

Коллоидный журнал, 2020, T. 82, № 1, стр. 66-74
К термодинамике тонких пленок. J-потенциал
А. И. Русанов *
Менделеевский центр, Санкт-Петербургский государственный университет
199034 Санкт-Петербург,
Университетская наб., 7, Россия
* E-mail: airusanov@mail.ru
Поступила в редакцию 17.06.2019
После доработки 28.06.2019
Принята к публикации 02.07.2019
Аннотация
Работа знакомит читателя с современным аппаратом химической термодинамики в науке о поверхностях и коллоидах. Новой величиной является J-потенциал, символизирующий целую серию термодинамических потенциалов и определяемый для флюидных систем как большой термодинамический потенциал в сумме с произведением объема системы на некое давление $p{\kern 1pt} '.$ Дана классификация с выделением классического J-потенциала (когда $p{\kern 1pt} '$ – внешнее давление) и специальных J-потенциалов ( при других значениях $p{\kern 1pt} '$). Для обоих классов рассмотрены гибридные виды потенциалов, когда для одной группы компонентов в качестве переменных выбираются химические потенциалы, а для другой – число молекул или молей. Основное внимание уделено приложению всех рассмотренных видов J-потенциала к системе при наличии в ней плоской тонкой пленки. В частности, проанализирован случай, когда в качестве $p{\kern 1pt} '$ фигурирует давление в материнской фазе тонкой пленки. Результирующие фундаментальные уравнения сформулированы в терминах расклинивающего давления.
ВВЕДЕНИЕ
Из двух составляющих термодинамики – тепла и работы – последняя несравненно сложнее и разнообразнее в описании, а потому можно сказать, что тип термодинамики определяется, прежде всего, тем, как рассчитывается работа. Термодинамика Гиббса отличается от предыдущих методов тем, что работа вычисляется с помощью термодинамических потенциалов – специальных термодинамических функций с размерностью энергии, изменение которых в заданных внешних условиях и определяет работу. Еще более крупным шагом было развитие Гиббсом, помимо тепла и работы, химической составляющей термодинамики, символизируемой химическим потенциалом. Тем самым была создана химическая термодинамика, в рамках которой мы и работаем.
Вначале термодинамические потенциалы формулировались для гомогенных (однофазных) систем. Это, например, энергия
свободная энергия большой термодинамический потенциал и энергия Гиббса где T – температура, S – энтропия, p – внешнее давление, V – объем, ${{\mu }_{i}}$ и ${{N}_{i}}$ – химический потенциал и число молекул (или молей) компонента i. Термодинамические потенциалы (1)–(4) особенно популярны в термодинамике растворов.Нужно также отметить гибридный термодинамический потенциал
(5)
$\tilde {\Omega } \equiv F - \sum\limits_i {{{\mu }_{i}}{{N}_{i}}} = - pV + \sum\limits_j {{{\mu }_{j}}{{N}_{j}}} ,$Впервые такой потенциал был введен в термодинамике твердого тела [1], где под i понимались подвижные, а под j – неподвижные компоненты твердого тела. Но в общем случае разбиение компонентов на группы i и j может быть совершенно произвольным, и именно так нужно понимать гибридный потенциал (5). Тогда можно сказать, что для компонентов группы i потенциал $\tilde {\Omega }$ играет роль большого термодинамического потенциала, а для компонентов группы j – роль свободной энергии.
Мы заметили, что при переходе к большому термодинамическому потенциалу от свободной энергии отнимается сумма с химическими потенциалами, а при переходе к энергии Гиббса – член –pV. А что, если отнять и то и другое? Для обычного (не гибридного) потенциала будет нуль, и все потеряет смысл. Да, но это так только для гомогенной системы. В коллоидной науке мы имеем дело со сложными гетерогенными системами при наличии ряда фаз, межфазных поверхностей и линий, а потому введение нового термодинамического потенциала может оказаться полезным. Эта идея была реализована автором [2, 3]. Новый термодинамический потенциал пока не имеет названия, но для его обозначения использовалась буква J (это логично, ибо, если вспомнить и частое обозначение энергии E и энтальпию H, термодинамические потенциалы, в основном, обозначаются буквами в начале латинского алфавита). Для краткости его можно называть “J-потенциал (джей-потенциал)”.
Самое общее определение J-потенциала с учетом того, что система может содержать твердые фазы под произвольным внешним нагружением, имеет вид [2, 3]
где P – внешняя сила (напряжение) на единицу площади поверхности системы как функция положения на поверхности (A), u – локальный вектор смещения поверхности и A – площадь поверхности; интегрирование производится по всей поверхности системы. Если система окружена флюидной средой и единственным механическим воздействием является внешнее давление p, то определение J-потенциала можно упростить до [3] где V – объем всей системы (ревнителям строгости напомним, что любой термодинамический потенциал определяется с точностью до постоянной). Согласно (3), для гомогенной системы определение (7) действительно приводит к нулю, но мы-то будем оперировать со сложными системами. Некая апробация J-потенциала уже состоялась в [3], где он весьма эффектно упростил вывод классических уравнений Неймана (для механического поверхностного натяжения) и Юнга (для термодинамического поверхностного натяжения).В этом сообщении мы хотели бы инкорпорировать J-потенциал в термодинамику тонких пленок, но прежде чем сделать это, обобщим определение J-потенциала (7) к виду
где $p{\kern 1pt} '$ – некоторое выбранное давление. Это может быть внешнее давление по отношению к гетерогенной системе в целом, давление в одной из фаз гетерогенной системы или любое другое давление. В варианте (8) термин J-потенциал уже обозначает целую серию потенциалов определенного типа. В классическом подходе, как при переходе к энергии Гиббса, $p{\kern 1pt} '$ полагается равным внешнему давлению $(p{\kern 1pt} ' = p).$ Для краткости мы будем называть этот тип J-потенциала классическим (обозначение ${{J}_{\operatorname{c} }}),$ а все другие типы, происходящие от выражения (8), – специальными J-потенциалами (обозначение ${{J}_{\operatorname{s} }}).$ Теперь мы можем сказать, что введенное в [3] определение (7) относится лишь к классическому J-потенциалу, а определение (8) включает в себя и специальные J-потенциалы.Термодинамика тонких пленок – относительно новый раздел химической термодинамики, выходящий за рамки гиббсовской термодинамики и сформулированный ближе к середине 20 в. [4–9] (современный обзор см. в [10, 11]). Спецификой тонкой пленки является наличие расклинивающего давления [12]
где ${{p}^{\alpha }}$ – внешнее давление на пленку, а ${{p}^{\beta }}$ – давление в равновесной материнской объемной фазе β тонкой пленки. Фаза β может находиться в равновесии с пленкой при контакте с ее торцом, но реальное присутствие фазы β не обязательно (${{p}^{\beta }}$ может быть расчетным значением давления для фазы β при тех же значениях температуры и химических потенциалов, какие есть в пленке). Открытие расклинивающего давления и его последующие исследования школой Дерягина подробно описаны в монографии [13].ОБЩИЕ СООТНОШЕНИЯ ДЛЯ J-ПОТЕНЦИАЛА
Хотя в теории пленок часто фигурируют твердые подложки, в термодинамике это не так важно. Для простоты будем оперировать с термодинамическим формализмом жидких фаз и пользоваться определением (8). Запишем сначала выражение для большого термодинамического потенциала применительно к произвольной многофазной многокомпонентной системе:
(10)
$\Omega = - \sum\limits_k {{{p}^{k}}} {{V}^{k}} + \sum\limits_s {{{\sigma }^{s}}{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}{{L}^{t}}} ,$(12)
$\begin{gathered} d\Omega = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} - pdV + \\ + \,\,\sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $(13)
$\begin{gathered} d\tilde {\Omega } = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + \sum\limits_j {{{\mu }_{j}}d} {{N}_{j}} - \\ - \,\,pdV + \sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $(15)
$J = - \sum\limits_k {({{p}^{k}} - p{\kern 1pt} '){{V}^{k}}} + \sum\limits_s {{{\sigma }^{s}}{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}{{L}^{t}}} ,$(16)
$\begin{gathered} dJ = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} - (p - p{\kern 1pt} ')dV + Vdp{\kern 1pt} '\,\, + \\ + \,\,\sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $(17)
${{J}_{\operatorname{c} }} = - \sum\limits_k {({{p}^{k}} - p){{V}^{k}}} + \sum\limits_s {{{\sigma }^{s}}{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}{{L}^{t}}} ,$(18)
$\begin{gathered} d{{J}_{\operatorname{c} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + Vdp + \\ + \,\,\sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $(20)
$\begin{gathered} d{{J}_{\operatorname{c} }} = - \bar {S}dT - \sum\limits_i {{{{\bar {N}}}_{i}}d{{\mu }_{i}}} + \sum\limits_k {{{V}^{k}}d(p - {{p}^{k}})} + \\ + \,\,\sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $По аналогии с (8) можем определить гибридный J-потенциал через гибридный большой термодинамический потенциал $\tilde {\Omega }$ как
и записать фундаментальные уравнения(22)
$\begin{gathered} \tilde {J} = \sum\limits_j {{{\mu }_{j}}} {{N}_{j}} - \sum\limits_k {({{p}^{k}} - p{\kern 1pt} '){{V}^{k}}} + \\ + \,\,\sum\limits_s {{{\sigma }^{s}}{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}{{L}^{t}}} , \\ \end{gathered} $(23)
$\begin{gathered} d\tilde {J} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + \sum\limits_j {{{\mu }_{j}}d} {{N}_{j}} - \\ - \,\,(p - p{\kern 1pt} ')dV + Vdp{\kern 1pt} '\, + \sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $(24)
$\begin{gathered} {{{\tilde {J}}}_{\operatorname{c} }} = \sum\limits_j {{{\mu }_{j}}} {{N}_{j}} - \sum\limits_k {({{p}^{k}} - p){{V}^{k}}} + \\ + \,\,\sum\limits_s {{{\sigma }^{s}}{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}{{L}^{t}}} , \\ \end{gathered} $(25)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{c} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + \sum\limits_j {{{\mu }_{j}}d{{N}_{j}}} + \\ + \,\,Vdp + \sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $КЛАССИЧЕСКИЙ J-ПОТЕНЦИАЛ СИСТЕМЫ С ТОНКОЙ ПЛЕНКОЙ
Будем считать, что тонкая пленка располагается между фазами α и γ (обозначение β сбережем для материнской фазы пленки). Как межфазную границу αγ, так и находящуюся на ней тонкую пленку будем предполагать плоскими. Посмотрим, как классический J-потенциал такого объекта выглядит в различных подходах.
Стандартный подход Гиббса
Гиббсовская термодинамика не выделяет тонкую пленку как отдельный объект и учитывает ее как любую другую часть межфазной поверхности. Для двухфазной системы α–γ, содержащей тонкую пленку (рис. 1), J-потенциал формируется на основе уравнения (17) следующим образом. Индекс k принимает значения α и γ, причем ${{p}^{\alpha }} = {{p}^{\gamma }},$ так как единственная присутствующая в системе поверхность плоская. Очевидно, в этом случае ${{p}^{\alpha }}$ играет роль внешнего давления и, значит, равно р, но тогда весь первый член в правой части (17) исчезает. Линий в нашей системе нет, а поверхность одна, и согласно (17), можем теперь записать
или (поскольку площади параллельных разделяющих поверхностей одинаковы, индекс площади можно опустить)(27)
${{j}_{\operatorname{c} }} \equiv {{{{J}_{\operatorname{c} }}} \mathord{\left/ {\vphantom {{{{J}_{\operatorname{c} }}} A}} \right. \kern-0em} A} = {{\sigma }^{{\alpha \gamma }}},$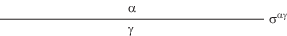
С другой стороны, согласно (20),
(28)
$d{{J}_{\operatorname{c} }} = - \bar {S}dT - \sum\limits_i {{{{\bar {N}}}_{i}}d{{\mu }_{i}}} + {{\sigma }^{{\alpha \gamma }}}dA.$(29)
$Ad{{\sigma }^{{\alpha \gamma }}} = - {{\bar {S}}^{{\alpha \gamma }}}dT - \sum\limits_i {\bar {N}_{i}^{{\alpha \gamma }}d{{\mu }_{i}}} $(30)
$d{{\sigma }^{{\alpha \gamma }}} = - {{\bar {s}}^{{\alpha \gamma }}}dT - \sum\limits_i {\Gamma _{i}^{{\alpha \gamma }}d{{\mu }_{i}}} ,$(31)
$d{{\sigma }^{{\alpha \gamma }}} = - \bar {s}_{{(j)}}^{{\alpha \gamma }}dT - \sum\limits_i {\Gamma _{{i(j)}}^{{\alpha \gamma }}d{{\mu }_{i}}} ,$Метод Гиббса с двумя разделяющими поверхностями
Чтобы сделать тонкую пленку явным объектом межфазной поверхности, необходимо введение двух разделяющих поверхностей [8–10]. Такой подход идет от представления о существовании некоей третьей объемной фазы (материнской фазы пленки β), утончением которой и образована тонкая пленка. Реальная или воображаемая фаза β должна быть обязательно в равновесии с тонкой пленкой (т.е. иметь одни и те же значения температуры и химических потенциалов), и отсюда ясно, что определение расклинивающего давления (9) носит термодинамический характер, хоть и включает только механические величины. Гиббсовские избытки берутся теперь следующим образом. Пространство между двумя параллельными разделяющими поверхностями мысленно заполняем фазой β, переходя таким путем к трехфазной системе с разделяющими поверхностями αβ и βγ (рис. 2). Избытки для разделяющей поверхности αβ берутся со стороны фаз α и β, а для поверхности βγ – со стороны фаз β и γ. Существенно, что в случае тонкой пленки избытки (например, адсорбции) по обеим сторонам фазы β не являются независимыми и должны рассматриваться совместно. Это, в частности, касается и поверхностных натяжений ${{\sigma }^{\alpha }}$ (между фазами α и β) и ${{\sigma }^{\gamma }}$ (между фазами γ и β), показанных на рис. 2.
Рис. 2.
Расположение фаз и поверхностные натяжения в методе двух разделяющих поверхностей, отстоящих на расстоянии H друг от друга.
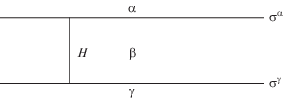
Теперь, в соответствии с (17), составим выражение для J-потенциала. Очевидно, не имеет значения, какую из фаз, α или γ, считать внешней, ибо обе они имеют одно и то же давление ${{p}^{\alpha }},$ которое и является внешним давлением p. Тогда объемные члены для фаз α и γ из выражения (17) исчезают, и остается только один член, относящийся к фазе β. Поверхностных же членов будет, наоборот, два, ибо теперь у нас две разделяющих поверхности. В результате из выражения (17) имеем
(32)
${{J}_{{\text{c}}}} = - ({{p}^{\beta }} - {{p}^{\alpha }}){{V}^{\beta }} + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})A$Сравнивая (26) и (34), приходим к соотношению
известному в термодинамике тонких пленок со времен работ [4–8] и полученному там другим путем. При $H \to \infty $ расклинивающее давление быстрее стремится к нулю, обращая в нуль и произведение $\Pi H,$ так что правая часть (34) и (35) переходит в сумму уже независимых поверхностных натяжений ${{\sigma }^{{\alpha \beta }}}$ и ${{\sigma }^{{\beta \gamma }}},$ а тонкая пленка превращается в толстую. Этот важный результат запишем как(36)
${{\left. {{{j}_{\operatorname{c} }}} \right|}_{{H \to \infty }}} = {{\left. {{{\sigma }^{{\alpha \gamma }}}} \right|}_{{H \to \infty }}} = {{\sigma }^{{\alpha \beta }}} + {{\sigma }^{{\beta \gamma }}}.$(37)
$\begin{gathered} d{{J}_{\operatorname{c} }} = - \bar {S}dT - \sum\limits_i {{{{\bar {N}}}_{i}}d{{\mu }_{i}}} + \\ + \,\,{{V}^{\beta }}d({{p}^{\alpha }} - {{p}^{\beta }}) + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA \\ \end{gathered} $(38)
$d{{J}_{\operatorname{c} }} = - \bar {S}dT - \sum\limits_i {{{{\bar {N}}}_{i}}d{{\mu }_{i}}} + {{V}^{\beta }}d\Pi + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA.$(39)
$Ad({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }}) = - \bar {S}dT - \sum\limits_i {{{{\bar {N}}}_{i}}d{{\mu }_{i}}} - \Pi d{{V}^{\beta }},$(40)
$d({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }}) = - \bar {s}dT - \sum\limits_i {{{\Gamma }_{i}}d{{\mu }_{i}}} - \Pi dH$(41)
$d{{\sigma }^{{\alpha \gamma }}} = - \bar {s}dT - \sum\limits_i {{{\Gamma }_{i}}d{{\mu }_{i}}} + Hd\Pi .$После стандартной операции перехода от абсолютных к относительным адсорбциям уравнения (40) и (41) принимают вид [10, 11]
(42)
$d({{\sigma }^{\beta }} + {{\sigma }^{\gamma }}) = - {{\bar {s}}_{{(j)}}}dT - \Pi d{{H}_{{(j)}}} - \sum\limits_{i \ne j} {{{\Gamma }_{{i(j)}}}d{{\mu }_{i}}} ,$(43)
$d{{\sigma }^{{\alpha \gamma }}} = - {{\bar {s}}_{{(j)}}}dT - \sum\limits_i {{{\Gamma }_{{i(j)}}}d{{\mu }_{i}}} + {{H}_{{(j)}}}d\Pi .$ГИБРИДНЫЙ КЛАССИЧЕСКИЙ J-ПОТЕНЦИАЛ
Рассмотрим теперь использование классического J-потенциала в гибридной форме. Общая запись, которую мы использовали выше, не исключает возможность проникновения всех компонентов системы во все фазы, и это самый сложный случай для идентификации тонкой пленки. Но по существу пленки в этом случае и нет, есть лишь поверхностный слой, т.е. неоднородная переходная межфазная зона. Понятие пленки подразумевает какую-то обособленность вещества. Простейший случай – фазы α и γ ни в чем не растворимы и сами не способны что-либо растворять, т.е. полностью инертны в отношении остального вещества системы. Такие подложки неспособны к проникновению и имеют четко выраженные граничные поверхности, расстояние между которыми задает толщину тонкой пленки. Такие подложки формально можно считать однокомпонентными. Пусть компонент фазы α имеет номер m, а компонент фазы γ – номер n. Как и в предыдущем разделе, посмотрим, как выглядят фундаментальные уравнения для гибридного классического потенциала при использовании одной и двух разделяющих поверхностей.
Одна разделяющая поверхность
В этом случае из (24) и (25) имеем
(44)
${{\tilde {J}}_{\operatorname{c} }} = {{\mu }_{m}}N_{m}^{\alpha } + {{\mu }_{n}}N_{n}^{\gamma } + {{\sigma }^{{\alpha \gamma }}}A,$(45)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{c} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + {{\mu }_{m}}dN_{m}^{\alpha } + \\ + \,\,{{\mu }_{n}}dN_{n}^{\gamma } + Vd{{p}^{\alpha }} + {{\sigma }^{{\alpha \gamma }}}dA, \\ \end{gathered} $(46)
$\begin{gathered} Ad{{\sigma }^{{\alpha \gamma }}} = - SdT + Vd{{p}^{\alpha }} - N_{m}^{\alpha }d{{\mu }_{m}} - \\ - \,\,N_{n}^{\gamma }d{{\mu }_{n}} - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} , \\ \end{gathered} $Для фаз α и γ действуют уравнения Гиббса–Дюгема
(47)
$\begin{gathered} {{V}^{\alpha }}d{{p}^{\alpha }} = {{S}^{\alpha }}dT + N_{m}^{\alpha }d{{\mu }_{m}}, \\ {{V}^{\gamma }}d{{p}^{\gamma }} = {{S}^{\gamma }}dT + N_{n}^{\gamma }d{{\mu }_{n}}, \\ \end{gathered} $(48)
$Ad{{\sigma }^{{\alpha \gamma }}} = - \bar {S}dT - \bar {N}_{m}^{\alpha }d{{\mu }_{m}} - \bar {N}_{n}^{\gamma }d{{\mu }_{n}} - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} ,$Две разделяющие поверхности
Для этого случая (см. рис. 2) из тех же самых уравнений (24) и (25) получаем
(49)
${{\tilde {J}}_{\operatorname{c} }} = {{\mu }_{m}}N_{m}^{\alpha } + {{\mu }_{n}}N_{n}^{\gamma } + \Pi {{V}^{\beta }} + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})A,$(50)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{c} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + {{\mu }_{m}}dN_{m}^{\alpha } + \\ + \,\,{{\mu }_{n}}dN_{n}^{\gamma } + Vd{{p}^{\alpha }} + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA. \\ \end{gathered} $(51)
$Vd{{p}^{\alpha }} = {{V}^{\alpha }}d{{p}^{\alpha }} + {{V}^{\gamma }}d{{p}^{\gamma }} + {{V}^{\beta }}d{{p}^{\beta }} + {{V}^{\beta }}d\Pi ,$(52)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{c} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + {{\mu }_{m}}dN_{m}^{\alpha } + {{\mu }_{n}}dN_{n}^{\gamma } + \\ + \,\,{{V}^{\alpha }}d{{p}^{\alpha }} + {{V}^{\gamma }}d{{p}^{\gamma }} + {{V}^{\beta }}d{{p}^{\beta }} + \\ + \,\,{{V}^{\beta }}d\Pi + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA. \\ \end{gathered} $(53)
$\begin{gathered} Ad({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }}) = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} - \\ - \,\,N_{m}^{\alpha }d{{\mu }_{m}} - N_{n}^{\gamma }d{{\mu }_{n}} - \Pi d{{V}^{\beta }}, \\ \end{gathered} $(54)
${{V}^{\beta }}d{{p}^{\beta }} = {{S}^{\beta }}dT + \sum\limits_i {N_{i}^{\beta }d{{\mu }_{i}}} .$(55)
$\begin{gathered} Ad({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }}) = - \bar {S}dT - \sum\limits_i {{{{\bar {N}}}_{i}}d{{\mu }_{i}}} - \\ - \,\,\bar {N}_{m}^{\alpha }d{{\mu }_{m}} - \bar {N}_{n}^{\gamma }d{{\mu }_{n}} - \Pi d{{V}^{\beta }}. \\ \end{gathered} $СПЕЦИАЛЬНЫЙ J-ПОТЕНЦИАЛ ТОНКОЙ ПЛЕНКИ
Вернемся к фундаментальным уравнениям (15) и (16) и положим $p{\kern 1pt} ' = {{p}^{\beta }}.$ Поскольку ${{p}^{\beta }}$ – давление в материнской фазе тонкой пленки, специфика данного выбора состоит в том, что он может относиться только к случаю существования тонкой пленки. Адресуясь к рис. 1 и 2, по-прежнему будем считать ${{p}^{\alpha }}$ внешним давлением (p = = ${{p}^{\alpha }} = {{p}^{\gamma }}).$ Соответственно перепишем (15) и (16) как
(56)
${{J}_{\operatorname{s} }} = - \sum\limits_k {({{p}^{k}} - {{p}^{\beta }}){{V}^{k}}} + \sum\limits_s {{{\sigma }^{s}}{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}{{L}^{t}}} ,$(57)
$\begin{gathered} d{{J}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} - ({{p}^{\alpha }} - {{p}^{\beta }})dV + \\ + \,\,Vd{{p}^{\beta }} + \sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $Одна разделяющая поверхность
На рис. 1 видно, что в данном случае имеются только две фазы (α и γ), и использование давления третьей фазы может показаться странным. Но мы уже отмечали, что фаза β может быть и воображаемой. Посмотрим все-таки, что получается. С учетом определения (9) и отсутствия линий, уравнения (56) и (57) принимают вид
(59)
$d{{J}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} - \Pi dV + Vd{{p}^{\beta }} + {{\sigma }^{{\alpha \gamma }}}dA$Две разделяющие поверхности
Теперь у нас две поверхности и три объемные фазы (рис. 2), в соответствии с чем уравнения (56) и (57) записываются в форме
(61)
${{J}_{\operatorname{s} }} = - \Pi ({{V}^{\alpha }} + {{V}^{\gamma }}) + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})A,$(62)
$\begin{gathered} d{{J}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} - \Pi dV + \\ + \,\,Vd{{p}^{\beta }} + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA, \\ \end{gathered} $(63)
$\begin{gathered} d{{J}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} - \Pi d{{V}^{\beta }} - \\ - \,\,d[\Pi ({{V}^{\alpha }} + {{V}^{\gamma }})] + {{V}^{\alpha }}d{{p}^{\alpha }} + {{V}^{\gamma }}d{{p}^{\gamma }} + \\ + \,\,{{V}^{\beta }}d{{p}^{\beta }} + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA. \\ \end{gathered} $ГИБРИДНЫЙ СПЕЦИАЛЬНЫЙ J-ПОТЕНЦИАЛ ТОНКОЙ ПЛЕНКИ
Вернемся к общим фундаментальным уравнениям для гибридного J-потенциала (22) и (23) и применим их к гибридному специальному J-потенциалу при $p = {{p}^{\alpha }} = {{p}^{\gamma }}$ и $p{\kern 1pt} ' = {{p}^{\beta }}{\text{:}}$
(65)
$\begin{gathered} {{{\tilde {J}}}_{\operatorname{s} }} = \sum\limits_j {{{\mu }_{j}}} {{N}_{j}} - \sum\limits_k {({{p}^{k}} - {{p}^{\beta }}){{V}^{k}}} + \\ + \,\,\sum\limits_s {{{\sigma }^{s}}{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}{{L}^{t}}} , \\ \end{gathered} $(66)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + \sum\limits_j {{{\mu }_{j}}d} {{N}_{j}} - \\ - \,\,\Pi dV + Vd{{p}^{\beta }} + \sum\limits_s {{{\sigma }^{s}}d{{A}^{s}}} + \sum\limits_t {{{\kappa }^{t}}d{{L}^{t}}} . \\ \end{gathered} $(67)
$\sum\limits_j {{{\mu }_{j}}{{N}_{j}}} = {{\mu }_{m}}N_{m}^{\alpha } + {{\mu }_{n}}N_{n}^{\gamma },$Одна разделяющая поверхность
Учитывая определение (9), условие (67) и наличие только двух фаз ($V = {{V}^{\alpha }} + {{V}^{\gamma }}),$ имеем
(68)
${{\tilde {J}}_{\operatorname{s} }} = {{\mu }_{m}}N_{m}^{\alpha } + {{\mu }_{n}}N_{n}^{\gamma } - \Pi V + {{\sigma }^{{\alpha \gamma }}}A,$(69)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + {{\mu }_{m}}dN_{m}^{\alpha } + {{\mu }_{n}}dN_{n}^{\gamma } - \\ - \,\,\Pi dV + Vd{{p}^{\beta }} + {{\sigma }^{{\alpha \gamma }}}dA. \\ \end{gathered} $(70)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + {{\mu }_{m}}dN_{m}^{\alpha } + {{\mu }_{n}}dN_{n}^{\gamma } - \\ - \,\,d(\Pi V) + Vd{{p}^{\alpha }} + {{\sigma }^{{\alpha \gamma }}}dA. \\ \end{gathered} $Две разделяющие поверхности
В этом случае ($V = {{V}^{\alpha }} + {{V}^{\gamma }} + {{V}^{\beta }}$) уравнения (65) и (66) принимают вид
(71)
${{\tilde {J}}_{\operatorname{s} }} = {{\mu }_{m}}N_{m}^{\alpha } + {{\mu }_{n}}N_{n}^{\gamma } - \Pi ({{V}^{\alpha }} + {{V}^{\gamma }}) + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})A,$(72)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + {{\mu }_{m}}dN_{m}^{\alpha } + {{\mu }_{n}}dN_{n}^{\gamma } - \\ - \,\,\Pi dV + Vd{{p}^{\beta }} + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA. \\ \end{gathered} $Подобно переходу от (62) к (63), уравнение (72) можно записать как
(73)
$\begin{gathered} d{{{\tilde {J}}}_{\operatorname{s} }} = - SdT - \sum\limits_i {{{N}_{i}}d{{\mu }_{i}}} + {{\mu }_{m}}dN_{m}^{\alpha } + {{\mu }_{n}}dN_{n}^{\gamma } - \\ - \,\,\Pi d{{V}^{\beta }} - d[\Pi ({{V}^{\alpha }} + {{V}^{\gamma }})] + {{V}^{\alpha }}d{{p}^{\alpha }} + {{V}^{\gamma }}d{{p}^{\gamma }} + \\ + \,\,{{V}^{\beta }}d{{p}^{\beta }} + ({{\sigma }^{\alpha }} + {{\sigma }^{\gamma }})dA. \\ \end{gathered} $ЗАКЛЮЧЕНИЕ
Данная работа знакомит читателя с современным аппаратом химической термодинамики, куда теперь вошел J-потенциал, символизирующий целую серию термодинамических потенциалов. Известно, что физический результат не должен зависеть от выбора термодинамического потенциала. В данной работе мы это многократно продемонстрировали на примере уравнения адсорбции Гиббса, но в нашем случае это имело значение еще и со стороны проверки правильности конструирования нового термодинамического потенциала. Ценность последнего определяется удобством его использования и внешними условиями, в которых оно проявляется. J-потенциал удобен для коллоидной науки, ибо, не будучи сам избыточной величиной, он уже включает в себя гиббсовские избытки термодинамических параметров по отношению к поверхностям и линиям.
Естественно, представленная совокупность J-потенциалов не является исчерпывающей. Мы сосредоточились на гетерогенных системах, содержащих тонкие пленки, но ограничились случаем плоских пленок. Но и в этих рамках можно спросить, почему не рассматривался вариант построения J-потенциала при величине ${{p}^{\beta }}$ в качестве внешнего давления. Ведь реально существуют системы, где внешней фазой является материнская фаза тонкой пленки (например, вокруг прилипшего к стенке пузырька со смачивающей тонкой пленкой [14]). Ответ следующий. Поскольку при определении J-потенциала мы стартуем от большого термодинамического потенциала, пространственная область (от нее зависит и объем) системы может быть очерчена из соображений удобства. Например, границы системы могут быть выбраны так, чтобы всегда внешней фазой была фаза α (или γ), как показано на рис. 1 и 2. Тогда ${{p}^{\alpha }}$ будет неизменно внешним давлением.
Список литературы
Rusanov A.I. // J. Colloid Interface Sci. 1978. V. 63. P. 330.
Rusanov A.I. // J. Chem. Phys. 2013. V. 138. P. 246 101.
Rusanov A.I. // Colloids Surf. A. 2014. V. 443. P. 363.
Русанов А.И. // Коллоид. журн. 1966. Т. 28. С. 718.
Русанов А.И. // Коллоид. журн. 1967. Т. 29. С. 142.
Русанов А.И. // Коллоид. журн. 1967. Т. 29. С. 149.
Русанов А.И. // Коллоид. журн. 1967. Т. 29. С. 237.
Русанов А.И. Фазовые равновесия и поверхностные явления. Л.: Химия, 1967.
Rusanov A.I. Phasengleichgewichte und Grenzflächenerscheinungen. Berlin: Akademie-Verlag, 1978.
Русанов А.И. // Коллоид. журн. 2007. Т. 69. С. 43.
Русанов А.И. // Коллоид. журн. 2019. Т. 81. С. 767.
Дерягин Б.В. // Коллоид. журн. 1955. Т. 17. С. 207.
Дерягин Б.В., Чураев Н.В., Муллер В.М. Поверхностные силы. М.: Наука, 1985.
Есипова Н.Е., Русанов А. И., Соболев В.Д., Ицков С.В. // Коллоид. журн. 2019. Т. 81. С. 563.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал