

Коллоидный журнал, 2020, T. 82, № 3, стр. 324-333
Синтез наночастиц гидроксиапатита методом контролируемого осаждения в присутствии додецилсульфата натрия
М. Ю. Королева 1, *, Е. Ю. Каракатенко 1, Е. В. Юртов 1
1 Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., д. 9, Россия
* E-mail: m.yu.kor@gmail.com
Поступила в редакцию 20.11.2019
После доработки 11.12.2019
Принята к публикации 16.12.2019
Аннотация
Синтез наночастиц (НЧ) гидроксиапатита проводился методом контролируемого осаждения в присутствии поверхностно-активного вещества (ПАВ) – додецилсульфата натрия. Размеры, форма и структура НЧ зависели от порядка смешивания компонентов в реакционной среде. При первоначальном добавлении в раствор ПАВ фосфата натрия, а затем нитрата кальция происходило образование стержнеобразных НЧ гидроксиапатита, длина которых уменьшалась с увеличением концентрации ПАВ. С ростом концентрации додецилсульфата натрия ζ-потенциал НЧ уменьшался до нулевых значений, а затем возрастал по абсолютной величине до –55 мВ. Это свидетельствует об образовании бислоя ПАВ на поверхности НЧ, который экранировал поверхность растущих кристаллов, что приводило к образованию более мелких НЧ при высоких концентрациях ПАВ. При первоначальном добавлении в раствор ПАВ нитрата кальция, а затем фосфата натрия, вероятнее всего, синтез происходил на поверхности мицелл, частично содержащих додецилсульфат кальция. Поэтому в синтезируемых частицах сохранялась сульфатная фаза, что подтверждают данные рентгенофазового анализа. При более высоких концентрациях ПАВ в реакционной среде, по-видимому, присутствовали гибкие цилиндрические мицеллы. Осаждение гидроксиапатита на поверхности таких мицелл приводило к образованию волнообразно изогнутых нитевидных частиц.
1. ВВЕДЕНИЕ
Для ограничения скорости роста и снижения степени агрегации наночастиц (НЧ) синтез проводят в среде, содержащей ПАВ. Для этих целей могут быть использованы ПАВ различной природы – анионные, катионные или неионные. В качестве анионного ПАВ, способного стабилизировать НЧ, достаточно часто применяется доцецилсульфат натрия (ДСН). С помощью этого ПАВ стабилизировали НЧ золота [1], серебра [2, 3], меди [4], TiO2 [5, 6], V2O5/TiO2 [6], Fe3O4 [7], WO3 [8], MnO2 [9], NiCo2O4 [10], Zn3V2O8 [11], нанокомпозитов Mg(OH)2/графен [12] и SnO2/графен [13], несквегонита [14] и др.
При синтезе гидроксиапатита в присутствии ДСН в зависимости от концентрации данного ПАВ изменялись как размеры, так и форма синтезированных НЧ. Так, при гидротермальном синтезе происходило образование стержнеобразных НЧ гидроксиапатита [15, 16]. Стержни были более короткими и более тонкими, чем при синтезе в отсутствие ПАВ [16]. Если сравнить влияние ДСН и других ПАВ, таких как бромид цетилтриметаммония или Triton X-100, то в гидротермальных условиях и при наличии ДСН в реакционной среде формировались более длинные стержни (до 137 нм) и с более высокой степенью анизодиаметрии (отношением длина/диаметр) по сравнению с НЧ, синтезированными в присутствии других ПАВ.
Механизм стабилизации НЧ, синтезируемых в присутствии ДСН, недостаточно ясен в настоящее время. Так, при синтезе НЧ золота на основании данных, полученных с помощью ИК-фурье-спектроскопии, авторы [1] заключили, что сульфатные группы ДСН взаимодействуют с поверхностными атомами НЧ золота.
Авторы [7], изучавшие синтез НЧ магнетита, полагают, что молекулы ДСН могут адсорбироваться на поверхности НЧ в виде гемимицелл [17, 18], т.е. в виде монослоя, в котором молекулы ПАВ ориентированы полярной группой к поверхности НЧ, или адмицелл с бислойной структурой.
В работе [2] на основании данных, полученных с помощью ЯМР, высказано предположение, что в случае синтеза НЧ серебра отдельные молекулы и субмицеллярные агрегаты ДСН являются переносчиками ионов серебра к растущим НЧ. В тоже время мицеллы ДСН концентрируются вблизи НЧ серебра, препятствуя подходу к ним отдельных молекул и субмицелл, т.е. препятствуют росту НЧ и их агрегации. Это приводит к тому, что, по мнению авторов [2], при более высоких концентрациях ПАВ образуются более мелкие НЧ с узким распределением по размерам.
Авторы работы [15] считают, что при синтезе НЧ гидроксиапатита при смешивании прекурсоров происходит сначала образование промежуточного комплекса с участием аниона додецилсульфата натрия и Ca2+. Именно в таких, обогащенных кальцием доменах и происходит последующий рост НЧ гидроксиапатита [19].
Возможно, что имеет место специфическое взаимодействие молекул ДСН с различными гранями растущего кристалла гидроксиапатита, которое может существенно изменяться вдоль базальных плоскостей (перпендикулярных c-оси) и латеральных плоскостей (параллельных c-оси), что и приводит к асимметричному росту НЧ гидроксиапатита [20].
В статье [21] предположено, что при синтезе НЧ гидроксиапатита додецилсульфат-анионы адсорбируются на их поверхности за счет электростатического притяжения к ионам кальция или за счет изоморфного замещения поверхностных фосфат-ионов. По мнению авторов, после адсорбции углеводородные радикалы анионов ПАВ ориентируются либо в сторону водной фазы, либо параллельно поверхности НЧ. В отсутствие ДСН ζ-потенциал НЧ гидроксиапатита был равен –14 мВ. При увеличении концентрации ДСН до 20 мМ ζ-потенциал монотонно увеличивался по абсолютной величине до –52 мВ.
Показано, что при синтезе НЧ гидроксиапатита в присутствии ДСН большое количество этого ПАВ встраивается в поверхностный слой кристаллов, что было подтверждено данными ИК-спектроскопии [22]. С другой стороны, в работе [23] установлено, что в условиях, моделирующих ротовую полость человека, ДСН быстро адсорбируется на поверхности кристаллов гидроксиапатита и также быстро десорбируется.
При концентрации ДСН ниже критической концентрации мицеллообразования (ККМ) рост кристаллов CaHPO4 · H2O замедлялся [24]. Происходило образование более тонких пластинчатых частиц по сравнению с частицами, синтезируемыми в отсутствие ПАВ, что свидетельствовало об адсорбции анионов ПАВ на боковых гранях кристаллов. При концентрациях ДСН выше ККМ изменялись состав и морфология частиц: CaHPO4 · H2O осаждался в виде призм, Ca8H2(PO4)6 · 5H2O – в виде сферических агрегатов, состоящих из тонких пластинок.
Авторы статьи [25] полагают, что додецилсульфат-анионы взаимодействуют с катионами кальция с образованием цилиндрических структур. Затем катион кальция реагирует с фосфат-анионом внутри таких цилиндрических капсул, что и приводит к образованию НЧ игольчатой формы.
В статье [26] описано получение полых микросфер CaCO3 в присутствии ДСН и аспарагиновой кислоты. Авторы [26] считали, что благодаря электростатическому притяжению между гидратированными группами ПАВ и ионами кальция на поверхности сферических мицелл возникали зоны, обогащенные кальцием, в которых протекала активная нуклеация, так же как и при синтезе гидроксиапатита в работах [24, 27]. Однако предположение о том, что мицеллы ДСН в описанном в статье синтезе выступали в качестве матрицы при получении микросфер, является маловероятным, так как размер получаемых микросфер (2 мкм) существенно превышал размеры мицелл ДСН, даже с учетом ассоциации их с молекулами аспарагиновой кислоты.
При гидротермальном извлечении гидроксиапатита из раковин моллюска абалон в присутствии ДСН формировались частицы пластинчатой формы, более тонкие, чем в отсутствие ПАВ [28]. Авторы считают, что ДСН не выступает в роли темплата при таком синтезе. Однако следует отметить, что в данной работе были использованы достаточно высокие концентрации ДСН (примерно 0.35 М), при которых в гидротермальных условиях могут образовываться дискообразные мицеллы.
Таким образом, в литературе отсутствуют систематические данные о влиянии ДСН на процесс синтеза НЧ гидроксиапатита, их размер и морфологию. В данной работе изучено влияние концентрации ДСН на размер, форму и состав частиц гидроксиапатита в зависимости от состава ракционной среды и порядка смешивания компонентов.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1. Реактивы и материалы
Для синтеза НЧ гидроксиапатита были использованы следующие реактивы: Ca(NO3)2 · 4H2O (>99%, Sigma-Aldrich), Na2HPO4 (Химмед, ч. д. а.), NaOH (Химмед, х. ч.), ДСН (>98.5%, Sigma-Aldrich). Дополнительной очистке их не подвергали.
2.2. Синтез НЧ гидроксиапатита
Синтез НЧ гидроксиапатита проводили методом контролируемого осаждения по реакции:
Процесс осуществляли по одной из двух схем. В первом случае в реакционный сосуд, содержащий 20 мл водного раствора ДСН заданной концентрации, добавляли 20 мл 0.06 М водного раствора Na2HPO4 и нагревали до 40°C на магнитной мешалке. Затем при постоянном перемешивании со скоростью 350 об./мин в реакционную смесь добавляли 20 мл 0.1 М водного раствора Ca(NO3)2 с помощью перистальтического насоса со скоростью 1 мл/мин. Во втором случае в водный раствор ДСН сначала добавляли 20 мл 0.1 М водного раствора Ca(NO3)2 и нагревали до 40°C, а затем добавляли 20 мл 0.06 М водного раствора Na2HPO4 также с помощью перистальтического насоса. Количество водного раствора фосфата натрия во всех экспериментах было таковым, чтобы обеспечить мольное соотношение Ca/P = 1.67. pH реакционной среды поддерживали постоянным и равным 9.0 ± 0.2 добавлением 1 M раствора NH4OH. Полученную первым или вторым способом суспензию подвергали старению в течение 24 ч при 25°C. Затем осадок фильтровали, многократно промывали дистиллированной водой и сушили при 120°C в течение 2 ч. Образцы для рентгенофазового анализа и ИК-спектроскопии после такой сушки прокаливали при 400°C в течение 3 ч в муфельной печи.
2.3. Методы исследования НЧ гидроксиапатита
Размер и морфологию синтезированных НЧ гидроксиапатита определяли по микрофотографиям образцов, полученных с использованием сканирующего электронного микроскопа (СЭМ) TESCAN Vega 3 LMU с вольфрамовым катодом при ускоряющем напряжении 10 кВ и просвечивающего электронного микроскопа (ПЭМ) JEOL JEM-1011 при ускоряющем напряжении 80 кВ.
Фазовый состав образцов определяли по дифракционной картине рентгеновского излучения. Дифрактограммы снимали на приборе D2 PHASER Bruker, используя CuKα-излучение (λ = 1.54178 Å), Ni-фильтр и графитовый монохроматор. Ток на трубке – 10 мА, напряжение на аноде – 30 кВ. Диапазон значений угла 2θ – от 20° до 60°, шаг 0.025°, щель 0.6 мм, выдержка в точке – 0.5 с (несколько проходов с накоплением данных), дискриминатор по энергиям – 0.17–0.23 кэВ. Расшифровку спектров осуществляли с помощью библиотеки JCPDS-ICDD с использованием специализированного программного обеспечения Topas.
ИК-спектры регистрировали на фурье-спектрометре Nicolet 380 с разрешением 4 см–1 в диапазоне 4000–400 см–1; общее время регистрации каждого спектра составляло 1 мин. Управление спектрометром осуществлялось с помощью программного обеспечения OMNIC 7.3. Пробоподготовку осуществляли методом прессования таблеток с KBr.
3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
3.1. Размер и морфология синтезированных НЧ гидроксиапатита
При синтезе НЧ в присутствии ПАВ их состав, размер и морфология зачастую зависят от порядка смешивания компонентов. В данной работе синтез НЧ гидроксиапатита был проведен по двум методикам, которые различались порядком смешивания реагентов с раствором ДСН. В первой серии экспериментов в реакционный сосуд, содержащий раствор ДСН заданной концентрации, добавляли раствор Na2HPO4. Затем по описанной в разделе 2.2 методике в реакционную смесь добавляли раствор Ca(NO3)2. Во второй серии экспериментов раствор ДСН сначала смешивали с раствором Ca(NO3)2 и затем добавляли раствор Na2HPO4.
В процессе синтеза концентрация ПАВ не оставалась постоянной. После постепенного добавления второго реагента, т.е. в конце синтеза, концентрация ДСН уменьшалась в 1.5 раза по сравнению с его концентрацией в начале синтеза. Так как наиболее сильное влияние на размер и морфологию синтезируемых частиц оказывает структура ассоциатов ПАВ в начале процесса нуклеации, то ниже везде будут указываться значения концентрации ДСН в начале синтеза, т.е. перед добавлением второго реагента.
На рис. 1 представлены ПЭМ-изображения частиц гидроксиапатита, полученные при добавлении в раствор ДСН сначала фосфата натрия, а затем нитрата кальция.
Рис. 1.
ПЭМ-изображения НЧ гидроксиапатита, синтезированных при концентрации ДСН в реакционной смеси 0 (а), 25 (б), 50 (в) и 100 мМ (г). Последовательность смешивания компонентов: раствор ПАВ + фосфат натрия + нитрат кальция.

При такой последовательности смешивания компонентов в диапазоне концентраций ДСН от 0 до 125 мМ происходило образование частиц стержнеобразной формы. В отсутствие ПАВ частицы представляли собой стержни длиной 200–280 нм (рис. 1а), состоящие из сросшихся НЧ диаметром примерно 20 нм. На рис. 2а приведено распределение стержней гидроксиапатита по длине. Такие стержни срастались в агломераты размером от одного до нескольких десятков микрометров. Как следствие, суспензия таких частиц была неустойчива к седиментации.
Рис. 2.
Распределение по длине стержней гидроксиапатита, синтезированных при концентрации ДСН в реакционной смеси 0 (а) и 100 мМ (б). Последовательность компонентов: раствор ПАВ + фосфат натрия + нитрат кальция.
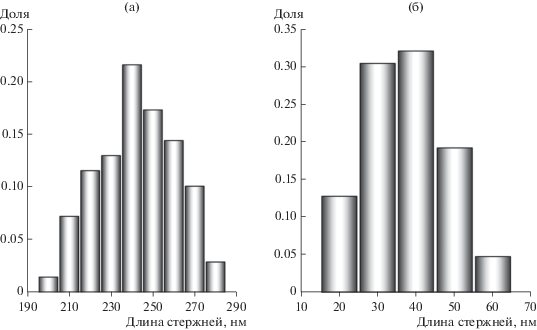
С увеличением концентрации ДСН в реакционной смеси длина стержней гидроксиапатита уменьшалась. На рис. 2б в качестве примера показано распределение по длине наностержней, синтезированных при концентрации ДСН 100 мМ. Средняя длина стержней составляла примерно 35 нм. Как будет показано в разделе 3.3, при концентрации ДСН ≥100 мМ заряд НЧ был относительно высоким. Агрегация таких НЧ была замедлена из-за электростатического отталкивания, поэтому суспензии НЧ гидроксиапатита были устойчивы к седиментации более 15 сут.
Во второй серии экспериментов, когда к водному раствору ДСН добавляли водный раствор нитрата кальция, наблюдалось помутнение раствора и его окрашивание в белый цвет, что, вероятнее всего, связано с образованием додецилсульфата кальция. После добавления в систему раствора Na2HPO4 белая окраска становилась интенсивнее, и постепенно выпадал осадок. При концентрации ДСН, равной 25 мМ, суспензии НЧ были устойчивы к седиментации в течение примерно 2 сут, при более высокой концентрации ПАВ частицы оседали в течение 1 ч.
Микрофотографии частиц гидроксиапатита, синтезированных при первоначальном добавлении в раствор ПАВ нитрата кальция, а затем фосфата натрия, представлены на рис. 3.
Рис. 3.
СЭМ-изображения НЧ гидроксиапатита, синтезированных при концентрации ДСН в реакционной смеси 0 (а), 25 (б), 50 (в), 75 (г), 100 (д) и 125 мМ (е). Последовательность смешивания компонентов: раствор ПАВ + нитрат кальция + фосфат натрия.
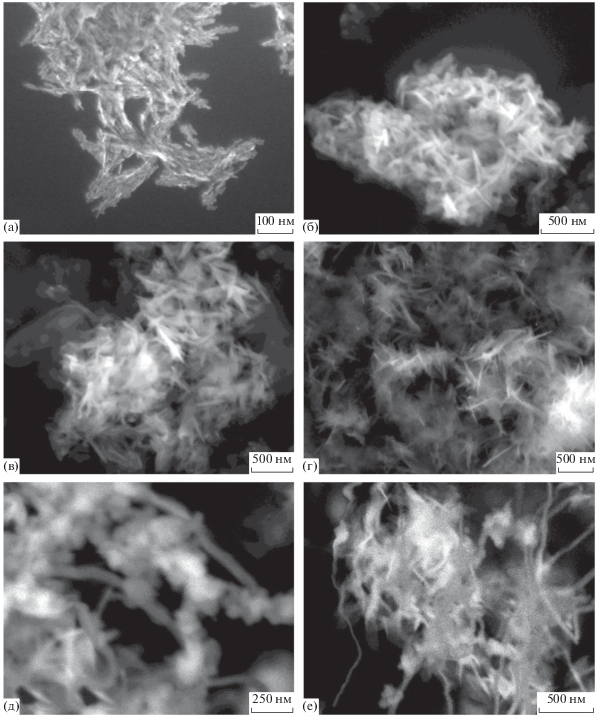
С увеличением концентрации ДСН от 0 до 75 мМ происходило образование стержней, длина которых возрастала с ростом концентрации ПАВ от 240 до 400 нм.
При концентрации ПАВ, равной 100 мМ, морфология синтезируемых частиц начала существенно изменяться. В системе формировались наностержни длиной 150–200 и нанонити диаметром от 40 до 80 нм и длиной до 1 мкм. Повышение концентрации ПАВ до 125 мМ привело к преимущественному образованию нанонитей диаметром 30–50 нм и длиной до 2 мкм.
Размеры и форма частиц, полученных при различной последовательности смешивания реагентов, представлены в табл. 1.
Таблица 1.
Влияние концентрации ДСН на длину и форму синтезированных частиц в зависимости от последовательности смешивания компонентов
| Концентрация ДСН, мМ | Последовательность смешивания компонентов | |||
|---|---|---|---|---|
| Раствор ДСН + фосфат натрия + нитрат кальция | Раствор ДСН + нитрат кальция + фосфат натрия | |||
| Форма | Длина, Нм | Форма | Длина, Нм | |
| 0 | Стержни | 240 ± 25 | Стержни | 240 ± 25 |
| 25 | Стержни | 120 ± 15 | Стержни | 280 ± 25 |
| 40 | Стержни | 90 ± 10 | Стержни | 300 ± 30 |
| 50 | Стержни | 70 ± 8 | Стержни | 350 ± 50 |
| 75 | Стержни | 55 ± 5 | Стержни | 400 ± 50 |
| 100 | Стержни | 45 ± 5 | Стержни + нити | 600–1000 |
| 125 | Стержни | 35 ± 5 | Нити | 1000–2000 |
3.2. Исследование состава синтезированных НЧ
Был выполнен рентгенофазовый анализ синтезированных НЧ, которые после промывки сушили при температуре 120°C и затем прокаливали при 400°C.
Рентгеновские дифрактограммы образцов, синтезированных при добавлении в раствор ПАВ сначала фосфата натрия, а затем нитрата кальция, как после сушки (рис. 4а), так и после сушки с последующим прокаливанием (рис. 4б) соответствовали гексагональной фазе гидроксиапатита (пространственная группа P63/m). Параметры кристаллической решетки были равны a = 9.432 Å, c = 6.881 Å; эти значения близки к справочным данным для гидроксиапатита (PDF2 09-0432). Широкие пики в области углов 2θ от 30° до 35° свидетельствуют о высокой дисперсности порошков.
Рис. 4.
Рентгенограммы образцов, синтезированных в присутствии 40 мМ ДСН. Последовательность смешивания компонентов: раствор ПАВ + фосфат натрия + нитрат кальция (а) и (б), раствор ПАВ + нитрат кальция + фосфат натрия (в) и (г); образцы после сушки при 120°C (а) и (в) и после сушки с прокаливанием при 400°C (б) и (г). Черными точками обозначена фаза Ca10(PO4)6(OH)2.
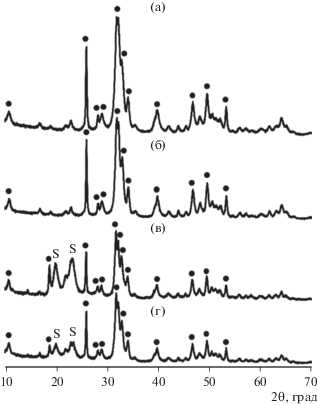
В образцах, синтезированных при добавлении в раствор ПАВ сначала нитрата кальция, а затем фосфата натрия, были обнаружены 2 фазы (рис. 4в и 4г). Основной кристаллической фазой являлся гидроксиапатит с параметрами кристаллической решетки a = 9.438 Å, c = 6.867 Å. Рефлексы на углах дифракции 2θ = 20.04° и 23.50°, вероятнее всего, относятся к сульфатной фазе. После прокаливания интенсивность пиков сульфатной фазы значительно уменьшалась, что обусловлено частичным удалением этой фазы при воздействии высокой температуры (рис. 4г).
Оценка размеров областей когерентного рассеяния по уравнению Шеррера показала, что средний размер кристаллитов во всех четырех образцах составляет 17–22 нм. Это соответствует результатам, полученным с помощью ПЭМ: на микрофотографиях видно, что наностержни состоят из сросшихся НЧ диаметром примерно 20 нм.
В ИК-спектрах образцов, синтезированных при добавлении в раствор ДСН сначала фосфата натрия, а затем нитрата кальция, как после сушки, так и после сушки и прокаливания (рис. 5а и 5б) наблюдались основные полосы, характерные для гидроксиапатита. Частоты полос валентных (964, 1029 и 1114 см–1) и деформационных (563, 599 см–1) колебаний групп PO4 соответствуют полосам мелкокристаллического гидроксиапатита. Для образца, подвергнутого сушке, регистрировались пики поглощения низкой интенсивности групп ${\text{SO}}_{4}^{{2 - }}$ при 1216 и 1249 см–1 (рис. 5а). Полосы поглощения при 2850 и 2917 см–1 отвечают валентным колебаниям связей С–Н в СН3-группах ДСН [29]. Слабый пик при 1473 см–1 свидетельствует о присутствии карбонат-ионов в образце.
Рис. 5.
ИК-спектры образцов, синтезированных в присутствии 40 мМ ДСН. Последовательность смешивания компонентов: раствор ПАВ + фосфат натрия + нитрат кальция (а) и (б), раствор ПАВ + нитрат кальция + фосфат натрия (в) и (г); образцы после сушки при 120°C (а) и (в), и после сушки с прокаливанием при 400°C (б) и (г).
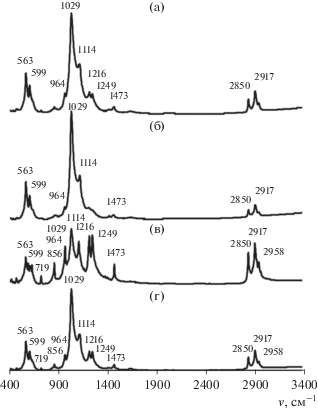
После прокаливания образца на его ИК-спектре не наблюдали полос поглощения сульфатных групп (рис. 5б), что свидетельствует об удалении ДСН, адсорбированного на внешней поверхности частиц, при температурном воздействии, что согласуется результатами РФА.
На ИК-спектре образца, синтезированного при добавлении в раствор ДСН сначала нитрата кальция, а затем фосфата натрия, после сушки (рис. 5в) также наблюдали частоты полос валентных (964, 1029 и 1114 см–1) и деформационных (563, 599 см–1) колебаний групп PO4, однако их интенсивность была ниже. Полосы поглощения при 1216, 1249 см–1 характерны для колебаний групп ${\text{SO}}_{4}^{{2 - }}{\text{.}}$ Их сравнительно высокая интенсивность может говорить о частичном замещении фосфатных групп на сульфатные в молекуле гидроксиапатита на начальном этапе синтеза [30]. На спектре присутствовали полосы поглощения ${\text{CO}}_{3}^{{2 - }}$ с максимумами при 1473 см–1 (ν3) и 856 см–1 (ν2). Интенсивность полос карбонатных групп уменьшалась после прокаливания НЧ (рис. 5г). Интенсивность полос поглощения сульфатных групп (1216, 1249 см–1) также значительно снижалась (рис. 5г), что свидетельствует о частичном удалении этих групп из образца. Возможно, в результате синтеза при добавлении в раствор ДСН сначала нитрата кальция, а затем фосфата натрия происходило образование нестехиометрического сульфат-замещенного гидроксиапатита Са10(PO4)(6 – x)(SO4)x(OH)2 –x.
3.3. Предполагаемый механизм синтеза НЧ при различной концентрации додецилсульфата натрия
При синтезе НЧ в присутствии ПАВ различные его структуры (мицеллы, жидкие кристаллы) могут выступать в качестве матрицы (темплата), на поверхности которой или в ее внутренних полостях протекают процессы нуклеации и роста частиц. Кроме того, молекулы ПАВ могут преимущественно адсорбироваться на определенных гранях кристалла, приводя к его анизотропному росту.
Первая критическая концентрация мицеллообразования (ККМ1) ДСН в воде равна 8.2–8.5 мМ [15, 31, 32]. В присутствии электролитов ККМ1 уменьшается. Например, при концентрации NaCl 0.01 М значение ККМ1 снижается до 3.3 мМ [33], а в 0.1 М растворе NaCl – до 1.5 мМ [34]. С повышением температуры до 40°C значение ККМ1 возрастает до 9.7 мМ [26]. В данной работе минимальная концентрация ДСН в начале синтеза была равна 25 мМ, т.е. синтез НЧ гидроксиапатита во всех случаях проводили при концентрации ПАВ выше ККМ1.
При концентрации ДСН, равной примерно 27 мМ, существующие в растворе сферические мицеллы объединяются в периодическую коллоидную структуру с кубической решеткой; расстояние между мицеллами равно 4.1 нм. При более высокой концентрации ПАВ мицеллы сближаются и образуют кластеры с более плотной упаковкой [35]. Вторая критическая концентрация мицеллообразования (ККМ2) ДСН при 40°C равна 80 мМ [26].
В случае синтеза частиц гидроксиапатита при первоначальном добавлении в раствор ПАВ фосфата натрия не происходило химического взаимодействия ДСН и фосфата натрия. Образование кристаллической фазы происходило только после добавления в раствор нитрата кальция. Так как произведение растворимости фосфата кальция меньше, чем додецилсульфата кальция, то имела место кристаллизация с образованием гидроксиапатита.
В отсутствие ДСН частицы гидроксиапатита были заряжены отрицательно из-за образования двойного электрического слоя присутствующими в реакционной системе электролитами и продуктами реакции (рис. 6). При синтезе в присутствии ПАВ додецилсульфат-анионы адсорбировались своими полярными группами на гранях растущего кристалла, а их неполярные углеводородные радикалы ориентировались в сторону водной фазы. Это сопровождалось уменьшением ζ-потенциала частиц до нулевых значений. После заполнения первого адсорбционного слоя начинал формироваться второй адсорбционный слой, состоящий из анионов ДСН, ориентированных углеводородными радикалами в сторону углеводородных радикалов первого адсорбционного слоя и полярными группами наружу, в сторону водной фазы, т.е. происходило образование бислоя ПАВ. При этом ζ-потенциал частиц был отрицательным и возрастал по абсолютной величине до –55 мВ с увеличением концентрации ПАВ, а затем оставался практически постоянным. Полученные результаты согласуются с данными об образовании аналогичного бислоя ДСН на поверхности НЧ диоксида кремния [36].
Рис. 6.
Зависимость ζ-потенциала частиц гидроксиапатита от концентрации ДСН в реакционной среде в начале синтеза.
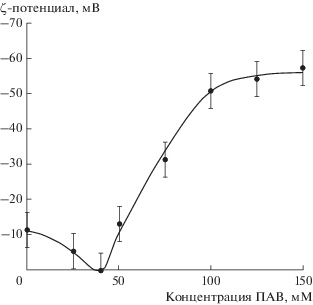
При адсобции ДСН на гранях кристаллов гидроксиапататита, молекулы ПАВ экранировали поверхность, что снижало скорость роста и приводило к образованию более мелких НЧ. Так как при концентрации ДСН ≥100 мМ ζ-потенциал НЧ был достаточно высоким, их суспензии были устойчивы к агрегации и последующей седиментации.
В случае синтеза частиц гидроксиапатита при первоначальном добавлении в раствор, содержащий сферические мицеллы ПАВ, нитрата кальция происходило частичное образование додецилсульфата кальция, о чем свидетельствует появление у раствора белой окраски. Возможно, что при этом структура мицелл сохранялась, и дальнейший синтез гидроксиапатита протекал на поверхности мицелл, которые выступали в качестве матрицы. Определенная часть додецилсульфата была инкорпорирована в растущие частицы, поэтому в конечном продукте сохранялась сульфатная фаза, что подтверждалось данными рентгенофазового анализа (рис. 4в).
Как показано в работе [37], в присутствии катионного мономера ККМ додецилсульфата натрия резко снижалась. Вероятно, в исследуемых нами системах также уменьшалась и величина ККМ2. Т.е. при концентрациях ДСН, меньших 80 мМ, уже происходило образование несферических мицелл, которые служили матрицей при синтезе частиц гидроксиапатита. С ростом концентрации ПАВ длина мицелл возрастала, соответственно увеличивалась длина стержней гидроксиапатита.
Безусловно, концентрации ДСН, равные 100 и 125 мМ, слишком низки для возникновения червеобразных мицелл. Возможно, при этих концентрациях ПАВ происходило образование гибких цилиндрических мицелл, на поверхности которых осаждался гидроксиапатит. Косвенно это подтверждают волнообразные изгибы формирующихся нитевидных частиц гидроксиапатита (рис. 3е), обусловленные тем, что мицеллы изгибались и переплетались в реакционной среде в первые моменты синтеза.
ЗАКЛЮЧЕНИЕ
Проведенные в данной работе исследования по влиянию додецилсульфата натрия на синтез НЧ гидроксиапатита показали, что размеры, форма и структура частиц зависели от порядка смешивания компонентов в реакционной среде. При добавлении в раствор ПАВ сначала фосфата натрия, а затем нитрата кальция имело место образование НЧ гидроксиапатита, имеющих стержнеобразную форму. С ростом концентрации ПАВ ζ-потенциал НЧ уменьшался до нуля, а затем возрастал по абсолютной величине до –55 мВ. Это свидетельствует об адсорбции ДСН на поверхности частиц и о последующем формировании бислоя ПАВ, экранирующего поверхность растущих кристаллов. В результате при высоких концентрациях ПАВ происходило образование более коротких стержнеобразных НЧ гидроксиапатита.
При первоначальном добавлении в раствор ДСН нитрата кальция, а затем фосфата натрия имело место частичное образование додецилсульфата кальция. Вероятнее всего, синтез происходил на поверхности мицелл, содержащих додецилсульфат кальция, молекулы которого служили центрами нуклеации. Именно поэтому в синтезируемых НЧ сохранялась сульфатная фаза, что подтверждено рентгенофазовым анализом. При более высоких концентрациях ДСН в реакционной среде присутствовали гибкие цилиндрические мицеллы. Осаждение гидроксиапатита на поверхности таких мицелл приводило к образованию волнообразно изогнутых нитевидных частиц.
Список литературы
Cabrera G.F.S., Balbin M.M., Eugenio P.J.G., Zapanta C.S., Monserate J.J., Salazar J.R., Mingala C.N. // Biochem. Biophys. Res. Commun. 2017. V 484. P. 774.
Shah V., Bharatiya B., Mishra M.K., Ray D., Shah D.O. // J. Mol. Liq. 2019. V. 273. P. 222.
Bhaduri G.A., Little R., Khomane R.B., Lokhande S.U., Kulkarni B.D., Mendis B.G., Šiller L. // J. Photochem. Photobiol. A. 2013. V. 258. P. 1.
Zhang X., Yin H., Cheng X., Hu H., Yu Q., Wang A. // Mater. Res. Bull. 2006. V. 41. P. 2041.
Sun B., Zhou G., Shao C., Jiang B., Pang J., Zhang Y. // Powder Technol. 2014. V. 256. P. 118.
Mohamed M.M., Bayoumy W.A., Khairy M., Mousa M.A. // Micropor. Mesopor. Mater. 2006. V. 97. P. 66.
Shakerian F., Chelongar Y., Shabani A.M.H., Dadfarnia S. // Microchem. J. 2019. V. 146. P. 234.
Anithaa A.C., Asokan K., Sekar C. // Electrochim. Acta. 2017. V. 237. P. 44.
Dai Y., Li J., Yan G., Xu G., Xue Q., Kang F. // J. Alloys Compd. 2015. V. 621. P. 86.
Wang H., Lu J., Yao S., Zhang W. // J. Alloys Compd. 2018. V. 744. P. 187.
Mazloom F., Masjedi-Arani M., Ghiyasiyan-Arani M., Salavati-Niasari M. // J. Mol. Liquids. 2016. V. 214. P. 46.
Sheikhhosseini E., Ranjbar M. // OpenNano. 2019. V. 4. Article 100028.
Jing S., Zheng H., Zhao L., Qu L., Yu L. // J. Electroanal. Chem. 2017. V. 787. P. 72.
Yang C., Song X., Sun S., Sun Z., Yu J. // Adv. Powder Technol. 2013. V. 24. P. 585.
Bricha M., Belmamouni Y., Essassi E.M., Ferreira J.M.F., Mabrouk K.E. // J. Nanosci. Nanotechnol. 2012. V. 12. P. 8042.
Wang A., Liu D., Yin H., Wu H., Wada Y., Ren M., Jiang T., Cheng X., Xu Y. // Mater. Sci. Eng. C. 2007. V. 27. P. 865.
Gaudin A.M., Fuerstenau D.W. // Trans. AIME. 1955. V. 7. P. 958.
Fuerstenau D.W. // J. Colloid Interface Sci. 2002. V. 256. P. 79.
Tari N.E., Motlagh M.M.K., Sohrabi B. // Mater. Chem. Phys. 2011. V. 131. P. 132.
Kannan S., Goetz-Neunhoeffer F., Neubauer J., Ferreira J.M.F. // J. Am. Ceram. Soc. 2008. V. 91. P. 1.
Shimabayashi S., Nishine S., Hino T., Uno T. // Phosphorus Res. Bull. 1998. V. 8. P. 49.
Donners J.J.J.M., Nolte R.J.M., Sommerdijk N.A.J.M. // Adv. Mater. 2003. V. 15. P. 313.
Jones S.B., Barbour M.E., Shellis R.P., Rees G.D. // Colloids Surf. B. 2014. V. 117. P. 193.
Bujan M., Sikirić M., Filipović-Vinceković N., Vdović N., Garti N., Füredi-Milhofer H. // Langmuir. 2001. V. 17. P. 6461.
Pon-On W., Meejoo S., Tang I.-M. // Mater. Chem. Phys. 2008. V. 112. P. 453.
Wang K., Wang Y., Zhao X., Li Y., Yang T., Zhang X., Wu X. // Mater. Sci. Eng. C. 2017. V. 75. P. 565.
Lim G.K., Wang J., Ng S.C., Gan L.M. // Langmuir. 1999. V. 15. P. 7472.
Zhong S., Wen Z., Chen J., Li Q., Shi X., Ding S., Zhang Q. // Mater. Sci. Eng. C. 2017. V. 77. P. 708.
Бондарева Е.В., Карпов С.И., Селеменев В.Ф., Вережников В.Н., Крохина Е.В. // Журн. физ. химии. 2009. Т. 83. С. 2339.
Alshemary A.Z., Goh Y.-F., Akram M., Razali I.R., Kadir M.R.A., Hussain R. // Mater. Res. Bull. 2013. V. 48. P. 2106.
Cheng Y., Yin L., Lin S., Wiesner M., Bernhardt E., Liu J. // J. Phys. Chem. C. 2011. V. 115. P. 4425.
Cappelletti G., Bianchi C.L., Ardizzone S. // Appl. Surf. Sci. 2006. V. 253. P. 519.
Решетняк Е.А., Чернышова О.С., Мчедлов-Петросян Н.О. // Коллоид. журн. 2016. Т. 78. С. 602.
Dunstan D.E., White L.R. // J. Colloid Interface Sci. 1990. V. 134. P. 147.
Усьяров О.Г. // Коллоид. журн. 2016. Т. 78. С. 634.
Binks B.P., Rodrigues J.A. // Langmuir. 2007. V. 23. P. 7436.
Мотякин М.В., Шулевич Ю.В., Захарова Ю.А., Вассерман А.М., Навроцкий А.В., Новаков И.А. // Коллоид. журн. 2009. Т. 71. С. 657.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал