

Коллоидный журнал, 2020, T. 82, № 2, стр. 155-162
Электростатическая стабилизация гидрозолей наночастиц карбоната кальция, синтезированных матричным методом
Д. А. Еуров 1, *, А. В. Швидченко 1, Д. А. Курдюков 1
1 Физико-технический институт им. А.Ф. Иоффе
194021 Санкт-Петербург,
Политехническая ул., 26, Россия
* E-mail: edan@mail.ru
Поступила в редакцию 06.06.2019
После доработки 12.08.2019
Принята к публикации 21.08.2019
Аннотация
Разработан метод синтеза наноразмерных частиц карбоната кальция с использованием мезопористых частиц кремнезема в качестве матрицы. Гидрозоли синтезированных частиц CaCO3 агрегативно неустойчивы при нейтральных значениях pH и коагулируют с образованием агрегатов субмикронного размера. Для формирования агрегативно устойчивых гидрозолей предложена методика электростатической стабилизации посредством добавления водного раствора FeCl3. Показано, что эта методика стабилизации приводит к пептизации агрегатов и позволяет получить индивидуальные наночастицы карбоната кальция размером 50−80 нм. Полученные наночастицы CaCO3 являются пористыми, что обуславливает перспективы медицинского применения их стабильных гидрозолей.
ВВЕДЕНИЕ
Карбонат кальция находит широкое применение в различных областях науки и техники – от производства пластмасс, красок и бумаги [1], до фармацевтической промышленности [2]. Интерес к использованию CaCO3 в медицине обусловлен его низкой стоимостью, биосовместимостью, биорастворимостью и остеокондуктивными свойствами. Различные соединения кальция являются основными составляющими костной ткани и зубов [3], что делает перспективным их использование для лечения костных поражений [4], кариеса [5], а также несовершенного остеогенеза [6]. Кроме того, кальций требуется для нормального роста и развития скелета. Алиментарный кальций необходим для здоровья костей в процессе роста человека, а также в старости [7]. При использовании в качестве пищевой добавки коммерчески доступных микрочастиц карбоната кальция происходит их растворение желудочной кислотой прежде, чем они попадают в кровь в виде ионов Ca2+ [8]. Эффективность поглощения кальция при этом крайне мала (около 4%) и зависит от pH [9]. Для увеличения биодоступности кальция при пероральном введении предпринимаются попытки создания наноразмерного CaCO3 [10, 11].
Благодаря медленной биодеградации и зависимости растворимости от pH наноразмерные частицы карбоната кальция перспективны для использования в системах доставки лекарств, в частности, в раковые опухоли [12]. Медленная деградация CaCO3 позволяет дольше удерживать лекарства после внутривенного введения в организм [13], а увеличение растворимости карбоната кальция с уменьшением рН ускоряет выход препарата именно в местах новообразований [12], поскольку в опухоли, как правило, более кислая среда по сравнению со здоровыми тканями.
Одной из проблем получения частиц карбоната кальция, в особенности наноразмерных, является низкая агрегативная устойчивость дисперсий таких частиц в воде [14]. Для того чтобы избежать коагуляции и получить индивидуальные частицы CaCO3, требуемые для медицинских применений, необходимо их стабилизировать. Как правило, для предотвращения коагуляции наночастиц используют полимерную (стерическую) стабилизацию [15], связывая органические молекулы с функциональными группами на поверхности частиц. Часто для стабилизации используют не одно органическое вещество, а несколько, что увеличивает количество технологических операций и усложняет процесс получения стабильных водных суспензий наночастиц.
В данной работе матричным методом синтезированы пористые наночастицы карбоната кальция. В качестве матрицы использованы сферические мезопористые частицы кремнезема. Показано, что после удаления материала матрицы синтезированные наночастицы CaCO3 образуют агрегаты субмикронного размера. Для пептизации образующихся агрегатов проведена электростатическая стабилизация коллоидной системы с использованием хлорида железа(III). Продемонстрировано, что используемая методика стабилизации позволяет получить индивидуальные наноразмерные частицы карбоната кальция, агрегативно устойчивые в водных средах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Сферические мезопористые частицы кремнезема (mSiO2) диаметром 190 ± 20 нм, использованные в качестве матрицы, синтезировали путем гидролиза тетраэтоксисилана (ТЭОС) в спирто-водно-аммиачной среде с добавлением поверхностно-активного структурообразующего агента (темплата). Подробно методика синтеза частиц кремнезема описана в [16, 17]. По данным адсорбционной порометрии в частицах имеются цилиндрические мезопоры диаметром 3.10 ± 0.15 нм, объем пор составляет около 50% от объема частиц (0.6 см3/г).
Для синтеза CaCO3 в порах частиц mSiO2 на первом этапе использовали капиллярную пропитку частиц водным раствором хлорида кальция. Для этого навеску частиц помещали в 2 М раствор CaCl2 (объем раствора рассчитывали исходя из массы навески mSiO2) и оставляли пропитываться в течение суток. После пропитки частицы высушивали при 60°C, полученный порошок заливали 4 М водным раствором (NH4)2CO3 и вновь выдерживали в течение суток. Карбонат аммония брали в избытке для обеспечения полноты превращения CaCl2 в CaCO3. После этого частицы многократно центрифугировали для очистки от непрореагировавших солей и высушивали на воздухе. Описанную процедуру повторяли трижды. В результате формировались частицы кремнезема, содержащие в порах карбонат кальция (mSiO2/CaCO3).
Для получения наночастиц карбоната кальция материал матрицы – SiO2 – вытравливали в водном растворе гидроксида калия. Для этого навеску частиц mSiO2/CaCO3 заливали 4 М раствором КОН. Травление проводили в течение 2 ч при температуре 85°C. После этого частицы центрифугировали, надосадочную жидкость сливали, а полученный осадок многократно промывали водой. Отмытые частицы CaCO3 редиспергировали в деионизованной воде под действием ультразвука.
Для стабилизации водной дисперсии наночастиц карбоната кальция использовали водный раствор хлорида железа(III). С этой целью к ней добавляли 0.1 М раствор FeCl3 и выдерживали в течение суток. Далее полученные частицы многократно центрифугировали и промывали деионизованной водой.
Гидродинамический диаметр и электрофоретическую подвижность наночастиц карбоната кальция определяли методом динамического рассеяния света (ДРС) на анализаторе Malvern Zetasizer Nano ZS при температуре 25°С. Среднеквадратичное отклонение диаметров частиц принималось равным W1/2/2.36, где W1/2 – значение ширины кривой распределения частиц по размерам на полувысоте, полученное с помощью встроенного программного обеспечения прибора. Относительная ошибка измерений составляла менее 2%. Расчет электрокинетического потенциала также осуществлялся с помощью программного обеспечения анализатора.
Величину pH исследуемых водных суспензий частиц измеряли с помощью иономера И-160 (Гомельский завод измерительных приборов, Белоруссия) с H+-чувствительным комбинированным стеклянным электродом ЭСЛ-43-07.
Для исследования методом ИК-спектроскопии частицы осаждали из водной суспензии на поверхность кремниевой подложки с последующей сушкой при комнатной температуре в течение дня. Измерения проводили в режиме многократного нарушенного полного внутреннего отражения на ИК-фурье-спектрометре Инфралюм ФТ-08 (Люмэкс, Россия).
Фазовый состав нанокомпозитов определяли методом рентгеновской дифракции на установке Bruker D2 Phaser (CuKα-излучение).
Спектры поглощения золей измеряли в кварцевых кюветах с длиной оптического пути 10 мм на спектрофотометре Unico 2800 (Unico System, США) с использованием коллимированного излучения в спектральном диапазоне 190−1100 нм.
Исследования методом адсорбционной порометрии проведены на анализаторе ASAP 2020 (Micromeritics Instruments) при температуре 77 K с применением азота в качестве адсорбата. Удельную поверхность материала рассчитывали в интервале давлений 0.05 ≤ P/P0 ≤ 0.15 по методу Брунауэра–Эммета–Теллера (БЭТ), объем пор вычисляли из изотермы адсорбции при относительном давлении P/P0 → 1. Распределение пор по размерам для синтезированных частиц рассчитывали методом Баррета–Джойнера–Халенды (БДХ) с помощью программного обеспечения прибора.
Микроскопические исследования проводили с помощью атомно-силового микроскопа (АСМ) NT-MDT Smena в полуконтактном режиме и просвечивающего электронного микроскопа (ПЭМ) Jeol JEM-2100F, оборудованного энергодисперсионным рентгеновским (EDX) спектрометром Oxford Instruments INCA. Образцы для АСМ готовили так же, как и для ИК-спектроскопии. Размер частиц CaCO3 определяли, анализируя изображения 80 частиц; затем строили гистограммы распределения их размеров с последующей аппроксимацией функцией Гаусса. Для приготовления образца для ПЭМ-исследования частицы наносили на поддерживающую пленку из ультратонкой графеновой бумаги.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 1 представлены гистограммы распределения частиц по гидродинамическим диаметрам, определенные методом ДРС. Видно, что средний размер синтезированных частиц CaCO3 больше размера исходных частиц mSiO2 (190 нм) и составляет 265 нм. По-видимому, регистрируемые объекты (рис. 1б) являются агрегатами частиц CaCO3. В свою очередь, размер наночастиц, образующих агрегаты, тоже отличается от размера mSiO2, средний размер стабилизированных частиц CaCO3 составляет 75 ± 15 нм (рис. 1в). По-видимому, разработанная методика заполнения пор с использованием водных растворов солей в качестве прекурсоров не позволяет получить взаимосвязанную реплику пор матрицы, состоящую из CaCO3, как при заполнении пор mSiO2 аморфным Si c использованием газофазного прекурсора SiH4 [18].
Рис. 1.
Определенные методом ДРС распределения по размерам сферических частиц мезопористого SiO2 (а) и наночастиц CaCO3 после синтеза (б) и после стабилизации (в).
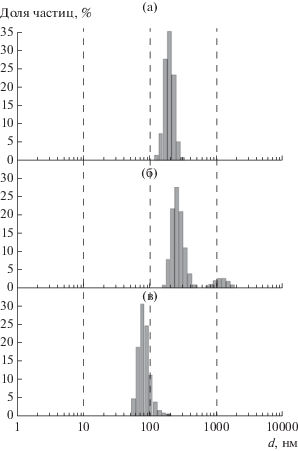
Частицы mSiO2 обладают разветвленной системой пор [16, 19], что обеспечивает эффективный массоперенос реагентов и продуктов. На первом этапе происходит полное заполнение пор mSiO2 раствором хлорида кальция под действием капиллярных сил. После удаления растворителя твердый CaCl2, возможно, закупоривает некоторые поры, но заполняет объем пор mSiO2 не полностью (примерно 15 об. %, судя по изменению массы навески), поэтому водный раствор (NH4)2CO3 может проникнуть внутрь, двигаясь по открытым взаимосвязанным порам.
Реакция CaCl2 с раствором (NH4)2CO3 с образованием CaCO3 идет в порах матрицы с образованием “наполнителя”, имеющего меньший мольный объем (Vm(CaCl2) ≈ 51 см3/моль, Vm(CaCO3) ≈ 37 см3/моль). Вследствие уменьшения Vm наполнителя увеличиваются количество и объем незаполненных областей внутри композитных частиц mSiO2/CaCO3 по сравнению с mSiO2/CaCl2. Таким образом, формирующийся карбонат кальция не образует сплошного взаимосвязанного каркаса по всему объему частиц mSiO2. Поэтому при травлении материала матрицы раствором KOH частицы распадаются на фрагменты, размер которых меньше размера исходных частиц mSiO2. Было установлено, что максимальная степень заполнения пор частиц кремнезема CaCO3, равная примерно 45 об. % (определена гравиметрически), достигается при трехкратной поочередной пропитке частиц mSiO2 растворами прекурсоров.
Результаты, полученные с использованием АСМ (рис. 2а, 2б), коррелируют с данными ДРС. Видно (рис. 2а), что синтезированные частицы карбоната кальция при нанесении на кремниевую подложку собираются в крупные агрегаты, состоящие из более мелких структурных элементов. На рис. 2б представлено типичное АСМ-изображение наночастиц CaCO3 после стабилизации. Отчетливо видны индивидуальные частицы, средний размер которых по данным АСМ составляет 60 ± 10 нм. Отметим, что полученное значение соответствует результатам ДРС, учитывая, что данным методом анализируется гидродинамический диаметр частиц, равный удвоенному расстоянию от центра частицы до плоскости скольжения, т.е. включающий в себя часть диффузного слоя ДЭС.

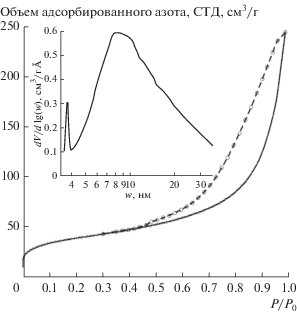
Изотермы адсорбции–десорбции азота стабилизированными частицами CaCO3 приведены на рис. 3. Значения удельной поверхности, рассчитанной методом БЭТ, и объема пор наночастиц CaCO3 при P/P0 = 0.994 составили 134 м2/г и 0.38 см3/г соответственно. Наблюдается широкая петля адсорбционного гистерезиса в области относительных давлений P/P0 = 0.5–1, свидетельствующая о наличии крупных мезопор. Расчет методом БДХ выявил широкое распределение пор по размерам (рис. 3, вставка) в диапазоне 3–35 нм с максимумом вблизи 9 нм. Мезопоры размером в несколько нанометров, вероятно, расположены внутри частиц CaCO3, тогда как более крупные мезопоры, по-видимому, находятся между частицами. Поскольку размеры пор между частицами обычно составляют 20–40% от их диаметра [20], данные адсорбционной порометрии коррелируют с результатами ДРС и АСМ.
Рис. 3.
Изотермы адсорбции–десорбции азота при 77 K наночастицами CaCO3 после стабилизации и распределение пор по размерам, рассчитанное методом БДХ.
Для определения состава и структуры полученных наночастиц проведен анализ ИК-спектров, измеренных до и после стабилизации (рис. 4). Карбонат кальция имеет три полиморфные модификации: кальцит, арагонит и ватерит [21], различающиеся пространственным расположением ионов ${\text{CO}}_{3}^{{2--}}.$ Ватерит редко встречается в природе и является наименее стабильной фазой карбоната кальция, которая обычно переходит в наиболее стабильную фазу кальцита [21, 22]. Как правило [23], в ИК-спектрах кальцита и арагонита в области 600–500 см–1 проявляются четыре основные моды, соответствующие симметричным валентным колебаниям (ν1), внеплоскостным деформационным колебаниям (δ1), дважды вырожденным асимметричным валентным колебаниям (ν2) и дважды вырожденным плоскостным деформационным колебаниям (δ2).
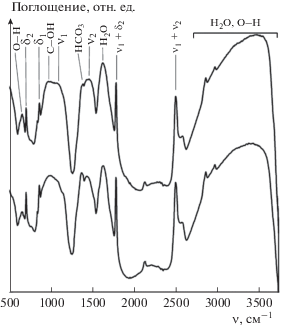
Видно, что ИК-спектр частиц после их стабилизации (рис. 4, кривая 2) практически совпадает со спектром синтезированных частиц (рис. 4, кривая 1). В обоих спектрах наблюдаются полосы при 713, 869, 1096 и 1460 см–1, типичные для ионов ${\text{CO}}_{3}^{{2--}}$ в CaCO3 и относящиеся к четырем основным модам – δ2, δ1, ν1 и ν2 соответственно [23]. Полоса при 1096 см–1 характерна для арагонита и не проявляется в спектрах чистого кальцита. Слабая интенсивность данной полосы может свидетельствовать о том, что доля фазы арагонита в полученных наночастицах незначительна и основной их составляющей является кальцит. Бикарбонат ${\text{HCO}}_{3}^{--}$ проявляется в спектрах в виде полосы при 1380 см–1, соответствующей симметричным валентным колебаниям CO2, и полосы при 1005 см–1, обусловленной валентными колебаниями C–OH [24]. Полосы на частотах 1795 и 2513 см–1 проявляются в результате совместных колебаний, соответствующих основным модам для молекулярных ионов ${\text{CO}}_{3}^{{2--}}.$ Так, полоса 1795 см–1 соответствует комбинации (ν1 + δ2), а полоса 2513 см–1 – комбинации (ν1 + ν2) [23]. В спектрах также присутствуют полосы, соответствующие колебаниям OH-групп в молекулах физически адсорбированной воды. Это полосы при 667 и 1635 см–1 (деформационные колебания H–O–H) и широкая полоса в области 2750–3600 см–1 (валентные колебания) [25]. Видно, что спектр частиц CaCO3 после стабилизации практически не изменяется, что свидетельствует о том, что используемая методика не влияет на качественный состав поверхностных функциональных групп. В результате сохраняется возможность дальнейшей функционализации поверхности стабилизированных частиц карбоната кальция.
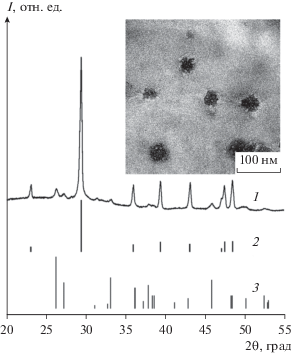
Наличие карбоната кальция в составе стабилизированных частиц подтверждено также методом порошковой рентгеновской дифракции. На дифракционной кривой наночастиц наблюдается набор рефлексов (рис. 5, кривая 1), соответствующих кальциту (рис. 5, кривая 2) и арагониту (рис. 5, кривая 3), примесных кристаллических фаз не обнаружено. На ПЭМ-изображениях (рис. 5, вставка) видны индивидуальные частицы CaCO3 размером 50−60 нм, внутри которых наблюдается пористая структура (области светлого контраста размером менее 10 нм). Элементный EDX-микроанализ показал наличие в составе наночастиц атомов Ca и O, железо не детектируется ввиду его малого содержания.
Рис. 5.
Дифрактограммы: 1 – наночастицы CaCO3 после стабилизации (на вставке приведено их ПЭМ-изображение), 2 – β-CaCO3, кальцит (JCPDS 01-071-3699), 3 – λ-CaCO3, арагонит (JCPDS 00-041-1475).
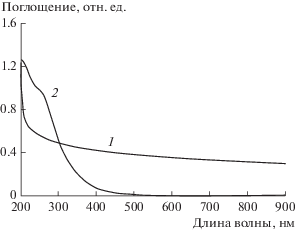
Для подтверждения наличия железа в частицах после стабилизации были измерены спектры поглощения (рис. 6) их водных суспензий. Для наночастиц CaCO3 до стабилизации (рис. 6, кривая 1) экстинкция во всем исследуемом спектральном диапазоне обусловлена рассеянием падающего излучения на их субмикронных агрегатах, полос поглощения не наблюдается. В спектре поглощения водной суспензии стабилизированных наночастиц CaCO3 (рис. 6, кривая 2) проявляется широкая полоса с максимумом на длине волны 260 нм, обусловленная поглощением трехвалентного железа [26], что подтверждает его наличие на поверхности этих наночастиц. Благодаря малому размеру стабилизированных частиц рассеяние не вносит значительного вклада в форму спектра.
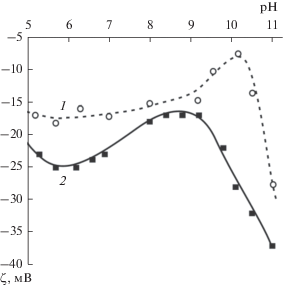
Рассмотрим подробно процесс стабилизации полученных наночастиц карбоната кальция. Агрегативная устойчивость наночастиц определяется их поверхностным зарядом, который, в свою очередь, характеризуется дзета-потенциалом (ζ). Результаты измерения ζ для частиц карбоната кальция сразу после синтеза показали (рис. 7, кривая 1), что частицы имеют отрицательный поверхностный заряд во всем исследуемом диапазоне рН. Отметим, что исходное значение pH водной суспензии частиц CaCO3 составило 8.3. Это обусловлено, по-видимому, гидролизом карбоната кальция, который в общем виде описывается уравнением
(1)
$2{\text{CaC}}{{{\text{O}}}_{3}} + 2{{{\text{H}}}_{2}}{\text{O}} \leftrightarrows {\text{Ca(HC}}{{{\text{O}}}_{{\text{3}}}}{{{\text{)}}}_{2}} + {\text{Ca(OH}}{{{\text{)}}}_{2}}.$(2)
$--{\text{CO}}_{3}^{{2--}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrows --{\text{HCO}}_{3}^{--} + {\text{O}}{{{\text{H}}}^{--}},$(3)
$--{\text{HCO}}_{3}^{--} \leftrightarrows --{\text{C}}{{{\text{O}}}_{2}} + {\text{O}}{{{\text{H}}}^{--}},$(4)
$--{\text{C}}{{{\text{a}}}^{{2 + }}} + {\text{O}}{{{\text{H}}}^{--}} \leftrightarrows --{\text{CaO}}{{{\text{H}}}^{ + }},$(5)
$--{\text{CaO}}{{{\text{H}}}^{ + }} + {\text{O}}{{{\text{H}}}^{--}} \leftrightarrows {\text{Ca(OH}}{{{\text{)}}}_{{2{\text{aq}}}}}.$Рис. 7.
Зависимости дзета-потенциала от рН суспензии наночастиц CaCO3 после синтеза (1) и стабилизации (2).
После стабилизации синтезированных наночастиц карбоната кальция и удаления избытка FeCl3 в исследуемом диапазоне pH наблюдается снижение величины ζ. По-видимому, на поверхности частиц CaCO3 адсорбируются продукты гидролиза хлорида железа(III), который в общем виде можно описать следующим образом [31]:
(6)
$x{\text{F}}{{{\text{e}}}^{{3 + }}} + y{{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrows {\text{F}}{{{\text{e}}}_{x}}\left( {{\text{OH}}} \right)_{y}^{{\left( {3x\,\,--\,\,y} \right)}} + y{{{\text{H}}}^{ + }}.$В работе [32] показано, что поверхность частиц оксида железа имеет отрицательное значение дзета-потенциала в нейтральной и основной областях pH. По-видимому, в этих областях проявляются амфотерные свойства железа, и присутствующие на поверхности наночастиц карбоната кальция после стабилизации группы =FeOH диссоциируют по кислотному типу, образуя =FeO−. В результате отрицательный заряд увеличивается. На рис. 7 видно, что в основной области происходит смещение экстремума кривой, наблюдавшегося для нестабилизированных частиц при значении pH, близком к 10 (рис. 7, кривая 1). Минимальный заряд поверхности по абсолютному значению в этой области теперь наблюдается при pH 8.5 (рис. 7, кривая 2). Значение pH водной суспензии стабилизированных наночастиц карбоната кальция до дзета-метрического титрования снижается с 8.3 (для только что синтезированных наночастиц) до 6.4. Вероятно, после стабилизации на поверхности наночастиц карбоната кальция наряду с появлением амфотерных групп =FeOH снижется концентрация основных групп –Ca–OH.
В результате, благодаря адсорбции ионов железа удалось снизить дзета-потенциал наночастиц карбоната кальция в области pH 5–9, важной для медицины, что привело к увеличению их агрегативной устойчивости и возможности получения индивидуальных наночастиц CaCO3.
ЗАКЛЮЧЕНИЕ
Матричным методом на основе сферических мезопористых частиц кремнезема синтезированы наночастицы карбоната кальция. Полученные частицы образуют в воде агрегаты субмикронного размера. Для получения агрегативно устойчивых гидрозолей CaCO3 разработана методика их электростатической стабилизации с использованием раствора хлорида железа FeCl3. Показано, что данная методика позволяет получить индивидуальные наночастицы карбоната кальция размером 60 ± 10 нм (по данным АСМ и ПЭМ) и 75 ± 15 нм (по данным ДРС). Гидрозоль стабилизированных наночастиц карбоната кальция агрегативно устойчив при pH > 5. Частицы CaCO3 являются пористыми, их удельная поверхность по БЭТ составляет 130 м2/г.
Полученные индивидуальные пористые наночастицы CaCO3 перспективны для использования в медицине в качестве биорастворимого носителя различных лекарственных препаратов.
Список литературы
Wilsen I. // Industrial Minerals and Rocks. 2006. P. 1287.
Ueno Y., Futagawa H., Takagi Y., Ueno A., Mizushima Y. // J. Control. Release. 2005. V. 103. P. 93.
Bandyopadhyay-Ghosh S. // Trends Biomater. Artif. Organs. 2008. V. 22. P. 116.
Fujihara K., Kotaki M., Ramakrishna S. // Biomaterials. 2005 V. 26. P. 4139.
Hannig M., Hannig C. // Adv. Dental Res. 2012. V. 24. P. 53.
Koroleva L.F., Dobrinskaya M.N., Kamantsev I.S. // Integr. Clin. Med. 2017. V. 1. № 2. P. 1.https://doi.org/0.15761/ICM.1000108
Rizzoli R. // Best Pract. Res. Clin. Endocrinol. Metab. 2008. V. 22. P. 813.
Bo-Linn G.W., Davis G.R., Buddrus D.J., Morawski S.G., Santa Ana C., Fordtran J.S. // J. Clin. Invest. 1984. V. 73. P. 640.
Hanzlik R.P., Fowler S.C., Fisher D.H. // J. Pharmacol. Exp. Ther. 2005. V. 313. P. 1217.
Huang S., Chen J.C., Hsu C.W., Chang W.H. // Nanotechnology. 2009. V. 20. P. 375102.
Shahnazari M., Martin B.R., Legette L.L., Lachcik P.J., Welch J., Weaver C.M. // J. Nutr. 2009. V. 139. P. 1308.
Maleki Dizaj S., Barzegar-Jalali M., Zarrintan M.H., Adibkia K., Lotfipour F. // Pharm. Sci. 2015. V. 20. P. 175.
Maleki Dizaj S., Barzegar-Jalali M., Zarrintan M.H., Adibkia K., Lotfipour F. // Expert Opin. Drug Deliv. 2015. V. 12. P. 1649.
Kim J., Bea S.K., Kim Y.H., Kim D.-W., Lee K.-Y., Lee C.-M. // Biotechnol. Bioprocess Eng. 2015. V. 20. P. 794.
Russel W., Saville D., Schowalter W. Colloidal Dispersions. Cambridge: Cambridge University Press, 1989. P. 310.
Trofimova E.Y., Kurdyukov D.A., Yakovlev S.A., Kirilenko D.A., Kukushkina Y.A., Nashchekin A.V., Sitnikova A.A., Yagovkina M.A., Golubev V.G. // Nanotechnology. 2013. V. 24. P. 155 601.
Еуров Д.А., Кириленко Д.А., Курдюков Д.А. // Коллоид. журн. 2017. Т. 79. С. 38.
Kurdyukov D.A., Eurov D.A., Shmakov S.V., Kirilenko D.A., Kukushkina J.A., Smirnov A.N., Yagovkina M.A., Klimenko V.V., Koniakhin S.V., Golubev V.G. // Micropor. Mesopor. Mat. 2019. V. 281. P. 1.
Kurdyukov D.A., Eurov D.A., Kirilenko D.A., Kukushkina J.A., Sokolov V.V., Yagovkina M.A., Golubev V.G. // Micropor. Mesopor. Mat. 2016. V. 223. P. 225.
Трофимова Е.Ю., Алексенский А.Е., Грудинкин С.А., Коркин И.В., Курдюков Д.А., Голубев В.Г. // Коллоид. журн. 2011. Т. 73. С. 535.
Meldrum F.C. // Int. Mater. Rev. 2003. V. 48. P. 187.
Zhou G.T., Yu J.C., Wang X.C., Zhang L.Z // New J. Chem. 2004. V. 28. P. 1027.
Linga Raju Ch., Narasimhulu K.V., Gopal N.O., Rao J.L., Reddy B.C.V. // J. Mol. Struct. 2002. V. 608. P. 201.
Su C., Suarez D.L. // Clays Clay Miner. 1997. V. 45. № 6. P. 814.
Еуров Д.А., Грудинкин С.А., Курдюков Д.А., Голубев В.Г. // ФТТ. 2015. Т. 57. С. 2031.
Siwach O.P., Sen P. // Mater. Sci. Eng. B. 2008. V. 149. P. 99.
Douglas H.W., Walker R.A. // Trans. Faraday Soc. 1950. V. 46. P. 559.
Predali J.J., Cases J.M. // J. Colloid Interface Sci. 1973. V. 45. P. 449.
Moulin P., Roques H. // J. Colloid Interface Sci. 2003. V. 261. P. 115.
Somasundaran P., Agar G.E. // J. Colloid Interface Sci. 1967. V. 24. P. 433.
Oriekhova O., Stoll S. // Colloids Surf. A. 2014. V. 461. P. 212.
Carlson J.J., Kawatra S.K. // Min. Proc. Ext. Met. Rev. 2013. V. 34. P. 269.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал