

Коллоидный журнал, 2020, T. 82, № 4, стр. 501-516
Зародыши конденсированной фазы воды в поле вакансии на поверхности кристаллической подложки
С. В. Шевкунов *
Санкт-Петербургский политехнический университет Петра Великого
195251 Санкт-Петербург, ул. Политехническая, 29, Россия
* E-mail: shevk54@mail.ru
Поступила в редакцию 06.03.2020
После доработки 10.03.2020
Принята к публикации 11.03.2020
Аннотация
Вакансия в кристаллической решетке образована удалением аниона йода из ближайшего к поверхности кристаллографического слоя на базовой грани β-модификации йодистого серебра. Методом Монте-Карло исследована молекулярная структура зародышей конденсированной фазы воды, растущих в поле вакансии из пара при температуре 260 K. Полученные изображения зародышей и пространственные корреляционные функции в районе поверхностного дефекта свидетельствуют об относительно глубоком проникновении молекул воды в первые кристаллографические слои и разрыве водородных связей между первыми адсорбированными молекулами. Рост зародыша в поле вакансии сначала развивается в форме молекулярных цепочек, вытянутых в сторону от кристаллической поверхности, а затем продолжается в виде пятна мономолекулярной пленки, расползающегося по поверхности вокруг вакансии, причем механизм удержания пятна остается таким же, как и на бездефектной поверхности. Зародыши в поле вакансии представляют собой пятна мономолекулярной пленки с возвышающимися над ними вертикальными выбросами, напоминающими протуберанцы.
1. ВВЕДЕНИЕ
Адсорбция на кристаллических поверхностях лежит в основе широкого круга технологий – от химического катализа [1–3] и очистки газовых смесей [4] до воздействий на атмосферные осадки [5–8]. Взаимодействие паров воды с поверхностями занимает здесь особое место ввиду повсеместного присутствия воды в окружающей среде и ее широкого использования в качестве растворителя в химическом производстве и медицине. Вода известна как наиболее распространенная и в то же время уникальная по своим свойствам жидкость. Химическая нейтральность воды, обусловленная наличием в электронной оболочке ее молекул такого же количества электронов, как и в химически инертном газе неоне, в сочетании с относительно большим по величине собственным дипольным моментом молекул определяет высокое качество этой жидкости как растворителя. Все критические для живых организмов химические реакции происходят в водной среде.
Химическая прочность и электрические характеристики молекул воды определяют специфические свойства последней и во взаимодействии с ионными кристаллами. Благодаря своему относительно большому дипольному моменту молекулы воды прежде других частиц, входящих в состав атмосферы, втягиваются в неоднородное электрическое поле поверхности кристалла, а затем на контактных расстояниях образуют водородные связи с его ионами. В результате комбинации электростатических взаимодействий с сильно направленными водородными связями молекулы воды способны образовывать в контактном слое сильно упорядоченные структуры, напоминающие кристаллические [9–11], но, строго говоря, к кристаллическим не относящиеся. Фазовое состояние такого слоя нельзя отнести ни к жидкому, так как в нем ясно выражены дальний молекулярный порядок и замедленная диффузия, ни к кристаллическому, так как дальний молекулярный порядок зависит в нем от периодического поля подложки.
Контактный слой на границе воды и твердой кристаллической поверхности может обладать собственными уникальными свойствами, к числу которых относится явление доменообразования – образование областей спонтанной поляризации, внешне напоминающих области спонтанного намагничивания в ферромагнетиках, но с качественно иным механизмом формирования [12–15]. Контактный мономолекулярный слой воды на поверхности со структурой, комплементарной структуре льда, как правило, обладает выраженными гидрофобными свойствами, каковые представляют барьер для роста следующих слоев конденсата [16]. В подтверждение этого в [17] методом молекулярной динамики получено долгоживущее метастабильное состояние микрокапли воды, не растекающейся по поверхности мономолекулярной пленки, поддерживаемой твердой подложкой с гексагональной кристаллической структурой. Явление гидрофобности контактного мономолекулярного слоя воды подтверждено и в лабораторных экспериментах на поверхностях платины и палладия [18, 19].
В пионерских работах по изучению компьютерными методами неупорядоченных газообразных фаз в контакте с твердыми подложками главные усилия были направлены на исследование поверхностей без дефектов: в [20] моделировались частицы Леннард-Джонса на идеальной поверхности модельной подложки с гранецентрированной кристаллической решеткой, а в [21] – на гладкой модельной поверхности. Уже в первых работах усиленное внимание уделялось контакту твердых подложек с водой и парами воды. В [22] моделировалась адсорбция паров воды на бездефектной поверхности BaF2, в [23, 24] исследовалась структура пленки воды на поверхности графита, в [25–29] – на рутиле, TiO2, в [30] – на кварце, SiO2, в [31] – на кристаллическом оксиде церия CeO2, в [32] – на кристаллах Mg(OH)2, Mg3Si4O10(OH)2 и NaCl, в [33–38] – на платине, рутении и родии, а в [39] – на бездефектных полимерных пленках.
Способность молекул воды устанавливать прочные связи с ионами подложки может привести на поверхности некоторых кристаллов к диссоциации молекул. Возможность диссоциации на поверхностях подтверждена экспериментальными спектроскопическими данными [40–42]. Вероятность диссоциации адсорбированных на поверхностях молекул оценивают теоретически методом DFT (Density functional theory). В приближении DFT энергия системы записывается в виде объемного интеграла некоторой феноменологической формулы, в которую входит локальная плотность электронов. Предполагается, что пространственное распределение плотности, отвечающее минимуму этого функционала, близко к решению уравнения Шредингера. Хотя резонансные явления, составляющие основу дискретной природы электронных состояний в электронных оболочках атомов, находятся за рамками такого подхода и могут вноситься только как эмпирические поправки, метод DFT широко применяется для описания устойчивости химических связей и диссоциации молекул на различных твердых поверхностях. В частности, в [43] в приближении DFT моделировалась хемосорбция молекул воды на бездефектной поверхности MgO, в [44] – на поверхностях металлов Rh и Ni, а в [45] – на поверхности кристалла NaCl. Получены картины пространственного распределения молекул. Cделано заключение о диссоциированном состоянии молекул на поверхности окиси магния как более устойчивом, по сравнению с недиссоциированным.
Первые работы по моделированию адсорбции газов на неидеальных поверхностях были выполнены в начале двухтысячных и относились главным образом к протяженным дефектам типа ступеней [46, 47]. В меньшей степени исследовалось действие точечных поверхностных дефектов [10, 48]. К тому же времени относятся и первые работы по компьютерному моделированию воды в наноскопических полостях [49], клиновидных [50, 51] и плоских [52] разломах. В [53] методом молекулярной динамики моделировалось выдавливание воды из наноскопического плоского разлома между двумя поверхностями графита.
В современных публикациях продолжаются исследования молекулярных механизмов смачивания и формирования контактного угла воды на различных поверхностях. В [54] методом молекулярной динамики воспроизведены состояния микрокапли воды, лежащей на твердых поверхностях с различными типами кристаллической решетки. Обнаружено, что на подложках с кубической симметрией кристаллической решетки микрокапля воды лежит, не растекаясь, непосредственно на поверхности кристалла, а в случае гексагональной симметрии решетки поверхность кристалла покрывается мономолекулярным слоем воды, а капля лежит на этом слое.
В работах последнего времени возрос интерес к явлениям в контакте воды с графеном. Графен представляет собой устойчивый мономолекулярный слой, состоящий из шестизвенных циклов атомов углерода с гексагональной симметрией, такой же, как в поверхностном слое базовой грани β-модификации кристалла йодистого серебра и в кристаллической решетке льда Ih. В [55] контактный слой воды с графеном исследован методом молекулярной динамики. Получены пространственные распределения локальной плотности и корреляционные функции молекул. Отмечается формирование у поверхности графена двойного молекулярного слоя воды и преимущественная направленность одной из водородных связей молекулы в сторону графена, оценено поверхностное натяжение на границе с графеном.
Кроме контакта c чистой водой, в последнее время исследуется также переходный слой в контакте с водными растворами электролитов [56]. Структура контактного слоя анализируется в терминах корреляционных функций и распределений ориентаций водородных связей и дипольных моментов. К разряду редких исследований следует отнести опубликованную недавно работу по моделированию микрокапель воды на поверхности графена в сильном магнитном поле [57]. Обнаружена относительно слабая, но заметная на фоне флуктуаций зависимость угла смачивания от величины приложенного поля.
Последнее время усиленное внимание привлекает контакт воды с поверхностями, несущими функциональные группы, представляющие собой разновидности точечных дефектов. К таким исследованиям относится работа [58], в которой методом молекулярной динамики изучен контакт воды с поверхностью бурого угля (лигнита). Для адсорбированной воды приводятся данные о состоянии водородных связей, корреляционные функции и угловые распределения молекул. В [59] методом DFT анализируется контакт между водой и поверхностью минерала оливина (Mg,Fe)2[SiO4] с точечными включениями атомов Ca, Sr и Ba. Оценено влияние включений на энергетику связей с поверхностью.
В недавних работах можно наблюдать оживление интереса к переходному слою между водой и поверхностью наночастиц [60], хотя первые такие исследования выполнены еще в предыдущем десятилетии [61–63].
В естественных условиях, если не предпринимать специальных мер, формирующиеся кристаллические поверхности содержат дефекты разнообразных типов. Отклонения от идеальной поверхности могут иметь различные пространственные размеры и быть множественными (шероховатая поверхность), одиночными (точечные дефекты) или протяженными (ступени, разломы). Конкретная морфология адсорбирующей поверхности определяется условиями ее приготовления. Вариации последней позволяют контролировать свойства сорбентов и их активность в качестве стимуляторов нуклеации.
Йодистое серебро β-AgI [64, 65], являющееся в форме аэрозоля наиболее активным из известных агентов нуклеации атмосферной влаги при отрицательных температурах шкалы Цельсия, в форме макроскопических монокристаллов не проявляет каких-либо особенных свойств в контакте с водой, водой умеренно смачивается и в ней не растворяется. Аэрозоль йодистого серебра вводят в переохлажденную часть облака с помощью пиропатронов и ацетоновых горелок [66], подвешиваемых к фюзеляжу самолета, или подмешивая йодистое серебро в топливо метеорологических реактивных снарядов [67]. В пламени выхлопных газов происходит сублимация йодистого серебра с последующим быстрым охлаждением и конденсацией в микроскопические аэрозольные частицы, которые служат центрами конденсации переохлажденных паров атмосферной влаги и их кристаллизации с последующей коагуляцией в макроскопические поликристаллы льда и выпадением в виде града или дождя, если при падении под действием сил тяжести они успевают растаять.
Высокая эффективность аэрозольных частиц в качестве центров нуклеации определяется их способностью адсорбировать и удерживать атмосферную влагу на начальной стадии нуклеации, когда размер зародышей недостаточен для их замерзания. При ударном нагревании и быстром охлаждении на поверхности частиц йодистого серебра образуются кристаллические дефекты различного типа, которые неизбежно влияют на адсорбционную способность аэрозоля. После кристаллизации в лед зародыши быстро растут, так как в кристаллическом льду существуют направления, наиболее благоприятные для присоединения молекул, однако именно начальная стадия, предшествующая замерзанию жидкой микрокапли, оказывается решающей для успешного развития процесса в целом.
Пространственные размеры поверхностных дефектов радикально влияют на механизм взаимодействия с сорбатом и адсорбционную способность поверхности. Если размеры дефектов значительно превышают радиус межмолекулярных корреляций в жидкой фазе, составляющий для воды примерно 1 нм, шероховатость поверхности может на порядки величины увеличить эффективную площадь поверхности адсорбции, перейти в пористость и превратить адсорбцию на поверхности в абсорбцию в объеме. Тем не менее, такая крупнозернистая структура принципиально не влияет на молекулярные механизмы удержания сорбата.
Качественно иные условия реализуются, если поверхностные дефекты имеют наноскопические размеры. Точечные дефекты или фрагменты протяженных поверхностных дефектов, будучи источниками сильно неоднородного электрического поля с характерными размерами неоднородности, сравнимыми с молекулярными, могут служить “ловушками” для молекул сорбата, предоставляя таким образом благоприятные условия для нуклеации газовой фазы. В то же время, слишком сильное и неоднородное поле дефектов, если структура контактного слоя плохо вписывается в его геометрию, может оказать на нуклеацию противоположное действие, в результате чего рост зародыша конденсированной фазы останавливается, и на его пути формируется барьер свободной энергии [68].
Рельеф с характерными размерами элементов, измеряемыми нанометрами и долями нанометра, способен вызывать усиление как гидрофобных, так и гидрофильных свойств поверхности. Проникновение жидкости в наноскопические поры сопряжено с разрывом межмолекулярных связей в адсорбате. Вместе с разрушением межмолекулярных связей в области контакта кардинально изменяется молекулярная структура жидкости. Если эта трансформация сопровождается повышением свободной энергии, проникновения жидкости в нанопору не происходит, и формируется так называемое состояние Касси–Бакстера [69], а наноструктурирование приводит к усилению гидрофобности поверхности. В случае полного проникновения жидкости в нанопоры образуется состояние, которое обозначают как состояние Венцеля [70]. В таком состоянии площадь контакта с поверхностью за счет наноструктурирования увеличивается, а вместе с ней может увеличиваться и гидрофильность поверхности. Как первое, так и второе состояние могут быть долгоживущими метастабильными состояниями, разделенными высоким барьером свободной энергии [71–77].
Следует ожидать, что наиболее благоприятные условия микроконденсации на поверхности обеспечиваются некоторым удачным сочетанием пространственных размеров и структуры поверхностного дефекта. Не последнюю роль здесь играют коллективные эффекты в виде совместного действия нескольких или множественных дефектов, которые могут быть распределены на поверхности регулярным [78, 79] и нерегулярным [80] образом, иметь упорядоченную [81–83] или неупорядоченную [84] структуру.
В представленной работе исследуется структура зародыша конденсированной фазы, который растет в поле точечного дефекта, полученного удалением одного аниона йода из второго кристаллографического слоя базовой грани кристалла β-AgI. Первый (поверхностный) кристаллографический слой этой грани состоит из катионов серебра. В последующих слоях вглубь кристалла чередуются анионы йода и катионы серебра. В каждом таком слое реализуется характерное расположение ионов одного знака заряда с гексагональной структурой, близкой по своим пространственным параметрам к структуре льда Ih. Близость структуры базовой грани кристалла йодистого серебра к структуре льда рассматривается как одна из возможных, но не единственная причина высокой активности этого материала в качестве стимулятора нуклеации паров воды. Другими причинами могут служить высокая поляризуемость анионов йода, явление доменообразования в контактном слое [12, 14, 85] и действие кристаллических дефектов.
В [86] методом Монте-Карло моделировалось зародышеобразование в парах воды на поверхности кристалла β-AgI в поле поверхностного дефекта в виде лишнего катиона Ag+. В [80] методом молекулярной динамики исследовалось коллективное действие вакансий на базовой грани кристалла β-AgI на микрокристаллизацию воды в контактном слое. В продолжение начатых исследований в представленной работе для той же поверхности получены данные об особенностях в структуре зародышей конденсированной фазы в поле дефекта в виде недостающего аниона I–.
2. УСЛОВИЯ МОДЕЛИРОВАНИЯ
Главной целью представленных компьютерных расчетов было получение свободной энергии как функции размера зародышей конденсированной фазы. Структурные же характеристики, обсуждаемые в данной статье, явились “побочным” продуктом вычислений. Для расчета свободной энергии применен метод биканонического статистического ансамбля (БСА), развитый автором в предыдущих работах [62, 87]. В методе БСА моделируются два канонических статистических ансамбля с числом молекул N = n и N = n + 1 и случайные переходы между ними с вероятностями большого канонического статистического ансамбля [88]. Рассчитанные в этих условиях средние величины, включая структурные характеристики, являются средними по этим двум статистическим ансамблям и относятся не к дискретным значениям N, а к нецелым средним значениям 〈N〉 между n и n + 1. Фактически происходит интерполяция между соседними величинами N = n и N = n + 1 и таким образом расширение значений N на всю вещественную числовую ось.
Расчет произведен методом Монте-Карло и состоял в выполнении на компьютере последовательности шагов двух типов: сдвигов и поворотов случайно выбранных молекул без изменения их количества и вбрасывания в систему или удаления из нее одной случайно выбранной молекулы. Вероятности шагов разыгрывались с помощью программного генератора случайных чисел. Шаги первого типа переводят систему из одного в другое микросостояние (молекулярную конфигурацию) в пределах одного канонического ансамбля, а шаги второго типа переводят систему из одного в другой ансамбль. Шаги обоих типов чередовались: на один шаг первого типа приходилось шесть шагов второго типа. Хотя для расчета корреляционных функций шаги по изменению количества молекул в системе не являются необходимыми, они, уменьшая длину корреляций между генерируемыми микросостояниями, оказывают благоприятное действие на скорость сходимости к средним значениям и уменьшение статистической погрешности расчетов в целом.
Для расчета равновесных средних генерировались случайные процессы длиной $2 \times {{10}^{8}}$ шагов, первые $5 \times {{10}^{7}}$ из которых отводились на термализацию и не использовались в расчете средних значений. Максимальная длина сдвига молекулы за один шаг составляла 0.25 Å, максимальный угол поворота вокруг каждой из декартовых осей – 18°. Шаги первого типа заканчивались принятием новой конфигурации с частотой ≈60%, шаги второго – с частотой ≈0.5%. Применялась процедура последовательного наращивания размеров системы, когда последняя конфигурация, полученная в процессе с N = n, использовалась как начальная для термализации системы с N = n + 1.
Расчеты выполнены при температуре 260 K для базовой грани кристалла ß-AgI с катионами серебра в первом кристаллографическом слое. В плоскости адсорбции на систему накладывались периодические граничные условия. Область моделирования представляла собой прямоугольную ячейку периодичности размером в плоскости адсорбции 9.519 × 10.99 нм и в перпендикулярном направлении 1.8 нм. Применена модель взаимодействий, описанная в [89]. В пределах ячейки периодичности учитывались взаимодействия каждой молекулы с каждой и каждой молекулы с каждым ионом кристаллической решетки подложки. Вглубь кристалла учитывались взаимодействия с двенадцатью кристаллографическими слоями. Ближние взаимодействия суммировались методом ближайшего образа. Дальние электростатические взаимодействия в плоскости адсорбции учитывались двумерным методом Эвальда, представляющим собой суммирование Фурье-образов потенциала электрического поля для дальнодействующей части и суммирования в прямом пространстве для короткодействующей части [90]. Кроме традиционного суммирования электростатической части, методом Эвальда суммировались и поляризационные взаимодействия [91], а также суммировались поправки на дисперсионные взаимодействия с ионами кристаллической решетки подложки решетки в шести слоях соседних ячейках периодичности.
Кристаллическая решетка β-AgI обладает гексагональной симметрией. Элементарная ячейка решетки представлена на рис. 1 в [89]. Вакансия в кристаллической решетке получена удалением одного аниона йода из второго кристаллографического слоя кристаллической решетки подложки, при этом релаксация в положениях соседних ионов не производилась. Ионы оставлены в тех же позициях, которые они занимают в объемном кристалле. После удаления отрицательно заряженного иона область в районе вакансии несет избыточный положительный заряд, влияющий на образование зародыша конденсированной фазы. В данной публикации представлены результаты только в части структуры зародыша.
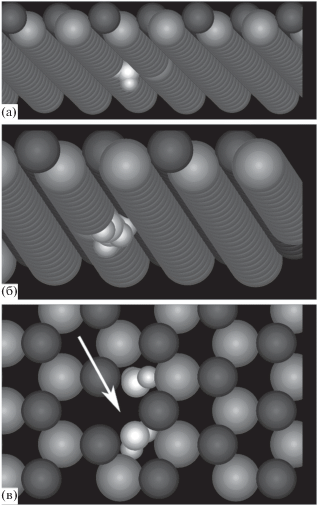
Рис. 1.
Компьютерные изображения одной (а) и двух (б, в) молекул пара, адсорбированных на поверхности кристаллического йодистого серебра в поле поверхностного дефекта, образованного удалением одного аниона йода из второго кристаллографического слоя, при температуре 260 K: (а, б) вид снизу на первые два кристаллографическиx слоя, (в) вид в направлении на адсорбирующую грань. Стрелка указывает положение вакансии, образованной удалением аниона.
3. МОДЕЛИРОВАНИЕ ОБМЕНА МОЛЕКУЛАМИ МЕЖДУ ПОВЕРХНОСТЬЮ И ПАРОМ
Расчеты в методе БСА выполняются в материальном и термическом контакте системы с виртуальным термостатом с заданным химическим потенциалом ${\tilde {\mu }}$. Расчет методом Монте-Карло состоит в последовательности случайных попыток вбрасывания или удаления из системы одной случайно выбранной молекулы, а также случайных попыток сдвинуть и повернуть одну случайно выбранную молекулу. Вероятности переходов разыгрываются с помощью генератора случайных чисел. Заданной температурой T, давлением пара p и величиной $\tilde {\mu } = \mu (p,T)$ определяются вероятности добавления молекул из термостата ${{p}_{{ij}}}$ или их изъятия из системы в термостат ${{p}_{{ji}}}$. Эти вероятности подчинены условию детального баланса и определены таким образом, чтобы обеспечить предельное равновесное распределение большого канонического статистического ансамбля ${{{\rho }}_{i}}$:
(1)
$\begin{gathered} \frac{{{{p}_{{ij}}}}}{{{{p}_{{j{\kern 1pt} i}}}}} = \frac{{{{{\rho }}_{j}}}}{{{{{\rho }}_{i}}}} = \frac{{\frac{1}{{n!}}{\kern 1pt} {{{(Z_{{{\text{tr}}}}^{{{\text{kin}}}})}}^{n}}{{{(Z_{{{\text{rot}}}}^{{{\text{kin}}}})}}^{n}}{\kern 1pt} \exp \left( {[n{\mu }(p,T)\, - \,{{{{U}_{j}}]} \mathord{\left/ {\vphantom {{{{U}_{j}}]} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right)\Delta \Omega \Delta V}}{{\frac{1}{{(n - 1)!}}{{{(Z_{{{\text{tr}}}}^{{{\text{kin}}}})}}^{{n - 1}}}{{{(Z_{{{\text{rot}}}}^{{{\text{kin}}}})}}^{{n - 1}}}\exp \left( {[(n - 1){\kern 1pt} {\mu }(p,T) - {{{{U}_{i}}]} \mathord{\left/ {\vphantom {{{{U}_{i}}]} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right){\kern 1pt} }} = \frac{1}{n}\exp \left( {\frac{{{{{\mu }}^{{{\text{conf}}}}}(p,T)\, - \,({{U}_{j}}\, - \,{{U}_{i}})}}{{{{k}_{{\text{B}}}}T}}} \right)v_{{{\text{ref}}}}^{{ - 1}}{\kern 1pt} \Delta \Omega {\kern 1pt} \Delta V = \\ = \frac{{\Delta \Omega }}{{\left( {\frac{{{\text{8}}{{{\pi }}^{{\text{2}}}}}}{{\sigma }}} \right)}}\frac{{\Delta V}}{V}\frac{V}{n}v_{{{\text{ref}}}}^{{ - 1}}\exp \left( {\frac{{{{{\mu }}^{{\text{c}}}}(p,T) - ({{U}_{j}} - {{U}_{i}})}}{{{{k}_{{\text{B}}}}T}}} \right) = \frac{{\Delta {\kern 1pt} \Omega }}{{\left( {\frac{{8{{\pi }^{2}}}}{\sigma }} \right)}}\frac{{\Delta V}}{V}\frac{{pV}}{{n{{k}_{{\text{B}}}}T}}\exp \left( { - \frac{{({{U}_{j}} - {{U}_{i}})}}{{{{k}_{{\text{B}}}}T}}} \right), \\ \end{gathered} $Вероятность $i$-го микросостояния, отвечающего элементарному конфигурационному объему ${\kern 1pt} {{(\Delta \Omega \Delta V)}^{n}}$, равна ${{{\rho }}_{i}} = {\rho }(n,q){\kern 1pt} {{(\Delta \Omega \Delta V)}^{n}}$, ${\rho }(n,q)$ – функция распределения в конфигурационном пространстве большого канонического статистического ансамбля, ${{v}_{{{\text{ref}}}}}$ – произвольная константа, имеющая размерность объема и задающая уровень отсчета конфигурационной части химического потенциала термостата, ${\kern 1pt} {{{\mu }}^{{\text{c}}}} \equiv {{{\mu }}^{{{\text{conf}}}}} - {{k}_{{\text{B}}}}T\ln \left( {\frac{{\sigma }}{{{\text{8}}{{{\pi }}^{{\text{2}}}}}}} \right)$, где ${\sigma } = 2$ – параметр вращательной симметрии молекулы воды. В последнем равенстве (1) конфигурационная часть химического потенциала записана в терминах отвечающего ему давления $p$идеального газа:
(2)
$\begin{gathered} {{{\mu }}^{{{\text{conf}}}}}(p,T) = {\mu }(p,T) + {{k}_{{\text{B}}}}T\ln \left( {Z_{{{\text{tr}}}}^{{{\text{kin}}}}Z_{{{\text{rot}}}}^{{{\text{kin}}}}{\kern 1pt} {{v}_{{{\text{ref}}}}}} \right) = \\ = {{k}_{{\text{B}}}}T\ln \left( {\frac{{\sigma }}{{{\text{8}}{{{\pi }}^{{\text{2}}}}}}\frac{p}{{{{k}_{{\text{B}}}}T}}{{v}_{{{\text{ref}}}}}} \right){\kern 1pt} . \\ \end{gathered} $Вероятности $\frac{{\Delta \Omega }}{{\left( {\frac{{{\text{8}}{{{\pi }}^{{\text{2}}}}}}{{\sigma }}} \right)}}$ и $\frac{{\Delta V}}{V}$ реализуются при вбрасывании молекулы в случайно выбранную точку области моделирования. Фактически расчет выполняется в непрерывном пространстве конфигураций в пределе $\Delta \Omega \to 0$ и $\Delta V \to 0$. Остальные множители в (1) разыгрываются с помощью генератора случайных чисел. Для этого сначала с помощью программы случайных чисел равновероятно назначаются точка внутри объема $V$, где поместится центр масс новой молекулы, и значения эйлеровых углов, задающих ориентацию молекулы. Значения первых двух эйлеровых углов выбираются равновероятно из интервалов $(0,{\pi })$ и $(0,2{\pi })$, а распределение по углу θ (углу между осью симметрии молекулы и осью z) задается пропорциональным $\sin ({\theta })$ в соответствии с выражением для якобиана перехода к сферическим координатам. Для этого из интервала $(0,{\pi })$ случайным образом равновероятно выбирается конкретное значение θ, и производится сравнение
где ξ1 – значение случайного числа, равномерно распределенного в интервале $(0,1)$. Если неравенство (3) не выполняется, попытка получить значение ${\theta }$ повторяется до тех пор, пока (3) не выполнится, при этом каждый раз численное значение ξ1 генерируется заново. Затем производится сравнение(4)
$\frac{V}{{{{v}_{{{\text{ref}}}}}}}\exp \left( {\frac{{{{{\mu }}_{{\text{c}}}} - ({{U}_{j}} - {{U}_{i}})}}{{{{k}_{{\text{B}}}}T}}} \right) > {{\xi }_{2}}n.$Если неравенство (4) выполняется, новая молекула добавляется в систему, при этом реализуется вероятность, равная ${1 \mathord{\left/ {\vphantom {1 n}} \right. \kern-0em} n}$, назначения молекуле конкретного номера. Если (4) не выполняется, в системе остается прежнее количество молекул. Попытка изъятия из системы одной молекулы начинается с назначения номера молекулы – претендента на удаление. Номер выбирается равновероятно с помощью программы случайных чисел. При этом реализуется вероятность, равная ${1 \mathord{\left/ {\vphantom {1 n}} \right. \kern-0em} n}$, назначения конкретного номера удаляемой частице. В процедуре добавления и изъятия молекулы считаются различимыми – состояния, отличающиеся нумерацией молекул, считаются различными. Квантово-механическая неразличимость молекул учитывается множителем ${1 \mathord{\left/ {\vphantom {1 {n{\kern 1pt} !}}} \right. \kern-0em} {n{\kern 1pt} !}}$ в равенстве (1).
На шаге изъятия молекулы случайным равновероятным образом выбирается номер удаляемой молекулы, генерируется равномерно распределенное в интервале $(0,1)$ случайное число ξ3 и производится сравнение
(5)
$n\exp \left( {\frac{{({{U}_{j}} - {{U}_{i}}) - {{{\mu }}_{{\text{c}}}}}}{{{{k}_{{\text{B}}}}T}}} \right) > {{\xi }_{3}}\frac{V}{{{{v}_{{{\text{ref}}}}}}}.$Если неравенство (5) выполняется, молекула удаляется из системы. В противном случае количество частиц не изменяется, и предыдущее микросостояние повторно учитывается при расчете средних.
Оптимальная с точки зрения скорости сходимости рассчитываемых средних задаваемая величина химического потенциала термостата ${\mu } = {\mu }(p,T)$ или соответствующее ему согласно формуле (2) значение давления $p$, отвечает условию ${{{\rho }}_{i}} \approx {{{\rho }}_{j}}$ и оценивается на начальном сегменте марковского случайного процесса методом итераций. Для этого применяется самонастраивающийся алгоритм, в котором параллельно с совершением шагов по вбрасыванию и удалению молекул производится плавная коррекция величины ${\mu }$ в ту или иную сторону с целью обеспечить равные средние частоты реализации микросостояний с N = n – 1 и N = n молекулами. Накопленная за предыдущие шаги статистика по этим микросостояниям учитывается с весовой функцией памяти, которая имеет вид такой затухающей зависимости, что влияние прошлых состояний учитывается с настолько уменьшенным весом, насколько дальше они отстоят от текущего состояния в последовательности конфигураций.
Поскольку в методе БСА разрешенное количество молекул ограничено как сверху, так и снизу, даже в отсутствие устойчивости, когда система в условиях контакта с паром должны была бы неограниченно расти или полностью испариться, в условиях моделирования методом БСА переполнения или испарения системы не происходит. Это позволяет воспроизводить не только устойчивые, но и термодинамически неустойчивые равновесные состояния. Первым отвечает минимум, а последним максимум работы образования как функции размера.
4. РЕЗУЛЬТАТЫ МОДЕЛИРОВАНИЯ
4.1. Локализация молекул в области вакансии
Удержание молекул воды на бездефектной поверхности осуществляется за счет водородных связей с анионами йода второго кристаллографического слоя [78]. Вакансия моделируется удалением из этого слоя одного аниона йода. Поскольку в месте образовавшейся вакансии локализован избыточный положительный заряд, следует ожидать, что молекулы воды будут втянуты в поле избыточного заряда, что должно дать стимулирующий толчок адсорбции и росту зародыша конденсированной фазы.
Полученные компьютерные изображения первых захваченных молекул свидетельствуют о глубоком проникновении молекул в кристаллическую решетку. На рис. 1а, 1б третий и четвертый слои не показаны, чтобы они не заслоняли вид снизу на первые два кристаллографических слоя. Видно, что молекулы проникли дальше первых двух слоев и оказались ориентированными своими атомами водорода вглубь кристалла, а на рис. 1в видно, что первые захваченные молекулы располагаются не в центре вакансии, а на ее периферии. Молекулы удерживаются, как и в случае бездефектной поверхности, связями атомов водорода с анионами второго кристаллографического слоя. Именно этим обусловлено их смещение к краю вакансии: в центре анион йода отсутствует. Поле избыточного положительного заряда вакансии оказывает дополнительное действие по втягиванию молекул вглубь кристалла, так что молекулы оказываются ниже поверхностного кристаллографического слоя подложки.
Таким образом, избыточный положительный заряд вакансии приводит к втягиванию молекул вглубь кристалла, однако удерживаются они на подложке так же, как и на подложке без вакансий – за счет связей с анионами йода второго кристаллографического слоя. Наличие вакансии в виде удаленного аниона йода не изменило механизм удержания молекул, но усилило их погружение в кристалл на глубину, превышающую толщину одного кристаллографического слоя.
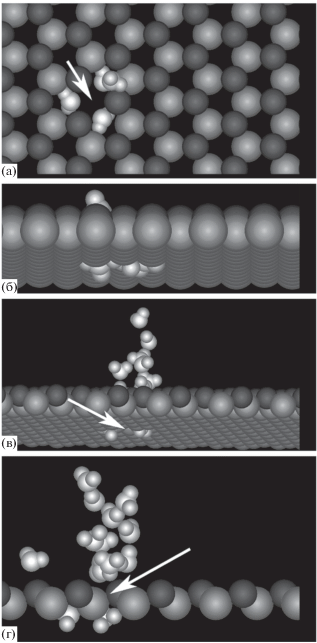
Третья молекула удерживается так же, как и первые две, на периферии вакансии за счет связей ее атомов водорода с анионами йода второго кристаллографического слоя, рис. 2а. Все первые три молекулы образуют треугольник, в вершинах которого они располагаются. Четвертая молекула присоединяется не к подложке непосредственно, а к одной из трех захваченных молекул. На рис. 2б видно, что первые три молекулы втянуты под первый кристаллографический слой, а четвертая располагается существенно выше над ними.
Рис. 2.
Изображения 4 (а, б), 10 (в) и 14 (г) молекул пара в тех же условиях, что на рис. 1: (а) вид в направлении, перпендикулярном к адсорбирующей поверхности, (б, в, г) вид сбоку к адсорбирующей поверхности, показаны только первые два кристаллографических слоя. Поверхность адсорбции расположена сверху.
Следующие молекулы тоже не связываются с подложкой непосредственно, а образуют цепочку, вытянутую в направлении, перпендикулярном к подложке, рис. 2в, причем ориентация дипольных моментов молекул в них в направлении от поверхности подложки соответствует направлению силовых линий избыточного положительного заряда вакансии. Формирование аналогичных сильно поляризованных цепочек в сильных локальных электрических полях в плоских порах наблюдалось в [92]. Сильно вытянутая вдоль силовых линий форма таких структур обеспечивается еще и тем обстоятельством, что молекулы с параллельными дипольными моментами отталкиваются, что препятствует их слиянию в более толстые цепочки.
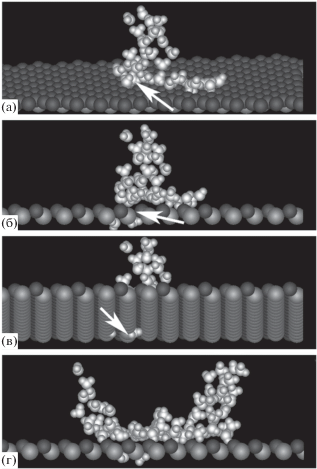
На последующих стадиях роста зародыша прежде, чем начнется “расползание” молекул по поверхности кристалла, в области вакансии формируются вытянутые в сторону от поверхности подложки структуры высотой в 5–7 молекулярных размеров, рис. 3а–3в. В основании этих образований находятся вакансия и проникшие вглубь кристалла молекулы, а над ними – до 2–3 молекулярных “протуберанцев”, рис. 3г.
Рис. 3.
Изображения 40 (а, б, в) и 84 (г) молекул пара в тех же условиях, что на рис. 1: (а) вид в направлении на адсорбирующую поверхность, (в) под двумя первыми кристаллографическими слоями, (б, г) вид с торца адсорбирующей поверхности. Показаны только первые два кристаллографических слоя.
Расползание молекул по поверхности в области вакансии начинается после накопления в зародыше более 30–40 молекул, рис. 4а–4в, при этом “протуберанцы” могут смещаться относительно вакансии на расстояния в несколько молекулярных размеров, рис. 4в. В результате расползания формируется пятно мономолекулярной пленки, состоящее из шестизвенных циклов с катионами серебра в центре. Молекулы пленки удерживаются на подложке за счет водородных связей молекул с анионами серебра второго кристаллографического слоя. В идеальной конфигурации три атома водорода каждого такого цикла задействованы в связях с подложкой, еще шесть атомов обеспечивают устойчивость цикла как целого, а оставшиеся три – связи с соседними циклами. В результате термических флуктуаций эта идеальная картина подвергается разрушению и восстановлению. Шестизвенные циклы присутствуют главным образом во внутренней области пятна. На его периферии превалируют пятизвенные циклы. Следует отметить, что и в отсутствие кристаллических дефектов на базовой грани кристалла пятизвенные циклы обладают более высокой устойчивостью, чем шестизвенные [78].
Рис. 4.
Изображения 26 (а), 40 (б), 84 (в) и 307 (г) молекул пара в тех же условиях, что на рис. 1. Вид в направлении, перпендикулярном к адсорбирующей поверхности.
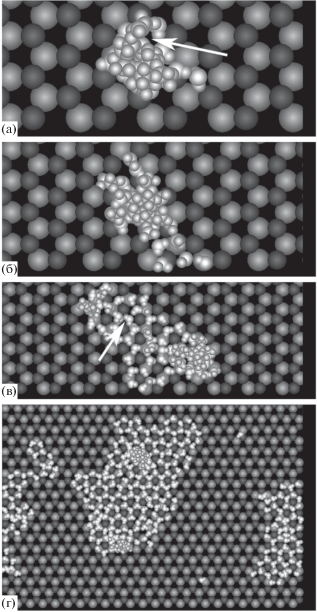
При превышении размера зародыша в 100–200 молекул он распадается на несколько образований, причем в тех образованиях, которые не связаны с вакансией, “протуберанцы” отсутствуют, рис. 4г. Таким образом, следует заключить, что электрическое поле вакансии способно стимулировать молекулярные “выбросы” над адсорбирующей поверхностью не обязательно непосредственно над вакансией, но и в стороне от нее, но над пятном, которое покрывает вакансию.
4.2. Пространственные корреляции молекул относительно подложки
Адсорбированные на поверхности частицы вовлечены в хаотическое тепловое движение. Статичные компьютерные изображения поверхности не дают исчерпывающего представления о структуре молекулярных комплексов с учетом флуктуаций. Сведения о внутреннем устройстве системы в таких условиях содержатся в усредненных пространственных распределениях и корреляционных функциях молекул.
Пространственная корреляционная функция катионы серебра–атомы кислорода молекул ${{{\rho }}_{{ + {\text{O}}}}}(r)$ имеет смысл усредненной по направлениям и тепловым флуктуациям локальной плотности молекул воды на расстоянии $r$ от произвольно выбранного катиона серебра кристаллической решетки подложки. Функция ${{{\rho }}_{{ + {\text{O}}}}}(r)$ нормирована следующим образом: $\int_0^\infty {{{{\rho }}_{{ + {\text{O}}}}}(r)4{\pi }r_{{}}^{2}d{\kern 1pt} r} = \left\langle N \right\rangle $. На рис. 5 представлены корреляционные функции ${{{\rho }}_{{ + {\text{O}}}}}(r)$ на начальной стадии адсорбции молекул пара на бездефектной поверхности базовой грани кристалла йодистого серебра (кривые 1) и на той же грани в поле вакансии (кривые 2).
Рис. 5.
Пространственная корреляционная функция катионы серебра кристаллической решетки подложки–атомы кислорода молекул воды ${{{\rho }}_{{ + {\text{O}}}}}$ при температуре 260 К на базовой кристаллической поверхности β-AgI на различных стадиях роста зародыша: (а) 〈N〉 = = 2.5, (б) 9.5, (в) 39.5; 1 – на поверхности без дефектов, 2 – на поверхности с дефектом в виде вакансии на месте аниона серебра во втором кристаллографическом слое.
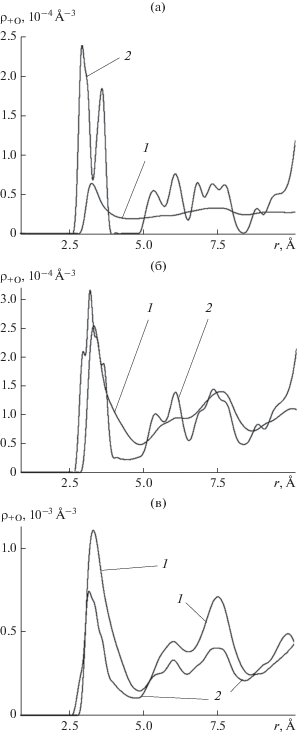
Из сравнения видно, что пространственное распределение молекул около катионов серебра в области вакансии гораздо менее однородно, чем на идеальной поверхности: корреляционная функция в поле вакансии имеет два острых максимума в области контактных расстояний и сохраняет нерегулярное поведение на расстояниях порядка 2–3-х молекулярных размеров. Резкие максимумы на контактных расстояниях отражают вклад первых трех молекул, втянутых в область вакансии, как это можно наблюдать на рис. 1в и 2а. Электрическое поле вакансии способствует сближению этих молекул с соседними катионами серебра, прочно закрепляя их в вершинах правильного треугольника с центром в районе узла, откуда удален анион йода, рис. 2а.
Если в начале поглощения плотность молекул около катионов серебра в поле вакансии выше, чем на бездефектной поверхности, рис. 5а, 5б, то с накоплением молекул их плотность около положительных ионов понижается в сравнении с плотностью на поверхности без дефектов, рис. 5в. Такое поведение корреляционной функции ${{{\rho }}_{{ + {\text{O}}}}}(r)$ является отражением того факта, что в поле вакансии молекулярный кластер не сразу расползается по поверхности адсорбции, а сначала растет в вертикальном к поверхности направлении (рис. 2г и 3), образуя характерный “протуберанец”. Молекулы в теле этого образования оказываются дальше от адсорбирующей поверхности и не дают вклад в локальную плотность на контактных расстояниях от катионов кристаллической решетки подложки.
В целом, форма кривой ${{{\rho }}_{{ + {\text{O}}}}}(r)$ в случае поверхности с вакансией при наращивании количества адсорбированных молекул приближается к форме аналогичной кривой для бездефектной поверхности: сходство становится очевидным, когда количество адсорбированных молекул превышает 30–40, рис. 5в.
Если бы между молекулами отсутствовали пространственные корреляции, то форма пространственных корреляционных функций ионы–молекулы при наращивании количества молекул оставалась бы неизменной: происходило бы только масштабирование кривой по оси ординат. Изменение формы кривой при варьировании количества молекул является мерой пространственных корреляций между ними. Сравнение показывает, что форма кривой 1, отвечающей бездефектной поверхности, меняется от рис. 5а к рис. 5в в сторону усложнения, в то время как форма кривой 2, полученной при наличии вакансии, наоборот, при увеличении количества адсорбированных молекул становится более регулярной, что означает в среднем ослабление межмолекулярных корреляций. Из этих наблюдений можно сделать общее заключение, что на бездефектной поверхности с ростом количества адсорбированных молекул корреляции между ними усиливаются, а на поверхности с дефектом в виде вакансии, наоборот, ослабевают. Эта закономерность указывает на сильные межмолекулярные корреляции в поле вакансии в самом начале адсорбции и объясняется повышенной неоднородностью поля в области вакансии – первые захваченные этим полем молекулы оказываются жестко фиксированными в соответствующих позициях относительно кристаллической решетки подложки и между собой.
Пространственная корреляционная функция ${{{\rho }}_{{ - {\text{O}}}}}(r)$ нормирована так же и имеет тот же смысл, что и в случае корреляционной функции ${{{\rho }}_{{ + {\text{O}}}}}(r)$, с точностью до замены катиона серебра на анион йода. Анион йода и атом кислорода молекулы воды несут одинаковые по знаку отрицательные электрические заряды. Следует ожидать, что силы отталкивания между этими зарядами приведут к смещению максимумов корреляционной функции ${{{\rho }}_{{ - {\text{O}}}}}(r)$ на контактных расстояниях к бóльшим значениям, по сравнению с аналогичными максимумами на кривой ${{{\rho }}_{{ + {\text{O}}}}}(r)$. Такое смещение, действительно, наблюдается, рис. 6.
Рис. 6.
Корреляционная функция анионы йода кристаллической решетки–атомы кислорода молекул воды ${{{\rho }}_{{ - {\text{O}}}}}$ в тех же условиях, что и на рис. 5, на тех же стадиях роста.
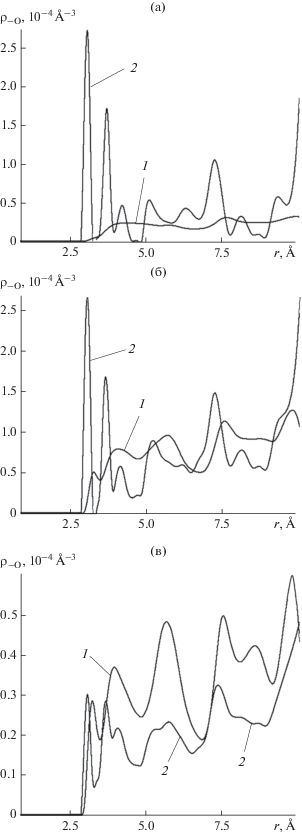
Первые два максимума на ${{{\rho }}_{{ - {\text{O}}}}}(r)$ – более узкие, рис. 6а, 6б, что свидетельствует о более жесткой фиксации молекул в радиальном направлении относительно анионов йода, по сравнению с катионами серебра. Это наблюдение подтверждает тот факт, что в области вакансии молекулы удерживаются подложкой за счет прямых сцеплений с анионами йода, а не с катионами серебра, то есть, механизм удержания молекул здесь тот же, что и на бездефектной поверхности. Как и на идеальной кристаллической поверхности базовой грани, в области вакансии разрушительному действию термических флуктуаций противостоят водородные связи молекул с отрицательными ионами второго кристаллографического слоя подложки.
4.3. Водородные связи между молекулами воды
Корреляционная функция атом кислорода–атом водорода молекул ${{{\rho }}_{{{\text{OH}}}}}(r)$ нормирована следующим образом: $\int_0^\infty {{{{\rho }}_{{{\text{OH}}}}}(r)4{\pi }{\kern 1pt} r_{{}}^{2}dr} = \left\langle {N - 1} \right\rangle $; она имеет смысл усредненной по направлениям локальной объемной плотности атомов кислорода молекул воды на расстоянии $r$ от атома водорода произвольно выбранной молекулы. Первый слева максимум этой корреляционной функции соответствует водородной связи с ближайшей соседней молекулой, а площадь под максимумом функции ${{{\rho }}_{{{\text{OH}}}}}(r)4{\pi }{\kern 1pt} r_{{}}^{2}$ определяет среднее количество водородных связей, приходящихся на один атом водорода. Второй максимум формируется атомом кислорода молекулы, присоединившейся ко второму атому водорода. Оба максимума ярко выражены в силу относительной прочности водородных связей на фоне термических флуктуаций при комнатной температуре. Положение первого максимума указывает на длину водородной связи, а его резкое смещение к большим расстояниям (кривая 2 на рис. 7а) свидетельствует о разрыве последней. Захваченные полем вакансии первые три молекулы воды в пространстве жестко фиксированы, рис. 1в. Об этом свидетельствуют контрастные максимумы на кривых 2 на рис. 7а, 7б.
Рис. 7.
Корреляционная функция атом кислорода–атом водорода молекул воды ${{{\rho }}_{{{\text{OH}}}}}$ в тех же условиях, что и на рис. 5, на различных стадиях роста зародыша: (а) 〈N〉 = 2.5, (б) 3.5, (в) 4.5.
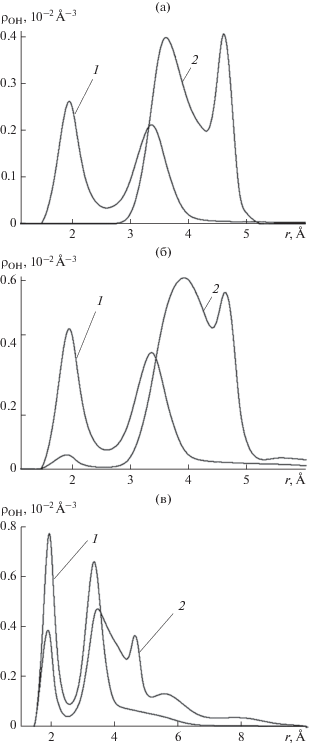
Захват четвертой и пятой молекул сопровождается формированием на кривой 2 максимума в районе r = 2 Å, указывающего на образование водородных связей, рис. 7в. Водородные связи с первыми тремя молекулами образуют четвертая и последующие молекулы, рис. 2а, 2б, а также остальные молекулы, выстраиваясь в цепочку, вытянутую вдоль нормали к поверхности, рис. 2в, 2г.
Таким образом, на рис. 7в кривая 2 обретает три максимума: первый слева свидетельствует об образовании водородных связей, второй максимум является результатом слияния двух максимумов – одного, образующегося за счет присоединения молекулы ко второму атому водорода, и другого – за счет первых трех молекул, жестко фиксированных в поле вакансии, третий же максимум обязан своим существованием первым трем молекулам, как на рис. 7б.
Из формы корреляционных функций следует, что сильное электрическое поле вакансии разрывает водородные связи между первыми тремя адсорбированными молекулами и разводит их на расстояние примерно в 4 Å. Водородные связи между молекулами формируются только, начиная с захвата четвертой молекулы. Когда размер кластера превышает десять молекул, в перпендикулярном направлении к поверхности выстраивается цепочка связанных между собой водородными связями молекул, относительный вклад первых трех адсорбированных молекул уменьшается, и корреляционная функция приобретает такую же форму с характерными двумя контрастными максимумами, как и на бездефектной поверхности, рис. 8.
Рис. 8.
Корреляционная функция атом кислорода–атом водорода молекул воды ${{{\rho }}_{{{\text{OH}}}}}$ в тех же условиях, что и на рис. 7, на последующих стадиях роста зародыша: (а) 〈N〉 = 9.5, (б) 13.5, (в) 39.5.
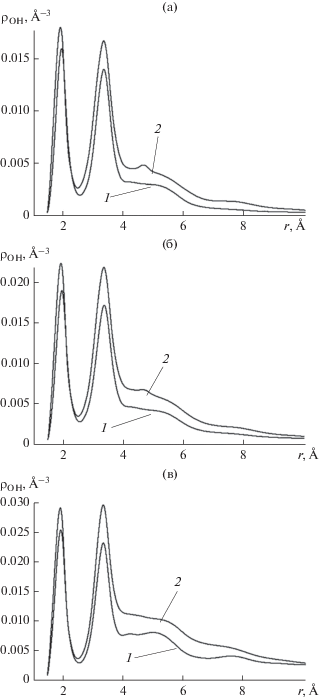
На рис. 7в первый максимум кривой 2 ниже соответствующего максимума на кривой 1, а после увеличения количества молекул в кластере, на рис. 8, наоборот, первый максимум кривой 2 оказывается выше соответствующего максимума кривой 1. Это означает, что в молекулярных цепочках, растущих в поле вакансии в направлении от адсорбирующей поверхности, вероятность образования водородных связей выше, чем на поверхности без дефектов. Объясняется этот эффект тем, что на бездефектной поверхности адсорбированные молекулы образуют мономолекулярный слой. Часть атомов водорода молекул в таком слое оказывается задействованной в образовании связей с анионами серебра второго кристаллографического слоя подложки. Эти атомы водорода оказываются исключенными из сетки водородных связей между молекулами – вероятность образования таких связей уменьшается, и первый максимум на кривой 1 на рис. 8 оказывается ниже, чем на кривой 2. Именно дефицитом молекул-доноров водородных связей в пленке на подложке со структурой, комплементарной структуре льда, объясняется гидрофобность поверхности пленки и торможение тоста следующих слоев. Выстраивание же молекул воды в цепочку над вакансией оказывает благоприятное действие на образование водородных связей.
Общая картина воздействия вакансии на образование водородных связей оказывается неординарной: на начальной стадии поле вакансии разрушает водородные связи между молекулами, а затем, наоборот, способствует их образованию.
4.4. Водородные связи с ионами кристаллической решетки подложки
Как установлено в предыдущих исследованиях [93], в отсутствие поверхностных кристаллических дефектов молекулы мономолекулярной пленки удерживаются на подложке за счет связующих взаимодействий с анионами йода. Атомы водорода молекул проникают вглубь кристалла, ко второму кристаллографическому слою и образуют с его анионами водородные связи. Взаимодействия же молекул с катионами поверхностного кристаллографического слоя носит разрыхляющий характер. Энергия взаимодействия с анионами отрицательна и по абсолютной величине превышает положительную энергию взаимодействия с катионами. Разность в абсолютных величинах на порядок величины меньше, чем каждое из этих двух составляющих, но именно она обеспечивает отрицательный знак результирующей энергии и связующий характер взаимодействия с подложкой в целом.
Нарастание относительного вклада водородных связей при увеличении количества адсорбированных молекул можно проследить по росту первого максимума на кривой 1 в сравнении со значениями на остальной части кривой ${{{\rho }}_{{ - {\text{H}}}}}(r)$ на рис. 9. Видно, что с ростом количества молекул максимум на расстоянии r ≈ 2 Å непрерывно растет не только по абсолютной величине, но и по сравнению со вторым и последующими максимумами. Это означает, что на поверхности без дефектов на начальной стадии адсорбции действие водородных связей в удержании молекул является относительно слабым и по мере накопления молекул и формирования мономолекулярной пленки усиливается.
Рис. 9.
Пространственная корреляционная функция анионы йода кристаллической решетки подложки–атомы водорода молекул воды ${{{\rho }}_{{ - {\text{H}}}}}$ в тех же условиях, что на рис. 5: (а) 〈N〉 = 2.5, (б) 9.5, (в) 39.5.
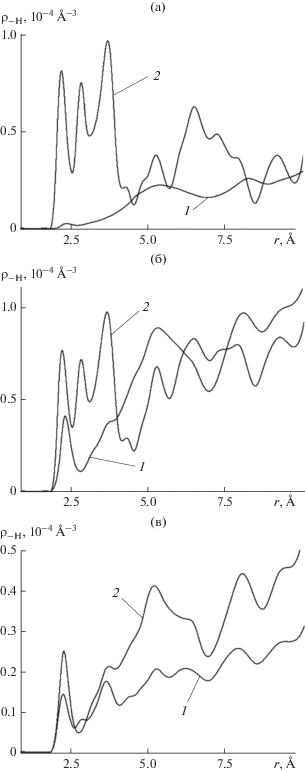
Противоположная картина наблюдается в области вакансии, кривые 2 на рис. 9. Здесь на начальной стадии максимум ${{{\rho }}_{{ - {\text{H}}}}}(r)$ на расстоянии r ≈2 Å – самый мощный, рис. 9а, но с накоплением адсорбированных молекул понижается в сравнении с остальной частью кривой, рис. 9б, и приближается к величине, характерной для бездефектной подложки, рис. 9в. Такое поведение корреляционной функции ${{{\rho }}_{{ - {\text{H}}}}}(r)$ указывает на высокую вероятность образования водородных связей с анионами подложки на начальной стадии и понижение среднего удельного количества таких связей в расчете на молекулу после присоединения следующих молекул. Такой результат представляется неожиданным, если учесть, что вакансия образована удалением из кристаллической решетки подложки аниона йода, за счет водородных связей с которыми и обеспечивается удержание молекул на поверхности кристалла. Удаление одного иона привело к упрочнению связей с оставшимися ионами решетки того же знака заряда.
При сравнении формы кривых 2 на рис. 9 обращает на себя внимание тот факт, что в состоянии с малым числом адсорбированных молекул, рис. 9а, 9б, рядом с первым максимумом, отвечающем водородной связи, присутствуют еще два максимума сопоставимой величины. По мере накопления адсорбированных молекул они исчезают, рис. 9в. Эти два дополнительных максимума расположены на расстояниях, превышающих длину водородной связи, и поэтому к ней не относятся, однако их присутствие говорит о таком расположении молекул воды в области вакансии, при котором атомы водорода оказываются на относительно близких расстояниях от соседних анионов йода. Возможность приблизиться к анионам второго кристаллографического слоя в районе вакансии появилась у молекул благодаря свободному пространству на месте удаленного аниона йода. Именно оно позволило молекулам проникнуть ниже первого кристаллографического слоя, как это можно видеть на рис. 1–3. Взаимодействие атомов водорода молекул с анионами на таких расстояниях носит преимущественно электростатический характер и обеспечивает дополнительные силы удержания молекул в их позициях. Благодаря сильной направленности этих взаимодействий ориентация молекул в поле вакансии оказывается жестко фиксированной.
5. ЗАКЛЮЧЕНИЕ
Неоднородное поле избыточного положительного заряда вакансии в кристаллической решетке подложки обеспечивает втягивание молекул воды в область кристаллического дефекта, однако удерживаются адсорбированные молекулы пара так же, как и на бездефектной поверхности, за счет водородных связей с анионами йода второго кристаллографического слоя. Вследствие сильной направленности водородных связей ориентация этих молекул жестко фиксирована.
Первые адсорбированные в области вакансии молекулы проникают в структуру подложки относительно глубоко, так что часть атомов водорода молекул оказывается ниже второго кристаллографического слоя. Проникновение в поверхностные слои обеспечивает в области дефекта жесткую пространственную локализацию первых трех молекул.
Следующие молекулы пара присоединяются не непосредственно к подложке, а к адсорбированным молекулам, образуя цепочку, растущую в перпендикулярном к подложке направлении. За счет ориентации дипольных моментов молекул вдоль силовых линий электрического поля вакансии цепочка сильно поляризована.
Только после достижения размера кластера в несколько десятков молекул рост цепочки сменяется “расползанием” адсорбированных молекул по поверхности кристалла вокруг вакансии. Расползающиеся по поверхности молекулы формируют пятно мономолекулярной пленки, состоящей из шестизвенных циклов. Структура пленки внешне напоминает пчелиные соты. На краях пятна присутствуют и пятизвенные циклы. Механизм удержания пленки на поверхности такой же, как и на бездефектной кристаллической поверхности, – за счет водородных связей с анионами йода.
На бездефектной поверхности с ростом количества адсорбированных молекул корреляции между ними усиливаются, а на поверхности с дефектом в виде вакансии, наоборот, ослабевают. Эта закономерность указывает на сильные межмолекулярные корреляции в поле вакансии в самом начале адсорбции. Пространственные радиальные корреляции молекул воды относительно анионов йода сильнее, чем относительно катионов серебра, что является следствием механизма удержания молекул за счет прямых связей с анионами йода.
Поле вакансии разрывает водородные связи между молекулами на начальной стадии адсорбции, а затем, наоборот, умеренно стимулирует их образование. Стимулирование водородных связей происходит на стадии роста молекулярных “протуберанцев” над вакансией и продолжается до начала расползания молекул по поверхности. В молекулярных цепочках, растущих над поверхностью подложки в поле вакансии, все атомы водорода могут быть задействованы в образовании водородных связей между молекулами, в то время как в мономолекулярной пленке на поверхности подложки часть атомов водорода участвует в водородных связях, обеспечивающих сцепление с подложкой.
В целом, поле вакансии в кристаллической решетке подложки в виде удаленного аниона йода кардинально влияет на форму зародыша конденсированной фазы, заставляя его расти не вдоль поверхности, как в отсутствие дефекта, а в перпендикулярном направлении. В месте расположения вакансии образуется характерный выброс, напоминающий “протуберанец”. Качественно меняется и механизм формирования водородных связей между молекулами в зародыше. Все эти особенности не могут не влиять на термодинамическое поведение адсорбата, его устойчивость и скорость роста.
Список литературы
Романовский Б.В. Основы катализа. М.: БИНОМ, 2012.
Чоркендорф И., Наймантсведрайт Х. Современный катализ и химическая кинетика. Долгопрудный: ИД “Интеллект”, 2010.
Колесников И.М. Катализ и производство катализаторов. М.: Техника, 2004.
Родионов А.И., Клушин В.Н., Систер В.Г. Технологические процессы экологической безопасности. Калуга: Издательство Н. Бочкаревой, 2000.
Heymsfield A.J., Kennedy P.C., Massie S., Schmitt C., Wang Z., Haimov S., Rangno A. // Bull. Amer. Meteor. Soc. 2010. V. 91. P. 753.
Mather G.K., Terblanche D.E., Steffens F.E., Flether L. // J. Appl. Meteorol. 1997. V. 36. P. 1433.
Gabriel K.R., Rosenfeld D. // J. Appl. Meteorol. 1990. V. 29. P. 1055.
Gabriel K.R., Avichai Y., Steinberg R. // J. Appl. Meteorol. 1967. V. 6. P. 323.
Шевкунов С.В. // Докл. АН 2005. Т. 402. С. 41.
Шевкунов С.В. // Журн. физ. химии 2005. Т. 79. С. 1860.
Шевкунов С.В. // Коллоид. журн. 2006. Т. 68. С. 391.
Шевкунов С.В. // Докл. АН. 2011. Т. 438. С. 752.
Шевкунов С.В. // Коллоид. журн. 2012. Т. 74. С. 612.
Шевкунов С.В. // Коллоид. журн. 2012. Т. 74. С. 634.
Шевкунов С.В. // Журн. физ. химии. 2013. Т. 87. С. 1678.
Шевкунов С.В. // Журн. физ. химии. 2006. Т. 80. С. 884.
Wang C., Lu H., Wang Z., Xiu P., Zhou B., Zuo G., Wan R., Hu J., Fang H. // Phys. Rev. Lett. 2009. V. 103. P. 137801.
Kimmel G.A., Petrik N.G., Dohnálek Z., Kay B.D. // J. Chem. Phys. 2006. V. 125. P. 044713.
Kimmel G.A., Petrik N. G., Dohnálek Z., Kay B.D. // J. Chem. Phys. 2007. V. 126. P. 114702.
Patrykiejew A., Sokołowski S., Binder K. // J. Chem. Phys. 2002. V. 117. P. 3369.
Toxvaerd S. // J. Chem. Phys. 2002. V. 117. P. 10303.
Nutt D.R., Stone A.J. // J. Chem. Phys. 2002. V. 117. P. 800.
Gordillo M.C., Marti J. // J. Chem. Phys. 2002. V. 117. P. 3425.
Jaffe R.L., Gonnet P., Werder T., Walther J.H., Koumoutsakos P. // Mol. Simulat. 2004. V. 30. P. 205.
Zhang C., Lindan P.J.D. // J. Chem. Phys. 2003. V. 118. P. 4620.
Jug K., Nair N.N., Bredow T. // Surf. Sci. 2005. V. 590. P. 9.
Skelton A.A., Walsh T.R. // Mol. Simulat. 2007. V. 33. P. 379.
Perron H., Vandenborre J., Domain C., Drot R., Roques J., Simoni E., Ehrhardt J.-J., Catalette H. // Surf. Sci. 2007. V. 601. P. 518.
Wahab H.S., Bredow T., Aliwi S.M. // J. Mol. Structure: THEOCHEM. 2008. V. 868. P. 101.
Puibasset J., Pellenq R.J.-M. // J. Chem. Phys. 2003. V. 119. P. 9226.
Kumar S., Schelling P.K. // J. Chem. Phys. 2006. V. 125. P. 204704.
Sakuma H., Tsuchiya T., Kawamura K., Otsuki K. // Mol. Simulat. 2004. V. 30. P. 861.
Konrad O., Lankau T. // Chem. Phys. Lett. 2002. V. 359. P. 35.
Lilach Y., Buch V., Asscher M. // J. Chem. Phys. 2003. V. 119. P. 11899.
Daschbach J.L., Peden B.M., Smith R.S., Kay B.D. // J. Chem. Phys. 2004. V. 120. P. 1516.
Vassilev P., van Santen R.A., Koper M.T.M. // J. Chem. Phys. 2005. V. 122. P. 054701.
Karlberga G.S., Wahnström G. // J. Chem. Phys. 2005. V. 122. P. 194705.
Meng S. // Surf. Sci. 2005. V. 575. P. 300.
Hirvi J.T., Pakkanen T.A. // J. Chem. Phys. 2006. V. 125. P. 144712.
Tiferet E., Zalkind S., Mintz M.H., Jacob I., Shamir N. // Surf. Sci. 2007. V. 601. P. 936.
Zhao X., Ma S., Hrbek J., Rodriguez J.A. // Surf. Sci. 2007. V. 601. P. 2445.
Kondo T., Kato H. S., Bonn M., Kawai M. // J. Chem. Phys. 2007. V. 127. P. 094703.
Jug K., Heidberg B., Bredow T. // Surf. Sci. 2007. V. 601. P. 1529.
Pozzo M., Carlini G., Rosei R., Alfè D. // J. Chem. Phys. 2007. V. 126. P. 164706.
Cabrera-Sanfelix P., Arnau A., Darling G.R., Sanchez-Portal D. // J. Chem. Phys. 2007. V. 126. P. 214707.
Schneemilch M., Quirke N. // Mol. Simulat. 2003. V. 29. P. 685.
Gong X.-Q., Selloni A. // J. Catalysis. 2007. V. 249. P. 134.
Ma Y., Foster A.S., Nieminen R.M. // J. Chem. Phys. 2005. V. 122. P. 144709.
Hendy S.C. // Curr. Appl. Phys. 2004. V. 4. P. 144.
Шевкунов С.В. // Коллоид. журн. 2007. Т. 69. С. 390.
Шевкунов С.В. // Коллоид. журн. 2007. Т. 69. С. 409.
Шевкунов С.В. // Журн. физ. химии. 2014. Т. 88. С. 326.
Shah K., Chiu P., Sinnott S.B. // J. Colloid and Interface Sci. 2006. V. 296. P. 342.
Qi C., Lei X., Zhou B., Wang C., Zheng Y. // J. Chem. Phys. 2019. V. 150. 234703.
Dreher T., Lemarchand C., Pineau N., Bourasseau E., Ghoufi A., Malfreyt P. // J. Chem. Phys. 2019. V. 150. P. 014 703.
Škvára J., Nezbeda I. // Mol. Simulat. 2019. V. 45. P. 358.
Zhang W., Li L., Zhang G., Zhang S. // J. Mol. Liquids. 2018. V. 269. P. 187.
Chen Z., Qiu H., Hong Z., Wang G. // Mol. Simulat. 2020. V. 46. P. 71.
Liu T., Luo W., Cole D.R., Asthagiri A. // J. Chem. Phys. 2019. V. 150. P. 044703.
Rajabpour A., Seif R., Arabha S., Heyhat M.M., Merabia S., Hassanali A. // J. Chem. Phys. 2019. V. 150. P. 114 701.
Шевкунов С.В. // Докл. АН. 2010. Т. 433. С. 761.
Шевкунов С.В. // ЖЭТФ. 2009. Т. 135. С. 510.
Шевкунов С.В. // ЖЭТФ. 2009. Т. 136. С. 282.
Aminoff G. // Zeit. Krist. 1922. V. 57. P. 180.
Majumdar A.J., Roy R. // J. Phys. Chem. 1959. V. 63, P. 1858.
Баханова Р.А., Киселев В.И., Куку Е.И., Ким Н.С., Школкин А.В. // Физика облаков и активные воздействия. Труды украинского регионального научно-исследовательского гидрометеорологического института / под ред. Бахановой Р.А., Осокиной И.Н. Вып. 242. М.: Московское отделение Гидрометеоиздата, 1991. С. 102.
Пермяков Г.Н., Безопасность применения активных средств воздействия. СПб.: Изд-во Балтийского государственного технического университета, 1996.
Шевкунов С.В. // Электрохимия. 1998. Т. 34. С. 860.
Cassie A.B.D. // Discuss. Faraday Soc. 1948. V. 3. P. 11.
Wenzel R.N. // J. Phys. Chem. 1949. V. 53. P. 1466.
Quere D., Lafuma A., Bico J. // Nanotechnology. 2003. V. 14. P. 1109.
Carbone G., Mangialardi L. // Eur. Phys. J. E. 2005. V. 16. P. 67.
Li W., Amirfazli A. // Soft Matter. 2008. V. 4. P. 462.
Koishi T., Yasuoka K., Fujikawa S., Ebisuzaki T., Ze-ng X.C. // Proc. Natl. Acad. Sci. USA. 2009. V. 106. P. 8435.
Richard D., Quere D. // Europhys. Lett. 2000. V. 50. P. 769.
Verho T., Korhonen J.T., Sainiemi L., Jokinen V., Bower C., Franze K., Franssila S., Andrew P., Ikkala O., Ras R.H. // Proc. Natl. Acad. Sci. USA. 2012. V. 109. P. 10210.
Boreyko J.B., Baker C.H., Poley C.R., Chen C.-H. // Langmuir. 2011. V. 27. P. 7502.
Шевкунов С.В. // Коллоид. журн. 2019. Т. 81. С. 367.
Шевкунов С.В. // Коллоид. журн. 2019. Т. 81. С. 382.
Prerna, Goswami R., Metya A.K., Shevkunov S.V., Singh J.K. // Mol. Phys. 2019. V. 117. P. 3651.
Шевкунов С.В. // Коллоид. журн. 2019. Т. 81. С. 86.
Шевкунов С.В. // Коллоид. журн. 2019. Т. 81. С. 101.
Shevkunov S.V., Singh J.K. // J. Mol. Liq. 2018. V. 264. P. 150.
Шевкунов С.В. // Коллоид. журн. 2017. Т. 79. С. 644.
Шевкунов С.В. // Журн. физ. химии. 2013. Т. 87. С. 1678.
Шевкунов С.В. // Журн. физ. химии. 2007. Т. 81. С. 2271.
Шевкунов С.В. // Журн. физ. химии. 2011. Т. 85. С. 1702.
Хилл Т. Статистическая механика. М.: ИЛ, 1960.
Шевкунов С.В. // Коллоид. журн. 2005. Т. 67. С. 548.
Шевкунов С.В. // Коллоид. журн. 2006. Т. 68. С. 691.
Шевкунов С.В. // ЖЭТФ. 2008. Т. 134. С. 1130.
Шевкунов С.В. // Коллоид. журн. 2013. Т. 75. С. 494.
Шевкунов С.В. // Коллоид. журн. 2018. Т. 80. С. 224.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал