

Коллоидный журнал, 2020, T. 82, № 3, стр. 334-338
Равновесная сорбция гистидина на клиноптилолите
Д. Л. Котова 1, Т. А. Крысанова 1, *, С. Ю. Васильева 1
1 Воронежский государственный университет
394006 Воронеж,
Университетская пл., 1, Россия
* E-mail: takrys@yandex.ru
Поступила в редакцию 17.06.2019
После доработки 15.08.2019
Принята к публикации 22.08.2019
Аннотация
Исследованы закономерности сорбции гистидина в виде катиона и цвиттер-иона на каркасном алюмосиликате клиноптилолите. Определены вклады обменной и необменной сорбции в сорбционную емкость. Показано, что монослойная сорбция гистидина на клиноптилолите протекает по механизму ионного обмена в результате электростатического взаимодействия ${\text{NH}}_{3}^{ + }$-группы аминокислоты с электроотрицательными центрами каркаса сорбента. В области эквивалентного обмена меньшая величина ионообменной составляющей сорбции для цвиттер-ионов гистидина обусловлена кулоновским отталкиванием диссоциированной карбоксильной группы аминокислоты отрицательно заряженным алюмокислородным каркасом клиноптилолита. Изотерма сорбции гистидина из разбавленных растворов описана с применением теории Ленгмюра. Определены равновесные параметры сорбции. Сделан вывод, что формирование полимолекулярных слоев аминокислоты в результате ассоциации протекает за счет образования водородных связей, диполь-дипольных и гидрофобных взаимодействий.
ВВЕДЕНИЕ
Природные алюмосиликаты являются перспективными сорбентами для решения задач селективного извлечения и концентрирования биологически активных веществ (БАВ) и обладают потенциалом для разработки на их основе носителей лекарственных средств и БАВ [1–3]. Свойства поверхности и текстурные характеристики алюмосиликатов оказывают существенное влияние на характер их взаимодействия с БАВ. Интерес к использованию одного из представителей нанопористых алюмосиликатов – клиноптилолита в качестве эффективного сорбента обусловлен его микро/мезопористой структурой, высокой сорбционной емкостью, доступностью пор для проникновения больших органических молекул [4–7].
Особое место среди БАВ занимают аминокислоты, которые являются важнейшими составляющими белковых структур, источником питания и жизнедеятельности организма и содержатся во многих лекарственных препаратах. Механизм сорбции аминокислот определяется их состоянием в зависимости от рН раствора (ионная форма) и способностью к ассоциации в объеме раствора и на поверхности сорбента [8–10]. В данной работе представлены результаты исследования закономерностей сорбции гистидина из водных растворов на клиноптилолите.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали гетероциклическую альфа-аминокислоту – L-гистидин (L-α-амино-β-имидазолилпропионовую кислоту, His) производства Японии. Гистидин – незаменимая аминокислота, которая входит в состав активных центров множества ферментов. Она является предшественником в биосинтезе гистамина и одной из “существенных” аминокислот, способствующих росту и восстановлению тканей. Молекулярная структура гистидина приведена на рис. 1.
В качестве сорбента был выбран каркасный алюмосиликат клиноптилолит (Люльинского месторождения Приполярного Урала Югры), используемый как энтеросорбент [11]. Мольное отношение Si/Al = 3.9 позволяет отнести его к высококремнистым и достаточно устойчивым к действию температур и кислот сорбентам.
Сорбционное равновесие в системе клиноптилолит (фракция 0.02–0.06 мм)/водный раствор гистидина исследовали при температуре 295 ± 2 К в статических условиях методом переменных концентраций аминокислоты [12] в интервале (1.0–25.0) × 10–3 моль/дм3. Предварительно определив влажность, алюмосиликат массой 0.10 ± ± 0.0002 г приводили в контакт с 200.0 см3 водного раствора аминокислоты известной концентрации и выдерживали до установления равновесия в системе при заданной температуре и постоянном перемешивании. Время установления равновесия в системе определено из предварительного кинетического эксперимента и составило 5 ч. Сорбцию аминокислоты осуществляли из растворов с рН 4.2 и 7.5, при которых гистидин присутствует в растворе соответственно в виде катиона (His+) и цвиттер-иона (His±).
Равновесные фазы разделяли фильтрованием и равновесный раствор анализировали на содержание гистидина с помощью спектрофотометра СФ-46, измеряя интенсивность полосы поглощения на длине волны 210 нм, положение которой сохраняется при изменении величины рН раствора от 3.5 до 7.5. Количество аминокислоты, сорбированной клиноптилолитом, устанавливали по разности концентраций раствора до и после контакта с сорбентом. В равновесном растворе определяли концентрацию внекаркасных катионов, принимающих участие в ионном обмене (калия и натрия методом пламенной фотометрии (Sr = 0.07), кальция и магния методом комплексонометрии, Sr = 0.03). Экспериментальные результаты обработаны методом математической статистики при доверительной вероятности 0.95.
Для получения информации о взаимодействии в системе клиноптилолит–гистидин использовали ИК-фурье-спектроскопию. Спектры регистрировали на приборе Bruker Equinox 55 в режиме диффузного отражения в диапазоне волновых чисел 400–4000 см–1 с разрешением 4 см–1. Ошибка метода составляла 1–3%. Спектры интерпретировали, используя данные [13–15].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Межфазное распределение гистидина в сорбционном процессе в виде однозарядного катиона (рН 4.2) и цвиттер-иона (рН 7.5) представлено на рис. 2. Изотермы сорбции His на клиноптилолите имеют S-образный вид и по классификации ИЮПАК отвечают 4-му типу [16]. Вид изотерм предполагает изменение механизма закрепления аминокислоты с ростом концентрации ее раствора, что характерно при сорбции из растворов БАВ, в которых имеют место сильные межмолекулярные взаимодействия [17].
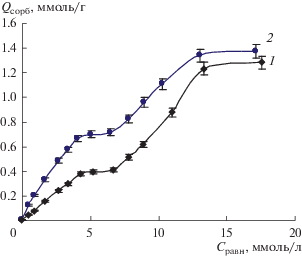
Анализ закономерностей сорбции аминокислоты проводили путем выделения на изотерме областей, соответствующих ионному обмену и необменной сорбции. В условиях равновесия при сорбции His+ и His± из разбавленных растворов (Cравн < 6.5 ммоль/дм3) количество сорбированной аминокислоты эквивалентно суммарному количеству вытесненных в равновесный раствор обменных катионов (K+, Na+, Ca2+ и Mg2+) (рис. 3). В этой области концентраций изотерма линейна, а затем кривая выходит на плато. Сорбция His в виде однозарядного катиона и цвиттер-иона на клиноптилолите по механизму ионного обмена возможна в результате электростатического взаимодействия ${\text{NH}}_{3}^{ + }$-группы аминокислоты с электроотрицательными центрами каркаса сорбента.
Рис. 3.
Зависимости обменной (а) и необменной (б) составляющих сорбции гистидина в виде His± (1) и His+ (2) на клиноптилолите от равновесной концентрации раствора.
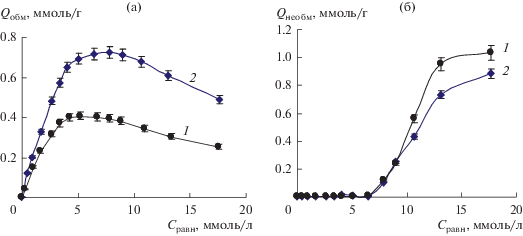
Порядок извлечения обменных катионов в процессе сорбции имеет вид K+ < Na+ < Са2+ < Мg2+ и обусловлен различием в доступности для ионов гистидина обменных центров в матрице клиноптилолита [14, 18]. Аналогичный ряд был получен при сорбции на клиноптилолите ионов аммония и алифатических аминокислот [19]. Меньшее количество ионов калия, участвующих в ионном обмене, является результатом их расположения в менее доступных местах кристаллической решетки сорбента, а также сравнительно большим размером (размер катионов K+ – 1.33 Å, Na+ – 0.98 Å, Са2+ – 1.06 Å, Мg2+ – 0.78 Å [20]).
Локализация гистидина на клиноптилолите проявляется на ИК-спектре в появлении дополнительных полос поглощения. Полоса поглощения при 1380 см–1 отвечает симметричным колебаниям СОО–-группы His±. Максимумы полос поглощения при 1555 и 1494 см–1 характеризуют валентные колебания имидазольного кольца. Его деформационным колебаниям отвечает полоса поглощения при 530 см–1. Электростатическое взаимодействие в системе проявляется в смещении в низкочастотную область спектра полос поглощения, относящихся колебаниям групп Si–O–Al (1040 → 1015 см–1) алюмосиликата и ${\text{NH}}_{3}^{ + }$-групп (3485 → 3465 см–1) аминокислоты.
Анализ ионообменной составляющей сорбции гистидина в виде катиона проводили, учитывая участие в реакции ионного обмена с внекаркасными катионами ионов водорода, присутствующих в растворе. Максимальное количество His+, сорбированного по ионообменному механизму, составляет 0.72 ммоль/г, что в 1.7 раза больше по сравнению с сорбцией His±. Меньшая величина ионообменной составляющей сорбции для цвиттер-ионов аминокислоты, возможно, обусловлена кулоновским отталкиванием диссоциированной карбоксильной группы аминокислоты отрицательно заряженным алюмокислородным каркасом клиноптилолита. Неполная реализация обменной емкости клиноптилолита по ионам аммония (1.90 ммоль/г) при сорбции катионов и цвиттер-ионов гистидина может быть следствием недоступности большей части заряженных центров сорбента для противоионов аминокислоты.
По мере роста концентрации раствора His± (Cравн > 6.5 ммоль/дм3) увеличивается сорбционная емкость, при этом уменьшается количество вытесненных в равновесный раствор обменных катионов (рис. 3). Максимальная сорбционная емкость по цвиттер-иону гистидина составляет 1.28 ммоль/г, обменная емкость – 0.25 ммоль/г. В разбавленных растворах гидратная оболочка His препятствует сближению ионов аминокислоты. При этом аминокислота частично экранирует ионогенные центры матрицы сорбента, что создает ограничения для реализации ионного обмена.
При определенной концентрации гистидин присутствует в растворе в виде ассоциатов, количество которых увеличивается с ростом концентрации аминокислоты в растворе [21]. Склонность к образованию гистидином ассоциатов проявляется при его концентрации выше 6.5 ммоль/дм3. Для цвиттер-иона гистидина в большей степени вероятно формирование ассоциатов линейной структуры за счет образования водородных связей, диполь-дипольного и гидрофобных взаимодействий [17, 22].
Ионы гистидина, формируя монослой в результате ионного обмена, выступают в роли модификатора поверхности, образуя новые сорбционные центры. Полимолекулярные слои, возникающие в результате ассоциации аминокислоты, включают от двух до четырех молекул, что позволяет им размещаться в микро- и мезопорах сорбента. Главную роль в расположении и количестве ионов аминокислоты, участвующих в образовании ассоциатов [1, 23], играет соответствие размера ассоциата объему внутренней полости сорбента. Полимолекулярный характер сорбции гистидина проявляется в смещении максимумов поглощения, характеризующих колебания имидaзольного кольца (1580 → 1555 см–1) и связи N–H (3445 → 3420 см–1), в низкочастотную область спектра. Образование связи СОО–···${\text{NH}}_{3}^{ + }$ в случае цвиттер-ионов гистидина приводит к появлению в спектре полосы поглощения при 3090 см–1.
Анализ участка изотермы в интервале концентраций, соответствующем монослойному закреплению гистидина, проводили, используя линейную форму уравнения Ленгмюра [24]. Определены значения коэффициента адсорбционного равновесия (K) и предельной емкости монослоя (Q∞) для ионов His+ и His± (табл. 1). По экспериментальным данным рассчитаны также величины свободной энергии Гиббса (ΔG) процесса сорбции ионов гистидина в ходе эквивалентного обмена (табл. 1).
Таблица 1.
Равновесные характеристики процесса сорбции His± и His+ на клиноптилолите
| Ионная форма гистидина | Q∞, ммоль/г |
K, дм3/ммоль |
ΔG, кДж/моль |
|---|---|---|---|
| His± | 0.46 | 0.98 | –26.7 |
| His+ | 0.78 | 1.37 | –27.6 |
Величины ΔG отрицательны, что свидетельствует об энергетической выгодности процесса. Близкие значения энергии Гиббса сорбции для цвиттер-иона и катиона гистидина подтверждают, что монослойное закрепление обеих форм аминокислоты протекает по одному и тому же механизму.
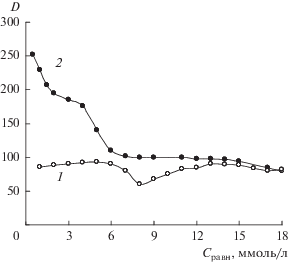
Рассчитан равновесный коэффициент распределения (D) катионов и цвиттер-ионов гистидина на клиноптилолите (рис. 4). Большее значение ионообменной емкости для катионов аминокислоты проявляется в более высоком значении коэффициента распределения в области концентраций, отвечающей мономолекулярной сорбции.
Список литературы
Titus E., Kalkar A.K., Gaikar V.G. // Colloids Surf. A. 2003. V. 223. P. 55.
Rivera A., Farías T., Ruiz-Salvador A.R., de Ménorval L.C. // Micropor. Mesopor. Mater. 2003. V. 61. P. 249.
Hartmann M., Vinu A., Chandrasekar G. // Chem. Mater. 2005. V. 17. P. 829.
Rivera A., Farıas T. // Micropor. Mesopor Mater. 2005. V. 80. P. 337.
Cristine E.A. // Astrobiology. 2011. V. 11. P. 409.
Lam A., Rivers A. // Micropor. Mesopor. Mater. 2006. V. 91. P. 181.
Котова Д.Л., Васильева С.Ю., Крысанова Т.А., Ресснер Ф., Бородина Е.В., До Т.Л. // Рос. нанотехнологии. 2014. Т. 9. № 9–10. С. 25.
Котова Д.Л., До Тхи Лонг, Крысанова Т.А., Селеменев В.Ф. // Журн. физ. химии. 2011. Т. 85. С. 2365.
Faghihian F.H., Nejati-Yazdinejad M. // Adsorpt. Sci. Technol. 2009. V. 27. P. 19.
Капуцкий Ф.Н., Юркштович Г.Л., Старобинец Г.Л., Бычковский П.М., Борщенская Т.И. // Журн. физ. химии. 2000. Т. 74. С. 277.
Жучков А. Н., Берлянд А.С., Алиханян А.С., Плесская Н.А. // Хим.-фарм. журн. 2011. Т. 45. № 2. С. 49.
Полянский Н.Г., Горбунов В.Г., Полянская Н.Л. Методы исследования ионитов. М.: Химия, 1976.
Наканиси К. Инфракрасная спектроскопия и строение органических молекул. М.: Мир, 1987.
Цицишвили Г.В. Природные цеолиты. М.: Химия, 1985.
Литтл Л. ИК-спектры адсорбированных молекул. М.: Мир, 1969.
Boekfa B., Pantu P., Limtrakul J. // J. Mol. Struct. 2008. V. 889. P. 81.
Munsch S., Hartmann M., Ernst S. // Chem. Commun. 2001.V. 19. P. 1978.
Kisler J.M. Stevens G.W., O’Connor A.J. // 13th Intern. Zeolite Conf., Recent Res. Rep., 18-R-08, Montpellier, Groupe Français des Zéolithes, 2001. P. 82.
Payra P., Dutta P.K. // In: Handbook of Zeolite Science and Technology. Ed. by Auerbach S.M, Carrado K.A., Dutta P.K. N.Y.: CRC Press, 2003. P. 1.
Galli E., Gottardi G. // Acta Crystallogr. 1983. V. 39. P. 189.
Котова Д.Л., Бейлина Д.С., Крысанова Т.А. // Журн. физ. химии. 2004. Т. 78. С. 458.
Krohn J.E., Tsapatsis M. // Langmuir. 2005. V. 21. P. 8743.
Krohn J.E., Tsapatsis M. // Langmuir. 2006. V. 22. P. 9350.
Фролов Ю.Г. Курс коллоидной химии. Поверхностные явления и дисперсные системы. М.: Химия, 1989.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал