярные ансамбли, в которых совокупность макромолекул образует динамические ассоциаты, стабилизированные гидрофобными взаимодействиями и водородными связями. При высоких концентрациях в растворе эти ассоциаты могут организовываться в мицеллоподобные агрегаты нанометрового размера [33–36]. Макромолекулы гуминовых соединений являются высокоароматическими сопряженными структурами, содержащими большое количество активных карбоксильных и гидроксильных групп, а также эфирные и аминогруппы, которые определяют их реакционную способность, физико-химические и биологические свойства.
Сочетание в структуре макромолекул гуминовых веществ гидрофильных (за счет большого количества функциональных групп) и гидрофобных (за счет алифатических и ароматических фрагментов) участков способствует проявлению ими дифильных свойств и выраженной поверхностной активности на межфазных границах [37, 38]. Нативные и модифицированные гуминовые вещества являются основой для получения эффективных ПАВ, которые могут использоваться в качестве моющих средств [39–41], добавок, регулирующих свойства дисперсных систем различного назначения [42–46], в том числе содержащих наночастицы [47–49], флотореагентов [50] и т.д.
Одним из способов разделения ГК на фракции является выделение ГК из органогенных субстратов щелочной экстракцией при разных значениях температуры [37, 51], а также выделение из этих фракций ГК экстракцией этиловым спиртом ГМК также при разной температуре.
Свойства ГК и ГМК на различных межфазных границах представляют особый интерес, однако детальному изучению межфазных характеристик фракций гуминовых веществ посвящено не так много работ [37, 51].
Целью настоящей работы было исследование тензиометрических свойств (динамическое и равновесное поверхностное натяжение) и реологических характеристик (модуль вязкоупругости, фазовый угол) поверхностных слоев растворов солей ГК и ГМК, полученных при разной температуре экстракции, на границе фаз жидкость–газ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ГК выделяли из образцов бурого угля Александрийского месторождения после механохимической обработки, согласно [52], однократной экстракцией при соотношении твердой и жидкой фаз 1 : 8 и двух значениях температуры – 100 и 20°C (ГК100 и ГК20 соответственно). Затем из “сырого” экстракта получали нерастворимые в воде ГК осаждением 5%-ным раствором HCl, который добавляли при постоянном перемешивании до pH 1−2. Выпавший осадок ГК отделяли от надосадочной жидкости центрифугированием и промывали дистиллированной водой до нейтральной реакции среды (pH 6−7). Промытые ГК сушили при 80°C до постоянной массы. Растворы гуматов натрия получали растворением промытых и сухих ГК в 0.1 н. растворе NaOH.
ГМК получали из ГК100 и ГК20 однократной экстракцией этанолом при соотношении твердой и жидкой фаз 1 : 10 и двух температурах – 20°C и температуре кипения спирта, 80°C. При этом получали следующие образцы ГМК: ГМК80 из ГК100 (ГМК80–100), ГМК20 из ГК100 (ГМК20–100), ГМК80 из ГК20 (ГМК80–20) и ГМК20 из ГК20 (ГМК20–20).
Экстракцию проводили при перемешивании смеси компонентов в интенсивном турбулентном режиме с использованием механической мешалки (900 об./мин, 2 ч). Спирт отделяли упариванием, остаток высушивали в сушильном шкафу до постоянной массы. Затем весовым методом из сухих образцов ГМК готовили растворы натриевых солей необходимой концентрации путем растворения в 0.1 н. растворе NаОН.
Поверхностное натяжение (γ) водных растворов фракций гуминовых веществ при фиксированной концентрации в зависимости от времени жизни поверхности (t) измеряли двумя методами. Методом кольца Дю Нуи (тензиометр TE-1, Lauda, Германия) исследовали поверхностное натяжение в интервале от 10 до 105 с. Принцип работы прибора описан в [53]. Значения γ рассчитывали с учетом поправочных коэффициентов Харкинса–Джордана [54]. Ошибка при измерении γ не превышала ±0.1 мН/м.
Для определения динамического и равновесного поверхностного натяжения в широком диапазоне времени жизни межфазной поверхности (от 1 до 105 с) использовали также метод формы висячей капли (тензиометр PAT-2P, SINTERFACE Technologies, Германия).
Дилатационные реологические характеристики поверхностных слоев на границе фаз жидкость–газ изучали методом формы осциллирующей капли (тензиометр PAT‑2P) [55, 56]. В этом методе капля заданного объема формируется на конце капилляра. После достижения адсорбционного равновесия площадь капли A подвергается периодической синусоидальной деформации (осцилляции) малой амплитуды (ΔA/A = ±7–8%), с частотой f в диапазоне 0.005–0.5 Гц. Результаты экспериментов с гармоническими осцилляциями поверхности капли были проанализированы с использованием преобразования Фурье [55, 56]:
где A0 – начальная площадь поверхности капли.Дилатационный модуль E характеризует вязкоупругие свойства поверхностных слоев ПАВ и учитывает все релаксационные процессы, влияющие на поверхностное натяжение γ. При малой амплитуде ΔA гармонических осцилляций поверхности с угловой частотой $\Omega = 2\pi f,$ ΔA = $ = \Delta \bar {A}\exp (i\Omega t),$ имеет место следующее выражение для дилатационного модуля вязкоупругости [57, 58]:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Поскольку гуминовые вещества являются биогеополимерами, их состав определяется набором сходных молекул, который отражает характерное поведение полимеров нерегулярного строения [59]. Несмотря на многочисленные исследования, проводимые в течение последних десятилетий, определение точного строения макромолекул гуминовых веществ и их конформаций является актуальной проблемой. В литературе приводятся экспериментально обоснованные данные об основных структурных фрагментах гуминовых веществ, представляющих собой минимальные по размеру части их молекул, которые содержат все важнейшие структурные составляющие [60–66]. Макромолекулы гуминовых веществ содержат гидрофобный центральный ароматический фрагмент и обогащенную функциональными группами периферическую часть.
На рис. 1 приведены вероятные структуры молекулярных фрагментов гуминовых веществ, которые наиболее полно отражают результаты экспериментальных исследований и ретробиосинтетического анализа [62–66].
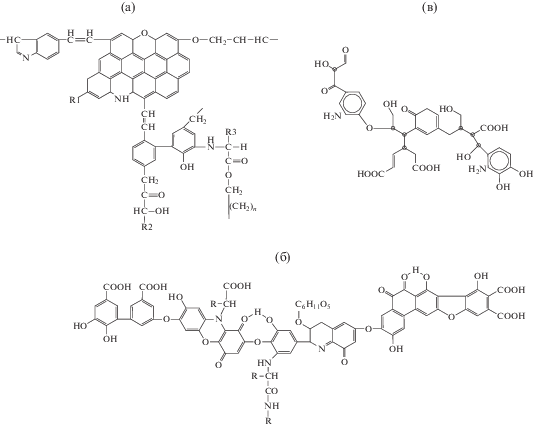
Тензиометрические характеристики растворов солей фракций гуминовых и гиматомелановых кислот на границе фаз жидкость–газ
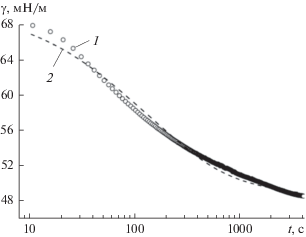
На рис. 2 и 3 приведены экспериментальные зависимости поверхностного натяжения от времени для растворов солей ГК и ГМК разной концентрации (метод кольца Дю Нуи). Время формирования поверхностных слоев в растворах солей ГК весьма значительно (>104 с), и минимальное значение γ существенно зависит от концентрации раствора. Время достижения равновесных значений γ для солей ГМК также зависит от концентрации их растворов, при этом для CГМК > > 0.5 мас. % поверхностный слой формируется гораздо быстрее, t ≈ 103 с. Кроме того, соли ГМК более эффективно снижают поверхностное натяжение по сравнению с солями ГК. Минимальное значение равновесного поверхностного натяжения их растворов на границе жидкость–газ составляет 38−40 мН/м. На рис. 3 (кривая 6) для сравнения приведена зависимость поверхностного натяжения от времени для раствора “остатка” ГК после экстракции ГМК. Такие соединения проявляет низкую поверхностную активность. Это значит, что фракции ГМК являются наиболее поверхностно-активными компонентами ГК.
Рис. 2.
Изменение поверхностного натяжения во времени для растворов гумата натрия (ГК100) различной концентрации: 1 – 7.2 × 10–6, 2 – 3.6 × 10–5, 3 – 7.2 × × 10–5, 4 – 3.6 × 10–4 и 5 – 7.2 × 10–4 моль/л (т.е. 0.01, 0.05, 0.10, 0.50 и 1.00 мас. %). Метод кольца Дю Нуи.
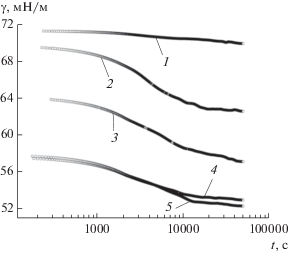
Рис. 3.
Изменение поверхностного натяжения во времени для растворов соли ГМК80–100 различной концентрации: 1 – 8.4 × 10–6, 2 – 4.2 × 10–5, 3 – 8.4 × 10–5, 4 – 4.2 × 10–4 и 5 – 5.9 × 10–4 моль/л (т.е. 0.01, 0.05, 0.10, 0.50 и 0.07 мас. %). 6 – Раствор остатка ГК100 (после экстракции ГМК80–100) концентрации 1.44 × × 10–4 моль/л (0.50 мас. %). Метод кольца Дю Нуи.
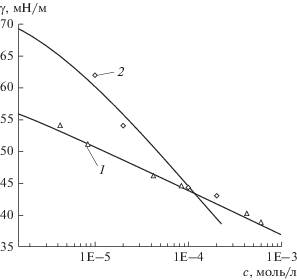
Зависимости равновесного поверхностного натяжения растворов солей ГМК от концентрации приведены на рис. 4. Для описания равновесного поверхностного натяжения этих растворов была использована модель неидеального двумерного раствора слабых полиэлектролитов белковой природы [67, 68]. Эта модель основана на предположении о том, что молекулы полиэлектролитов могут существовать в n состояниях с различной молярной площадью, изменяющейся от максимального значения, ωmax, при низкой степени заполнения ими поверхности до минимального значения, ωmin, при высокой степени заполнения поверхности. Молярная площадь полиэлектролита в i-ом состоянии равна ${{\omega }_{i}} = {{\omega }_{1}} + (i - 1){{\omega }_{0}}$ (1 ≤ i ≤ n), а инкремент молярной площади при переходе от одного состояния к другому принимается равным ω0. Таким образом, ${{\omega }_{1}} = {{\omega }_{{\min }}} \ll {{\omega }_{0}}$ и ${{\omega }_{{\max }}} = {{\omega }_{1}} + (n - 1){{\omega }_{0}}.$
Рис. 4.
Изотермы γ = f (СГМК), измеренные методом кольца Дю Нуи, для ГМК80–100 (1) и ГМК20–100 (2). Кривые рассчитаны по уравнениям модели реального двумерного раствора для случая бимолекулярной адсорбции полиэлектролитов.
Основными уравнениями модели неидеального двумерного раствора слабых полиэлектролитов являются уравнения состояния поверхностного слоя и изотермы адсорбции для каждого j-го состояния молекулы полиэлектролита в поверхностном слое:
(1)
$ - \frac{{\Pi {{\omega }_{0}}}}{{RT}} = \ln (1 - {{\theta }_{{\text{P}}}}) + {{\theta }_{{\text{P}}}}({{1 - {{\omega }_{0}}} \mathord{\left/ {\vphantom {{1 - {{\omega }_{0}}} {{{\omega }_{{\text{P}}}}}}} \right. \kern-0em} {{{\omega }_{{\text{P}}}}}}) + {{a}_{{\text{P}}}}\theta _{{\text{P}}}^{{\text{2}}},$(2)
${{b}_{{\text{P}}}}{{c}_{{\text{P}}}} = \frac{{{{\omega }_{{\text{P}}}}{{\Gamma }_{{{\text{P}}j}}}}}{{{{{(1 - {{\theta }_{{\text{P}}}})}}^{{{{{{\omega }_{j}}} \mathord{\left/ {\vphantom {{{{\omega }_{j}}} {{{\omega }_{{\text{P}}}}}}} \right. \kern-0em} {{{\omega }_{{\text{P}}}}}}}}}}}\exp \left[ { - 2{{a}_{{\text{P}}}}\frac{{{{\omega }_{j}}}}{{{{\omega }_{{\text{P}}}}}}{{\theta }_{{\text{P}}}}} \right],$(3)
${{{\Gamma }}_{{{\text{P}}j}}} = {{{\Gamma }}_{{\text{P}}}}\frac{{{{{(1 - {{\theta }_{{\text{P}}}})}}^{{({{{{\omega }_{j}} - {{\omega }_{1}})} \mathord{\left/ {\vphantom {{{{\omega }_{j}} - {{\omega }_{1}})} {{{\omega }_{{\text{P}}}}}}} \right. \kern-0em} {{{\omega }_{{\text{P}}}}}}}}}\exp \left[ {2{{a}_{{\text{P}}}}{{\theta }_{{\text{P}}}}\frac{{{{\omega }_{j}} - {{\omega }_{1}}}}{{{{\omega }_{{\text{P}}}}}}} \right]}}{{\sum\limits_{i = 1}^n {{{{(1 - {{\theta }_{{\text{P}}}})}}^{{{{({{\omega }_{i}} - {{\omega }_{1}})} \mathord{\left/ {\vphantom {{({{\omega }_{i}} - {{\omega }_{1}})} {{{\omega }_{{\text{P}}}}}}} \right. \kern-0em} {{{\omega }_{{\text{P}}}}}}}}}\exp \left[ {2{{a}_{{\text{P}}}}{{\theta }_{{\text{P}}}}\frac{{{{\omega }_{i}} - {{\omega }_{1}}}}{{{{\omega }_{{\text{P}}}}}}} \right]} }}.$Эта модель была использована ранее [37] применительно к растворам солей ГК, и показано, что для этих соединений изотерма поверхностного натяжения может быть хорошо описана в рамках модели реального двумерного раствора в предположении бимолекулярной адсорбции.
Кривые на рис. 4 также были рассчитаны по уравнениям модели реального двумерного раствора для случая бимолекулярной адсорбции солей ГМК [67, 68]. Значения параметров модели приведены в табл. 1. Наблюдается хорошее согласие между экспериментальными и расчетными зависимостями γ = f(CГМК).
Таблица 1.
Параметры модели двумерного раствора адсорбции ГМК
| Образец ГМК | а | α | ω0 × 105, м2/моль |
ωmin× 106, м2/моль |
ωmах × 106, м2/моль |
b × 104, м3/моль |
ωmax/ωmin | D × 10–10, м2/с |
$\overline М $* |
|---|---|---|---|---|---|---|---|---|---|
| ГМК80–100 | 1.0 | 0 | 7.25 | 3.00 | 20.00 | 1.50 | 5.7 | 1.20 | 12 000 |
| ГМК20–100 | 0 | 0 | 3.50 | 2.40 | 3.50 | 0.10 | 1.5 | 1.60 | 5000 |
| ГМК80–20 | 1.2 | 0 | 7.00 | 3.50 | 26.0 | 1.50 | 5.7 | 1.00 | 21 000 |
| ГМК20–20 | 1.1 | 0 | 6.00 | 3.00 | 20.0 | 1.00 | 8.7 | 1.10 | 17 000 |
Низкие значения параметра межмолекулярных взаимодействий a и константы адсорбционного равновесия b для фракции ГМК20–100 могут быть обусловлены как меньшей средней молекулярной массой этой фракции (табл. 1), так и меньшей ее активностью (рис. 4). Величина b зависит от степени заполнения поверхностного слоя и часто уменьшается с ростом степени заполнения.
Отношение ωmax/ωmin для трех фракций ГМК изменяется от 5.7 до 8.7, а для фракции ГМК20–100 оно равно 1.5.
Известно, что макромолекулы белков способны изменять молярную площадь на межфазной поверхности, причем для гибкоцепных белков в силу высокой способности их молекул к изменению конформации этот эффект проявляется в большей степени, чем для глобулярных: отношение ωmax/ωmin для гибкоцепного β-казеина равно 10. Для глобулярных белков набор конформаций ограничен по сравнению с гибкоцепными белками, поэтому отношение ωmax/ωmin для глобулярного β-лактоглобулина равно 2, что указывает на гораздо меньшую способность изменять молярную площадь при адсорбции [77].
В рамках модели двумерного адсорбционного раствора полиэлектролитов макромолекулы фракции ГМК20–100 слабо изменяют молярную площадь на межфазной поверхности и близки в этом смысле к β-лактоглобулину, тогда как фракции ГМК80–100, ГМК80–20, ГМК20–20 по способности к изменению молярной площади занимают промежуточное положение по сравнению с β-лактоглобулином и β-казеином.
Для выяснения механизма адсорбции солей ГМК на границе фаз жидкость–газ были использованы экспериментальные зависимости поверхностного натяжения от времени.
Известно, что зависимости динамической адсорбции ПАВ, Г(t), на свежеобразованной недеформируемой поверхности при диффузионном механизме процесса описываются соотношением Уорда и Тордея [78].
Если экспериментальные данные могут быть описаны с помощью этого уравнения, это указывает на формирование поверхностного слоя ПАВ по диффузионному механизму. При этом величина коэффициента диффузии должна составлять не менее 10–11 м2/с.
Коэффициенты диффузии молекул ГМК были рассчитаны по формуле Польсона, D = 2.74 × $ \times {{10}^{{ - 9}}}{{M}^{{{{ - 1} \mathord{\left/ {\vphantom {{ - 1} 3}} \right. \kern-0em} 3}}}}$ [79], и приведены в табл. 1. Следует отметить, что близкие значения коэффициентов диффузии для гуминовых соединений из различных природных источников были получены разными экспериментальными методами [80–82].
При совместном решении уравнения Уорда–Тордея с уравнениями, являющимися граничными условиями для диффузионного механизма адсорбции [83], показано (рис. 5), что экспериментальные данные описываются этой моделью только при очень низком значении коэффициента диффузии солей ГМК, D = 10–13 м2/с, тогда как реальные значения составляют порядка 10–10 м2/с. Для расчета были использованы программы, представленные в [73].
Рис. 5.
Изменение поверхностного натяжения во времени для раствора соли ГМК80–100 концентрации 4.2 × 10–5 моль/л; метод формы висячей капли. Кривая 1 – эксперимент, кривая 2 – расчет при D = 10–13 м2/с (пояснения в тексте). Различие результатов, полученных методами формы капли и кольца Дю Нуи (см. кривую 2 на рис. 3), обусловлено разной геометрией объектов, главным образом, – разным отношением объема раствора к площади его поверхности [84].
Порядок величины коэффициента диффузии для растворов солей гуминовых соединений указывает на недиффузионный (барьерный) механизм адсорбции, при котором скорость диффузии ПАВ из объема раствора велика по сравнению со скоростью установления равновесия между поверхностным и приповерхностным слоями. В этом случае концентрация ПАВ во всех частях раствора, в том числе и в приповерхностном слое, является постоянной величиной (C0). Согласно [85, 86], недиффузионный механизм адсорбции ПАВ объясняется существованием вблизи границы фаз энергетического адсорбционного барьера, который обусловлен электростатическими и неэлектростатическими взаимодействиями между молекулами ПАВ замедляет переход молекул ПАВ из приповерхностного слоя в поверхностный.
Реологические характеристики поверхностных слоев солей гиматомелановых кислот на границе фаз жидкость–газ
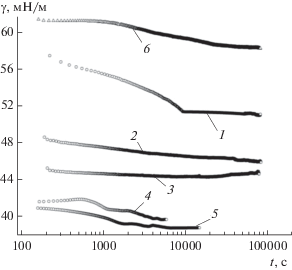
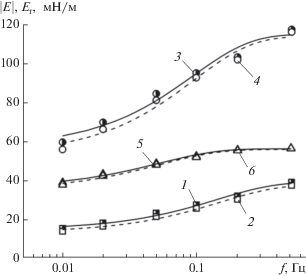
На рис. 6 представлены экспериментальные зависимости модулей вязкоупругости и упругости растворов солей ГМК от частоты осцилляций капли. Модули $\left| E \right|$ и Er монотонно возрастают при увеличении частоты осцилляции поверхности капли, а их абсолютные значения близки между собой для солей как ГМК, так и ГК.
Рис. 6.
Зависимости модулей вязкоупругости $\left| Е \right|$ (1, 3, 5) и упругости Еr (2, 4, 6) от частоты осцилляций f капель растворов ГМК с концентрацией 0.2 мас. %: 1, 2 – ГМК80–100, 3, 4 – ГМК20–100, 5, 6 – ГК100. Амплитуда – 6%.
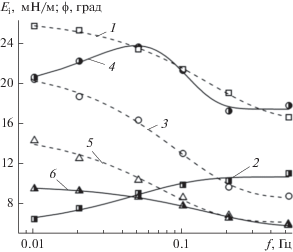
Значения фазового угла для растворов солей ГК и ГМК монотонно уменьшаются в диапазоне 6°−26° с ростом частоты колебаний поверхности капли (рис. 7, кривые 1, 3, 5). Величины модуля вязкости для солей ГМК80–100 и ГК100 – небольшие и изменяются с частотой в диапазоне 4−12 мН/м, а для ГМК20–100 величины Еi заметно выше и лежат в диапазоне 18−24 мН/м.
Рис. 7.
Зависимости фазового угла ϕ (1, 3, 5) и модуля вязкости Еi (2, 4, 6) от частоты осцилляций f капель растворов солей ГМК и ГК с концентрацией 0.2%. 1, 2 – ГМК80–100, 3, 4 – ГМК20–100, 5, 6 – ГК100.
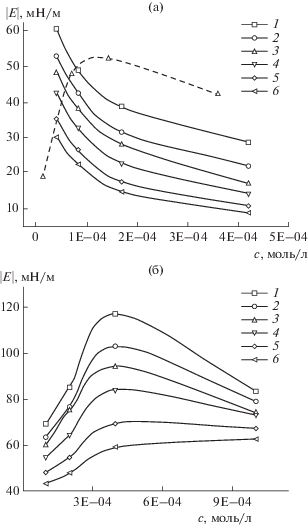
Зависимости модуля вязкоупругости от концентрации для двух фракций солей ГМК, полученные методом формы капли, приведены на рис. 8. Для ГМК80–100 наблюдается уменьшение величины $\left| E \right|$ для всех значений частоты осцилляции капли при увеличении концентрации раствора в выбранном диапазоне (рис. 8а). Для ГМК20–100 зависимость модуля вязкоупругости имеет выраженный максимум при концентрации 4 × × 10–4 моль/л (рис. 8б). Аналогичная картина была получена и для солей ГК [37].
Рис. 8.
Зависимости модуля вязкоупругости растворов фракций ГМК от концентрации: (а) ГМК80–100 (пунктирная линия – ГК100), (б) ГМК20–100. Частота осцилляций капли: 1 – 0.5, 2 – 0.2, 3 – 0.1, 4 – 0.05, 5 – 0.02, 6 – 0.01 Гц; амплитуда – 6%.
При высоких концентрациях растворы полиэлектролитов способны образовывать бислои (или полислои) на подвижных поверхностях раздела [68, 88]. Степень заполнения второго и последующих слоев пропорциональна константе адсорбционного равновесия b2. Теоретические основы равновесных адсорбционных свойств полиэлектролитов белковой природы для случая полимолекулярной адсорбции на межфазных границах приведены в работах [87–89]. Изотерма суммарной полимолекулярной адсорбции полиэлектролита в m слоях описывается уравнением [87–89]:
Рис. 9.
Аппроксимация экспериментальных концентрационных зависимостей E = f(C) уравнениями модели реального двумерного раствора для адсорбции полиэлектролитов (f = 0.1 Гц): (а) растворы ГМК80–100, (б) растворы ГМК20–100. 1 – Экспериментальные данные, 2, 3 и 4 – моно-, би- и 4 – тримолекулярная адсорбция. Пояснения в тексте.
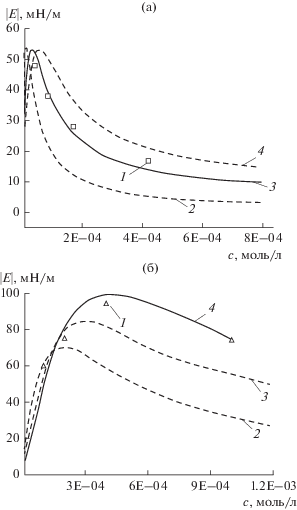
Наличие максимумов на зависимостях $\left| E \right|$ = = f(CПАВ) связано с влиянием обменных процессов как между поверхностным слоем и объемом раствора, так и в самом поверхностном слое и является следствием того, что молярная площадь полиэлектролита в поверхностном слое уменьшается с ростом адсорбции [67]. Экстремальные зависимости $\left| E \right|$ = f(CПАВ) характерны, например, для гибкоцепных белковых макромолекул, которые способны значительно изменять молярную площадь на межфазной границе [67, 77].
Согласно данным табл. 2, величина модуля вязкоупругости для фракции ГМК20–100 значительно выше, чем для других фракций ГМК и ГК и приведенных в качестве примеров других природных полиэлектролитов.
Таблица 2.
Максимальные значения реологических характеристик поверхностных слоев природных полиэлектролитов
| Полиэлектролит | $\left| {\rm E} \right|$, мН/м | Еr, мН/м | Еi, мН/м | ϕ, град |
|---|---|---|---|---|
| ГК100 | 55 | 54 | 6 | 7 |
| ГК20 | 50 | 49 | 8 | 10 |
| ГМК80–100 | 60 | 59 | 9 | 11 |
| ГМК20–100 | 117 | 115 | 18 | 9 |
| ГМК80–20 | 51 | 50 | 10 | 9 |
| ГМК20–20 | 51 | 50 | 8 | 7 |
| Альбумин [90] | 70 | 68 | 10 | 0 |
| β-Казеин [91] | 10 | 9 | 3 | 0.3 |
| Хитозан [92, 93] | 21−26* | 4−5 | 20−25 | 4−6* |
* Параметры рассчитаны нами, исходя из значений Еr и Еi [92, 93], по формулам ${{E}_{{\text{r}}}} = \left| E \right|\cos \phi ,$ ${{E}_{{\text{i}}}} = \left| E \right|\sin \phi ,$ $\left| E \right| = \sqrt {E_{{\text{i}}}^{{\text{2}}} + E_{{\text{r}}}^{{\text{2}}}} ,$ $\phi = {\text{arctg}}\left( {{{{{E}_{{\text{i}}}}} \mathord{\left/ {\vphantom {{{{E}_{{\text{i}}}}} {{{E}_{{\text{r}}}}}}} \right. \kern-0em} {{{E}_{{\text{r}}}}}}} \right).$
Высокие значения модулей вязкоупругости и упругости поверхностных пленок высокомолекулярных ПАВ (например, полиэлектролитов белковой природы) характеризуют высокую степень их упорядоченности и организованности, а также способность к сильным межмолекулярным взаимодействиям. Низкие значения фазового угла (определяющего запаздывание поверхностного натяжения относительно изменения площади капли) наблюдаются в случае образования прочных упругих поверхностных слоев на межфазной границе. При этом изменение площади капли при ее осцилляции приводит к незначительному изменению поверхностного натяжения. В этом случае замедление десорбции ПАВ из поверхностного в приповерхностный слой может быть обусловлено перераспределением макромолекул или их сегментов между приповерхностным и поверхностным слоями, т.е. приповерхностный слой слабо выражен. На это же указывают и низкие значения модуля вязкости. Максимальные значения модуля вязкоупругости, полученные экспериментально для солей ГК и ГМК, сопоставимы со значениями $\left| E \right|$ для альбумина [90] (табл. 2). Для гибкоцепного β-казеина [91] характерны низкие значения модулей вязкоупругости и упругости. Для другого класса природных полиэлектролитов – полисахаридов на основе хитина – имеет место обратная зависимость. Для поверхностных слоев хитозана на границе фаз жидкость–газ характерны высокие значения модуля вязкости (сопоставимые с величиной модуля вязкоупругости) и низкие – модуля упругости. Структурная упорядоченность таких поверхностных слоев выражена слабо [92, 93].
С учетом сказанного высокие значения модуля вязкоупругости для фракции ГМК20–100 могут быть связаны, прежде всего, с образованием тримолекулярного адсорбционного слоя на границе жидкость–газ, меньшей средней молекулярной массой этой фракции по сравнению с другими фракциями ГМК и, вероятно, более выраженными внутри- и межмолекулярными взаимодействиями в поверхностном слое. Вместе с тем, такие значения реологических параметров могут быть и следствием возможных различий в структуре макромолекул ГМК20–100 и других фракций ГМК и ГК.
Макромолекулы гуминовых соединений содержат конденсированные ароматические фрагменты и алифатические цепи разной длины и степени разветвленности. Для анализа молекулярных свойств гуминовых веществ широко используется УФ-спектроскопия. Соотношение между ароматической и алифатической частями в макромолекулах гуминовых соединений принято оценивать отношением коэффициентов поглощения при 465 и 665 нм, Е465/Е665 [61, 63, 94–97]. Низкие значения отношения Е465/Е665 указывают на превалирование конденсированных ароматических фрагментов, а его высокие значения означают преобладание алифатических фрагментов в структуре макромолекул гуминовых веществ. Как следует из данных, приведенных в табл. 3 (спектры записаны на спектрофотометре Genesys 10 SUV, Thermo Electron Corp.), для фракции ГМК20–100 отношение Е465/Е665 имеет наибольшее значение, что может указывать на более выраженную алифатическую природу их макромолекул. Кроме того, фракция ГМК20–100 содержит больше карбоксильных групп по сравнению с другими фракциями ГМК и ГК.
Таблица 3.
Характеристики фракций ГК и ГМК
| Образец | Е465/Е665 | [–СООН]*, мг-экв/г |
[–ОН]*, мг-экв/г |
|---|---|---|---|
| ГК100 | 5.92 | 2.8 | 2.4 |
| ГК20 | 6.43 | 2.4 | 2.0 |
| ГМК80–100 | 8.50 | 2.4 | 3.2 |
| ГМК20–100 | 12.13 | 3.2 | 2.8 |
| ГМК80–20 | 7.50 | 2.8 | 3.2 |
| ГМК20–20 | 8.22 | 2.8 | 3.2 |
* По данным потенциометрического титрования согласно [98].
ЗАКЛЮЧЕНИЕ
Методами кольца Дю Нуи и формы висячей капли исследовано динамическое и равновесное поверхностное натяжение водных растворов солей гиматомелановых и гуминовых кислот. Характер изотерм равновесного поверхностного натяжения растворов этих соединений соответствуют модели реального двумерного раствора, разработанной для полиэлектролитов белковой природы.
При анализе изотерм динамического поверхностного натяжения и расчетных значений коэффициентов диффузии ГМК, которые сопоставимы с имеющимися в литературе, показано, что для солей ГМК так же, как и ГК, характерен недиффузионный (барьерный) механизм адсорбции.
Методом формы осциллирующей капли исследована дилатационная реология поверхностных слоев фракций ГМК, выделенных при разной температуре экстракции из двух фракций ГК. Выявлена фракция ГМК20–100, которая характеризуется высокими значениями модулей вязкоупругости и упругости ($\left| E \right|$ = 117 мН/м, Еr = = 115 мН/м). В рамках модели полимолекулярной адсорбции полиэлектролитов зависимость $\left| E \right|$ = f(CГМК) для этой фракции ГМК подчиняется уравнению тримолекулярной адсорбции на границе фаз жидкость–газ. Фракция ГМК20–100 имеет низкую среднюю молекулярную массу и более выраженную алифатическую составляющую макромолекул.
Полученные экспериментальные данные позволяют характеризовать поверхностные слои солей ГМК и ГК на границе фаз жидкость–газ как упругие пленки, аналогичные пленкам полиэлектролитов белковой природы.
Список литературы
Кухаренко Т.А. Окисленные в пластах бурые и каменные угли. М.: Недра, 1972.
Глебова Г.И. Гиматомелановые кислоты почв и их место в системе гумусовых кислот. Автореф. дис. … канд. биол. наук. М.: МГУ, 1980.
Anastas P.T., Warner J.C. Green Chemistry: Theory and Practice. N.Y.: Oxford University Press, 1998.
Перминова И.В., Жилин Д.М. // Зеленая химия в России / Под ред. Лунина В.В., Тундо П., Локтевой Е.С. М.: Изд-во МГУ, 2004. С. 146.
Лунин В.В., Локтева Е.С., Голубина Е.В. Химия в интересах устойчивого развития – зеленая химия. М.: Изд-во МГУ, 2007.
Обзор рынка гуминовых удобрений в России и мире. М.: ООО “ИГ "Инфомайн”, 2018.
Terkhi M.C., Taleb F., Gossart P., Semmoud A., Addou A. // J. Photochem. Photobiol. A. 2008. V. 198. P. 205.
Aquino A.J.A., Tunega D., Pašalić H., Haberhauer G., Gerzabek M.H., Lischka H. // Chem. Phys. 2008. V. 349. P. 69.
Lu Y., Yan M., Korshin G.V. // Geochim. Cosmochim. Acta. 2017. V. 213. P. 308.
Baker H., Khalili F. // Anal. Chim. Acta. 2005. V. 542. P. 240.
Reiller B.P., Moulin V., Casanova F., Dautel C. // Radiochim. Acta. 2003. V. 91. P. 513.
Takahashi Y., Minai Y. // J. Nuclear Radiochem. Sci. 2004. V. 5. P. 37.
Giokas D.L., Antelo J., Paleologos E.K., Arce F., Karayannis M.I. // J. Environ. Monit. 2002. V. 4. P. 505.
Mishima S., Nakagawa T. // J. Membr. Sci. 2004. V. 228. P. 1.
Negre M., Schulten H.-R., Gennari M., Vindrola D. // J. Environ. Sci. Health. B. 2001. V. 36. P. 107.
Fukushima M., Tatsumi K. // Colloids Surf. A. 1999. V. 155. P. 249.
Struyk Z., Sposito G. // Geoderma. 2001. V. 102. P. 329.
Antilén M., González M.Á., Pérez-Ponce M., Gacitúa M., Valle M.A., Armijo F., Río R., Ramírez G. // Int. J. Electrochem. Sci. 2011. V. 6. P. 901.
Martínez C.M., Celis L.B., Cervantes F.J. // Appl. Microbiol. Biotechnol. 2013. V. 97. P. 9897.
Jiang L., Mao X., Yu J., Gan F. // Anti Corros. Meth. M. 2008. V. 55. P. 204.
Касаткина М.В., Федоров С.Е., Горохов М.В., Кураторов А.В. Пат. РФ 2221900C2, 2004.
Юдина Н.В., Писарева С.И., Пынченков В.И., Лоскутова Ю.В. // Химия растительного сырья. 1998. № 4. С. 33.
Хилько С.Л., Ефимова И.В., Смирнова О.В. // Химия твердого топлива. 2011. № 6. С. 3.
Ефимова И.В., Смирнова О.В., Хилько С.Л. // Журн. прикл. химии. 2012. Т. 85. С. 1436.
Ефимова И.В., Смирнова О.В., Хилько С.Л. // Журн. прикл. химии. 2012. Т. 85. С. 263.
Ефимова И.В., Хилько С.Л., Смирнова О.В., Бережной В.С. Рыбаченко В.И. // Химия твердого топлива. 2013. № 4. С. 3.
Федько И.В., Гостищева М.В., Исматова Р.Р. // Химия растительного сырья. 2005. № 1. С. 49.
Pant K., Singh B., Thakur N. // Int. J. Toxicol. Pharmacol. Res. 2012. V. 4. № 2. P. 17.
Akbas A., Silan C., Gulpinar M.T., Sancak E.B., Ozkanli S.S., Cakir D.U. // Inflammation. 2015. V. 38. P. 2042.
Беркович А.М. Применение гуминовых и гуминоподобных препаратов в ветеринарии и медицине // http://stomfaq.ru/53851/index.html
Хилько С.Л., Семенова Р.Г. // Химия твердого топлива. 2016. № 6. С. 66.
Aristilde L., Sposito G. // Environ. Toxicol. Chem. 2013. V. 32. P. 1467.
Sutton R., Sposito G. // Environ. Sci. Technol. 2005. V. 39. P. 9009.
Baalousha M., Motelica-Heino M., Galaup S., Le Coustumer P. // Microsc. Res. Tech. 2005. V. 66. P. 299.
Piccolo A. //Soil Sci. 2001. V. 166. P. 810.
Fedotov G.N., Shoba S.A. // Eurasian Soil Sci. 2015. V. 48. P. 1292.
Хилько С.Л., Ковтун А.И., Файнерман В.Б. // Коллоид. журн. 2011. Т. 73. С. 97.
Dmitrieva E., Efimova E., Siundiukova K., Perelomov L. // Environ. Chem. Lett. 2015. V. 13. P. 197.
Rozanova M.S., Mylnikova O.I, Klein O.I., Filippova O.I., Kholodov V.A., Listov E.L., Kulikova N.A. // Eurasian Soil Sci. 2018. V. 51. P. 1111.
Meng F., Yuan G., Wei J., Bi D., Ok Y.S., Wang H. // Chemosphere. 2017. V. 181. P. 461.
Soleimani M., Hajabbasi M.A., Afyuni M., Isfahan S.A., Jensen J.K., Holm P.E., Borggaard O.K. // J. Environ. Qual. 2010. V. 39. P. 855.
Лиштван И.И., Косаревич И.В. // Торфяная промышленность. 1984. № 1. С. 22.
Хилько С.Л., Титов Е.В. // Коллоид. журн. 1993. Т. 55. С. 117.
Хилько С.Л., Титов Е.В. // Журн. прикл. химии. 2000. Т. 73. С.1383.
Хилько С.Л., Титов Е.В. // Химия твердого топлива. 2001. № 1. С. 78.
Лотов В.А., Маслов С.Г., Чухарева Н.В. // Техника и технология силикатов. 2004. Т. 11. № 3–4. С. 26.
Gunsolus I.L., Mousavi M.P.S., Hussein K., Bühlmann P., Haynes C.L. // Environ. Sci. Technol. 2015. V. 49. P. 8078.
Tang Z., Zhao X., Zhao T., Wang H., Wang P., Wu F., Giesy J.P. // Environ. Sci. Technol. 2016. V. 50. P. 8640.
Касымова Э.Дж., Ли С.П. // Международный журн. прикладных и фундаментальных исследований. 2017. № 6-2. С. 219.
Шишмина Л.В., Чухарева Н.В., Кравцов А.В. // Кокс и химия. 2002. № 2. С. 7.
Юдина Н.В, Тихова В.И. // Химия растительного сырья. 2003. № 1. С. 93.
Попов А.Ф., Луцик А.И., Титов Е.В., Суйков С.Ю., Хилько С.Л. Пат. 5583 Украины // Б.И. 2005. № 3. С. 10.
Miller R., Makievski A.V., Fainerman V.B. // Studies in Interface Science / Ed. by Fainerman V.B., Möbius D., Miller R. Amsterdam: Elsevier, 2001. V. 13. P. 87.
Русанов А.И., Прохоров В.А. Межфазная тензиометрия. СПб.: Химия, 1994.
Loglio G., Pandolfini P., Miller R., Makievski A.V., Ravera F., Ferrari M., Liggieri L. Novel Methods to Study Interfacial Layers. Amsterdam: Elsevier, 2001.
Zholob S.A., Makievski A.V., Miller R., Fainerman V.B. // Adv. Colloid Interface Sci. 2007. V. 322. P. 134.
Ravera F., Liggieri L., Loglio G. // Progress in Colloid and Interface Science / Ed. by Miller R., Liggieri L. Boca-Raton: CRC Press, 2009. V. 1. P. 137.
Zholob S.A., Kovalchuk V.I., Makievski A.V., Kragel J., Fainerman V.B., Miller R. // Progress in Colloid and Interface Science / Ed. by Miller R., Liggieri L. Boca-Raton: CRC Press, 2009. V. 1. P. 77.
Cook R.L. Langford C.H. // Understanding Humic Substances. Advanced Methods, Properties and Applications / Ed. by Cihabbour E.A., Davies G. Sawston: Woodhead Publishing, 1999. P. 31.
Kleinhempel D. // Albrecht-Thaer- Arhiv. 1970. V. 14. № 1. P. 3.
Schnitzer M., Khan S.U. Humic Substances in the Environment. N.Y.: Marcel Dekker, 1972.
Орлов Д.С. // Соросовский образовательный журн. 1997. № 2. С. 56.
Stevenson F.J. Humus Chemistry. Genesis, Composition, Reactions. N.Y.: John Wiley & Sons Inc., 1982. P. 221.
Sein L.T., Varnum J.M., Jansen S.A. // Environ Sci. Technol. 1999. V. 33. P. 546.
Попов А.И. // Гуминовые вещества: свойства, строение, образование / Под ред. Ермакова Е.И. СПб.: Изд-во С.-Петерб. ун-та, 2004.
Orsi M. // Chemical and Biological Technologies in Agriculture. 2014. V. 1. 10.
Файнерман В.Б., Миллер Р. // Коллоид. журн. 2005. Т. 67. С. 437.
Fainerman V.B., Lucassen-Reynders E.H., Miller R. // Adv. Colloid Interface Sci. 2003. V. 106. P. 237.
Pranzas P.K., Willumeit R., Gehrke R., Thieme J., Knöchel A. // Anal. Bioanal. Chem. 2003. V. 376. P. 618.
Shang Ch., Rice J.A. // J. Colloid Interface Sci. 2007. V. 305. P. 57.
Рябова И.Н., Мустафина Г.А., Акулова 3.Г., Сатымбаева А.С. // Коллоид. журн. 2009. Т. 71. С. 716.
Парфенова Л.Н., Труфанова М.В., Селянина С.Б., Боголицын К.Г., Орлов А.С., Стригуцкий В.П. // Фундаментальные исследования. 2014. Т. 12. С. 1411.
http://www.sinterface.com
Сивакова Л.Г., Лесникова Н.П., Ким Н.М., Ротова Г.М. // Химия твердого топлива. 2011. № 1. С. 3.
Kawahigashi M., Sumida H., Yamamoto K. // J. Colloid Interface Sci. 2005. V. 284. P. 463.
Visser S. A. // Plant Soil. 1985. V. 87. P. 209.
Lucassen-Reynders E.H., Fainerman V.B., Miller R. // J. Phys. Chem. B. 2004. V. 108. P. 9173.
Ward A.F.H., Tordai L. // J. Chem. Phys. 1946. V. 14. P. 543.
Шервуд Т., Пигфорд Р., Уилки Ч. Массопередача. М.: Химия, 1982.
Cornel P.K., Summers R.S., Roberts P.V. // J. Colloid Interface Sci. 1986. V. 110. P. 149.
Lead J.R., Wilkinson K.J., Starchev K., Canonica S., Buffle J. // Environ. Sci. Technol. 2000. V. 34. P. 1365.
Otto W.H., Britten D.J., Larive C.K. // J. Colloid Interface Sci. 2003. V. 261. P. 508.
Miller R., Fainerman V.B., Aksenenko E.V., Leser M.E., Michel M. // Langmuir. 2004. V. 20. P. 771.
Хилько С.Л., Котенко А.А., Гребенюк С.А., Заречная О.М., Михайлов В.А. // Коллоид. журн. 2019. Т. 81. С. 344.
Файнерман В.Б. // Успехи химии. 1985. Т. 54. С. 1613.
Файнерман В.Б. // Журн. физ. химии. 1990. Т. 64. С. 1611.
Wustneck R., Fainerman V.B., Aksenenko E.V., Kotsmar Cs., Pradines V., Kragel J., Miller R. // Colloids Surf. A. 2012. V. 404. P. 17.
Dan A., Wustneck R., Kragel J., Aksenenko E.V., Fainerman V.B., Miller R. // Food Hydrocolloid. 2014. V. 34. P. 193.
Miller R., Aksenenko E.V., Zinkovych I.I., Fainerman V.B. // Adv. Colloid Interface Sci. 2015. V. 222. P. 509.
Pezennec S., Gauthier F., Alonso C., Graner F., Croguennec T., Brule G., Renault A. // Food Hydrocolloid. 2000. V. 14. P. 463.
Noskov B.A., Latnikova A.V., Lin S.-Y., Loglio G., Miller R. // J. Phys. Chem. C. 2007. V. 111. P. 16895.
Babak V.G., Desbrieres J. // Colloid Polym. Sci. 2006. V. 284. P. 745.
Дербиер Ж., Бабак В.Г. // Рос. хим. журн. 2008. Т. 52. № 1. С. 75.
Ширшова Л.Т., Гиличинский Д.А., Остроумова Н.В., Ермолаев А.М. // Криосфера Земли. 2017. Т. 21. № 2. С. 70.
Naidja A., Huang P.M., Anderson D.W., Kessel C.V. // Appl. Spectroscopy. 2002. V. 56. P. 318.
Chen J., Gu B., LeBoeuf E.J., Pan H., Dai S. // Chemosphere. 2002. V. 48. P. 59.
Silva R.R., Lucena G.N., Machado A.F., de Freitas G.A., Matos A.T., Abrahao W.A.P. // Comun. Sci. 2018. V. 9. P. 264.
Хилько С.Л., Ковтун А.И., Рыбаченко В.И. // Химия твердого топлива. 2011. № 5. С. 50.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал