

Коллоидный журнал, 2020, T. 82, № 5, стр. 576-584
Исследование дисперсности органозолей золота путем использования композитных пленок Au–AOT
А. Н. Колодин 1, *, И. В. Коростова 1, Е. А. Максимовский 1, А. Т. Арымбаева 1, А. И. Булавченко 1
1 Институт неорганической химии им. А.В. Николаева СО РАН
630090 Новосибирск, просп. Академика Лаврентьева, 3, Россия
* E-mail: kolodin@niic.nsc.ru
Поступила в редакцию 13.03.2020
После доработки 15.04.2020
Принята к публикации 21.04.2020
Аннотация
В работе предложены подходы к определению дисперсности органозоля золота с концентрацией AOT 0.25 М путем исследования поверхности композитной пленки Au–AOT методами сканирующей электронной микроскопии (СЭМ) и атомно-силовой микроскопии (АСМ). Пленка получена на основе концентрированного золя методом Doctor Blade с последующим термолизом при 523 K в течение 2 ч. Композит состоит из отдельных наночастиц Au, покрытых адсорбционным слоем ПАВ. Средний размер частиц составил 11 ± 2 нм (по данным СЭМ) и 12 ± 3 нм (по данным АСМ) и коррелирует с результатами классических методов определения дисперсности золей: фотон-корреляционной спектрометрии (13 ± 1 нм), просвечивающей электронной микроскопии (7 ± 2 нм) и спектрофотометрии плазмонного резонансного поглощения (15 нм).
ВВЕДЕНИЕ
Наночастицы золота активно используются в различных высокотехнологичных областях микроэлектроники [1, 2], катализа [3–6] и медицины [7, 8]. Частицы золота служат основой для различных биохимических сенсоров [9–11], а также электропроводящих покрытий в технологиях 2D- и 3D-печати [12]. Стабильные дисперсии наночастиц золота хорошо зарекомендовали себя в качестве средств доставки лекарственных препаратов [13, 14], а также нашли широкое применение в фототермической и фотодинамической противораковой терапии [15, 16].
Одной из важнейших характеристик органо- и гидрозолей частиц является их дисперсность. Различные физико-химические и биохимические свойства золей зависят от размеров наночастиц. Авторы работ [6, 17, 18] отмечают, что агрегативная устойчивость, а также каталитическая активность дисперсий Au во многом определяются размером наночастиц. По этой причине на данный момент широко распространены методы получения наночастиц Au и других материалов определенного размера с использованием углеводородного геля, полимерных матриц и других стабилизаторов [19–23].
Особое внимание уделяется синтезу наночастиц в прямых и обратных мицеллах. Поскольку размер указанных супрамолекулярных структур способен изменяться в процессе роста наночастиц, у исследователя появляется дополнительная возможность создавать системы с частицами кинетически контролируемого размера [6, 24, 25].
Тем не менее, использование прямых и обратных мицелл накладывает существенные ограничения при исследовании полученных золей на предмет их дисперсности. Наиболее распространенными методами определения размера наночастиц в дисперсных системах являются фотон-корреляционная спектрометрия (ФКС) и просвечивающая электронная микроскопия (ПЭМ). Однако измерение фактического диаметра частиц в золях данными методами крайне проблематично вследствие формирования развитых адсорбционных слоев молекул ПАВ на поверхности наночастиц [26, 27].
Помимо вышеуказанных подходов к определению дисперсности в литературе также представлены работы по оценке диаметра наночастиц в исследуемых системах с применением теории Ми и данных спектрофотометрии [28–30]. Данный способ предполагает наличие сложных алгоритмов расчета и позволяет найти оценочное значение диаметра наночастиц.
Перспективными инструментами исследования дисперсности синтезируемых органозолей могли бы стать классическая сканирующая электронная микроскопия (СЭМ), а также атомно-силовая микроскопия (АСМ), активно применяющиеся при изучении размеров наночастиц Au [31, 32] и других материалов [33]. Однако в случае органозолей данные методы не могут быть использованы напрямую, поскольку наличие больших количеств ПАВ препятствует корректному анализу образцов. Многократное промывание исследуемых систем различными растворителями данную проблему не решает. Удаление ПАВ из золей с высоким содержанием золота с помощью термообработки приводит к спеканию частиц, как это было показано в нашей предыдущей работе [34]. Использование сильно разбавленных золей золота не позволяет провести корректное исследование из-за сложностей при идентификации частиц по причине их низкой численной концентрации.
В данной работе предложены общие способы определения дисперсности органозолей на примере золей Au с использованием комбинации методов неводного электрофореза и СЭМ, а также неводного электрофореза и АСМ. Методики предполагают получение и изучение шероховатых композитных пленок Au–Аэрозоль OT (АОТ) на основе органозолей с низким содержанием этого стабилизатора и оптимальной концентрацией наночастиц Au.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы и материалы
В работе были использованы: дистиллированная вода, соляная и азотная кислоты (х.ч., ОАО Реактив), этанол (абсолютированный 99.6%, ОАО Биохим), ацетон (чистота не ниже 99%, ОАО Реактив), н-гексан (чистота не ниже 99%, ОАО Реактив), н-декан (чистота не ниже 99%, эталонный, ОАО Реактив), бис-(2-этилгексил)сульфосукцинат натрия (АОТ, Sigma-Aldrich, содержание основного вещества не менее 97%), моногидрат гидразина (20 М, чистота не ниже 99%, ЗАО Вектон), металлическое золото (проволока, содержание Au 99.9%), сульфат натрия, бромкрезоловый зеленый, метиловый красный (все – марки ч.д.а., АО ЛенРеактив). Подложками служили предметные стекла размером 25 × 76 × 1 мм (ПАО Стеклоприбор).
Эмульсионный синтез наночастиц Au
Методика синтеза частиц Au в эмульсиях, стабилизированных AOT, подробно описана в нашей предыдущей работе [34]. Исходный водный раствор золота(III) состава 0.25 М HAuCl4 + 3 М HCl получали растворением металлического золота в царской водке с последующим переводом в хлоридный комплекс и разбавлением соляной кислотой до соответствующей концентрации. Исходный 10 М водный раствор гидразина готовили путем двукратного разбавления раствора моногидрата гидразина водой. Точная конечная концентрация восстановителя установлена путем кислотно-основного титрования 1 М раствором HCl в присутствии смеси (3 : 1 по объему) индикаторов бромкрезолового зеленого и метилового красного.
В 10 мл мицеллярного раствора АОТ в н-декане при перемешивании на магнитной мешалке (500 мин–1) последовательно вводили 10 мл 0.02 М водного раствора HAuCl4 и 10 мл 10 М раствора гидразина. Фоновый электролит содержал 3 М HCl и 3.55 М Na2SO4. Итоговое соотношение объемов фаз составляло 2 : 1. Синтез наночастиц Au проходил в течение 1 ч при перемешивании и нормальных термодинамических условиях, после чего обратную эмульсию разрушали выдерживанием в термостате при 333 K в течение 35 мин и центрифугировали 10 мин со скоростью 1500 мин–1. Затем органическую фазу извлекали из реакционной смеси и перемешивали в открытом стакане на магнитной мешалке в течение 2 ч до полного испарения воды. Далее органозоль подвергали электрофоретическому концентрированию.
Электрофоретическое концентрирование органозоля
Концентрирование наночастиц золота осуществляли методом неводного электрофореза без добавления воды, хлороформа и других органических веществ.
Наличие AOT в микроэмульсиях способствовало формированию поверхностного заряда на частицах Au. Непосредственно перед концентрированием были проведены измерения электрокинетического потенциала наночастиц методом лазерного электрофореза с использованием опции фазового анализа рассеянного света (phase analysis light scattering, PALS) на спектрометре NanoOmni (Brookhaven, США). Рассеянные частицами фотоны детектировались под углом 15°. Для измерений была задействована специальная ячейка SRR2, устойчивая к действию органических растворителей, с плоскопараллельными палладиевыми электродами площадью примерно 45 мм2 и межэлектродным зазором 3.45 мм. Скорость движения наночастиц (v) определяли в ручном режиме в диапазоне напряжений U = 70–200 В. Для каждого напряжения среднее значение скорости рассчитывалось по результатам 50 измерений. Электрофоретическая подвижность (μе) определялась из тангенса угла наклона зависимости v = = f(U), а расчет ζ-потенциала производился по формуле Хюккеля–Онзагера
(1)
$\zeta = {{3\eta {{\mu }_{{\text{e}}}}} \mathord{\left/ {\vphantom {{3\eta {{\mu }_{{\text{e}}}}} {2\varepsilon {{\varepsilon }_{0}},}}} \right. \kern-0em} {2\varepsilon {{\varepsilon }_{0}},}}$Для концентрирования наночастиц конечную микроэмульсию заливали в электрофоретическую ячейку конденсаторного типа с горизонтально ориентированными плоскопараллельными медными электродами площадью 16 см2 и межэлектродным зазором 1 см и подвергали электрофорезу в течение 10 ч при постоянном напряжении 600 В. В процессе электрофореза жидкий концентрат наночастиц оседал на катоде и формировалась четкая граница между рафинатом и концентратом. По окончании электрофореза концентрат отбирался микродозатором. Объем аликвот составлял 50–200 мкл.
Определение содержания Au в органозолях
Содержание золота в микроэмульсиях до и после электрофореза определяли спектрофотометрически относительно н-декана. Электронные спектры поглощения регистрировали на спектрофотометре UV-1700 (Shimadzu, Япония) в кварцевых кюветах с длиной оптического пути 1 см. Коэффициент экстинкции на длине волны поглощения 520 нм составлял 4.4 × 103 M–1 см–1 [35].
Исследование дисперсности органозолей
Размер частиц золота в конечных органозолях определяли с помощью ФКС, ПЭМ, СЭМ и АСМ.
Способ 1 (ФКС). Эффективный гидродинамический диаметр частиц после концентрирования измеряли на спектрометре NanoOmni. Автокорреляционная функция обрабатывалась с использованием полимодального анализа при помощи алгоритма NNLS (Non-Negatively Constrained Least Squares). Перед измерениями органозоли очищали от пыли пятикратным циклическим фильтрованием через политетрафторэтиленовый мембранный фильтр с диаметром пор 0.2 мкм (Sartorius, Германия) непосредственно в измерительную 1-см ячейку из стекла (Brookhaven, США). Мощность твердотельного лазера с длиной волны 640 нм составляла 35 мВт, рассеянные частицами фотоны детектировались под углом 90° к источнику излучения. Время накопления фотонов в ходе одного измерения составляло 10–30 с. Определенный из данных по интенсивности рассеяния гидродинамический диаметр частиц (Dz) усредняли по результатам 50 измерений. Расчет производили в предположении сферической формы частиц по формуле Стокса–Эйнштейна
(2)
${{D}_{{\text{z}}}} = {{{{k}_{{\text{B}}}}T} \mathord{\left/ {\vphantom {{{{k}_{{\text{B}}}}T} {(3\pi \eta {{d}_{{\text{d}}}}),}}} \right. \kern-0em} {(3\pi \eta {{d}_{{\text{d}}}}),}}$Способ 2 (ПЭМ). Диаметр наночастиц после концентрирования определяли с помощью электронного микроскопа JEM-2010 с максимальным разрешением 0.2 нм на точку. Электрофоретический концентрат наночастиц Au разбавляли в 500 раз н-гексаном. Затем капли такого коллоидного раствора наносили на углеродную и безуглеродную подложки и высушивали при комнатной температуре. Функцию распределения наночастиц по размеру строили по результатам 50 измерений.
Способ 3 (СЭМ). Диаметр наночастиц после концентрирования определяли с помощью сканирующего электронного микроскопа Hitachi-3400N с приставкой для энергодисперсионной спектроскопии (Oxford Instruments).
Электрофоретический концентрат наночастиц Au порциями по 50 мкл наносили на стеклянную подложку по методу Doctor Blade. Суммарный объем концентрата составил 200 мкл. Стеклянные подложки были предварительно выдержаны в концентрированной азотной кислоте, обезжирены в этаноле и ацетоне, промыты дистиллированной водой и высушены в атмосфере чистого воздуха в течение 3 ч. Нанесенный концентрат высушивался 12 ч на воздухе, после чего был подвергнут термолизу при 523 K в течение 2 ч в программируемой лабораторной печи L 03/12 (Чехия). После термической обработки подложки многократно промывали водой для удаления продуктов разложения AOT и высушивали на воздухе в течение 3 ч.
Съемку образцов проводили при различных увеличениях под прямым углом к поверхности. Диаметр частиц определяли как среднее значение по результатам 50 измерений.
Способ 4 (АСМ). Исследование шероховатости образцов и измерение высоты наночастиц проводили с помощью атомно-силового микроскопа Ntegra Prima II (NT-MDT, Россия) в полуконтактном режиме при нормальных термодинамических условиях и относительной влажности 17%. Площадь области сканирования составляла 10 и 2 мкм2. Использовали зонды HA_NC (A) с номинальной константой жесткости 17 Н/м и резонансной частотой 230 кГц. Предварительно поверхность образца была обеспылена в токе чистого воздуха в течение 10–15 с. Расчет параметров шероховатости проводили с помощью программного обеспечения Nova SPM согласно стандартам ISO 4287-1 (параметры ГОСТ 25 142-82) [36], ISO 4287 [37], ASME B46 [38]. Высоту частиц определяли как среднее значение 10 измерений.
Исследование смачиваемости пленок
Измерения краевых углов воды на пленках Au–AOT проводили методом лежащей капли на приборе OCA 15 PRO (DataPhysics Instruments, Германия), оснащенном измерительной видеосистемой с USB-камерой, а также светосильным измерительным объективом с настраиваемым углом наблюдения. Все образцы находились в термостатированном боксе, оснащенном элементом Пельтье, при T = 298 ± 2 K и p = 1 атм. Диаметр иглы подающего шприца составлял 0.51 мм. В качестве тестовой жидкости использовали дистиллированную воду. Объем капель был постоянным и составлял примерно 2.0 мкл. Расчет краевых углов проводили по уравнению Юнга–Лапласа. Конечное значение краевого угла вычисляли как среднее значение по результатам 3-х измерений, сделанных на разных участках поверхности образца.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Органозоли золота
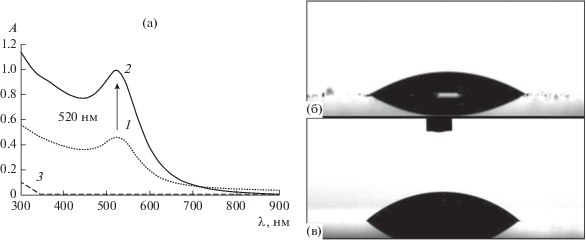
В результате эмульсионного синтеза получены стабильные органозоли золота. Согласно данным спектрофотометрии (рис. 1а), образцам соответствуют электронные спектры с характерной полосой плазмонного поглощения на длине волны 520 нм, что свидетельствует о получении наночастиц Au. Обработка спектров показала, что в результате электрофореза в течение 10 ч при напряжении 600 В и отсутствии воды концентрация Au возросла с 3.0 × 10–4 до 3.9 × 10–3 М. Таким образом, коэффициент концентрирования равен 13, что существенно уступает результатам электрофоретического концентрирования в присутствии 1 об. % воды [34]. Спектрофотометрическое исследование рафината показало, что мольная концентрация Au в рафинате меньше 1 × 10–6 М. Таким образом, степень извлечения составляет более 99%.
Композитная пленка Au–AOT
Методом Doctor Blade с последующим термолизом при 523 К в течение двух часов получена композитная пленка Au–AOT на основе органозоля наночастиц Au. Согласно измерениям смачиваемости, пленка является гидрофильной. Значение краевого угла воды составляет 27 ± 3° (рис. 1б). Гидрофильные свойства обусловлены наличием стабилизатора на поверхности образца. Значение краевого угла для контрольной системы без наночастиц составило 35 ± 3° (рис. 1в). Кроме того, хорошая смачиваемость пленки связана с шероховатостью ее поверхности.
В рамках данной работы проведено исследование поверхности образца с помощью АСМ. На рис. 2а–2в представлены 2D- и 3D-изображения, а также профилограмма поверхности. Композитная пленка имеет шероховатую поверхность. Поскольку образец не обладает четкой текстурой, а характеризуется сложной морфологией рельефа, расчет параметров шероховатости проводили по пяти профилограммам, полученным для разных участков поверхности (рис. 2а). Длина участков сканирования составляла 10 мкм. Рассчитанные параметры шероховатости приведены в табл. 1.
Рис. 2.
АСМ-анализ пленки Au–AOT: (а) 2D-изображение поверхности (линиями отмечены участки сканирования пяти профилограмм), (б) 3D-изображение поверхности, (в) профилограмма № 5, (г) функция распределения частиц Au–AOT по диаметру (N = 10).
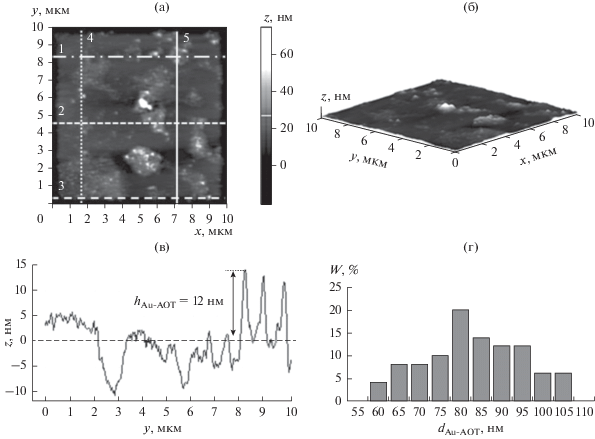
Таблица 1.
Параметры шероховатости пленки Au–AOT (N = 5, Pдов = 0.95)
| Стандарт | Ra, нм | Rq, нм | Rsk | Rku | Rmax, нм | Rp, нм | Rv, нм |
|---|---|---|---|---|---|---|---|
| ISO 4287-1 | 4 ± 2 | 5 ± 2 | 0.9 ± 0.7 | 4 ± 1 | 25 ± 9 | 17 ± 5 | 8 ± 4 |
| ISO 4287 | 4 ± 2 | 5 ± 2 | 0.9 ± 0.6 | 4 ± 1 | – | 17 ± 5 | 9 ± 4 |
| ASME B46 | 4 ± 2 | 5 ± 2 | 0.9 ± 0.6 | 4 ± 1 | 23 ± 8 | 17 ± 5 | 9 ± 4 |
Результаты расчетов по трем стандартам коррелируют между собой. Согласно полученным данным, среднеарифметическая шероховатость (Ra) составляет 4 ± 2 нм, среднеквадратичная шероховатость (Rq) – 5 ± 2 нм, асимметрия (Rsk) и эксцесс профиля (Rku) равны 0.9 ± 0.6 и 4 ±1 соответственно. Максимальная высота профиля шероховатости (Rmax) достигает 24 ± 9 нм, а максимумы высоты пика (Rp) и глубины долины профиля (Rv) – 17 ± 5 и 9 ± 4 нм соответственно.
Наличие шероховатости обусловлено вкладами от самой подложки и стабилизатора (АОТ), а также связано с размером частиц Au, покрытых слоем стабилизатора. Таким образом, морфология пленки и, как следствие, ее смачиваемость во многом зависят от дисперсности частиц Au.
Дисперсность органозолей Au
С помощью АСМ и СЭМ исследована дисперсность наночастиц Au в исходном органозоле посредством измерения их размеров в композитной шероховатой пленке Au–AOT.
Исследование с помощью АСМ. Напрямую корректно измерить диаметр наночастиц Au в композитной пленке с помощью АСМ не представляется возможным. Согласно литературным данным [34], эмульсионный синтез наночастиц золота в обратных мицеллах AOT в н-декане позволяет получить частицы размером не более 15–20 нм. В силу инструментальных особенностей метода (а именно, конечного значения радиуса закругления острия зонда) погрешность определения диаметра частиц как их “латерального” размера может достигать 50% и более [39]. Действительно, результаты обработки данных АСМ показывают, что среднее значение диаметра частиц (dAu-AOT) сильно завышено и составляет 80 нм (рис. 2г). Между тем, высота частиц, а также шероховатость поверхности определяются с помощью АСМ с хорошей точностью, поэтому данный метод широко используется при изучении, например, морфологии различных покрытий на наноуровне [40–42]. В данной работе дисперсность частиц оценивали именно по среднему значению их высоты.
Анализ профилограмм позволил оценить среднюю высоту наночастиц золота, покрытых слоем стабилизатора (hAu-AOT). Она составила 12 ± ± 3 нм (рис. 2в). Результаты расчета высоты выступов по всей площади сканирования другого участка образца подтверждают данные о высоте частиц Au (рис. 3a, 3б). Образцу соответствует узкое мономодальное распределение выступов по высоте. Среднее значение высоты составляет ~13 нм.
Рис. 3.
АСМ-анализ пленки Au–AOT: (а) 2D-изображение поверхности, (б) функция распределения выступов поверхности по высоте (N = 3136).
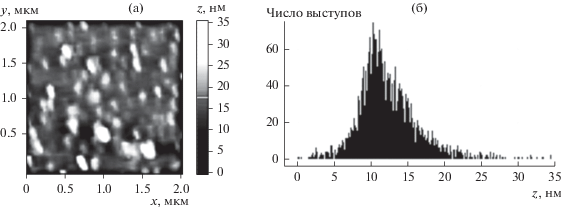
Тем не менее, этот подход к оценке дисперсности наночастиц имеет несколько недостатков. На значение их высоты влияет шероховатость подложки (средняя высота выступов для “контрольной” (без частиц Au) системы составила ~3 нм). Кроме того, частицы золота покрыты слоем стабилизатора, вследствие чего методика дает завышенные значения размера наночастиц. Преодолеть данные ограничения можно путем применения СЭМ для исследования дисперсности наночастиц.
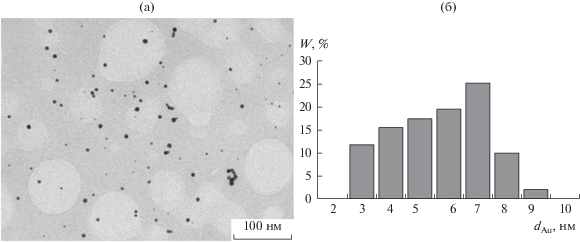
Исследование методом СЭМ. Съемка композитной пленки Au–AOT проводилась в режиме обратно-рассеянных электронов, что позволяло обнаружить фазу металла, инкапсулированную в твердом слое органических соединений.
На рис. 4a представлена микрофотография участка поверхности пленки. Видно, что поверхность образца равномерно покрыта тонким слоем ПАВ. При этом под слоем AOT отчетливо просматриваются отдельные наночастицы Au. Умеренная концентрация Au (4 × 10–3 М) и наличие ПАВ (0.25 М) в конечном концентрате позволили избежать коагуляции и спекания частиц в процессе получения композитной пленки Au–AOT. При этом наночастицы спекались с поверхностью подложки, что позволило избежать их редиспергирования при промывании подложки водой для удаления избытка ПАВ, а также продуктов его разложения.
Рис. 4.
(a) СЭМ-изображение пленки Au–AOT и (б) функция распределения частиц Au по диаметру (N = 50).
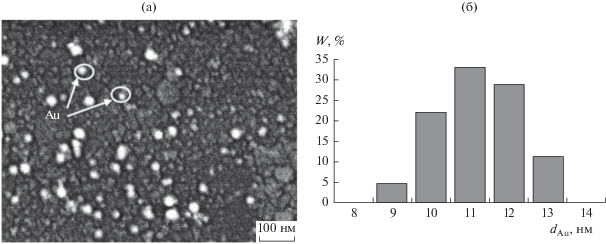
Результаты измерения диаметра частиц золота (dAu) приведены на гистограмме (рис. 4б). Согласно полученным данным, образец характеризуется узкой функцией распределения наночастиц по размеру, от 9 до 13 нм. Среднее и наиболее вероятное значения совпадают и составляют 11 ± 2 нм (табл. 2). Результаты СЭМ по определению размеров частиц Au согласуются с данными АСМ. Разница в значениях размера составляет ~1 нм и обусловлена наличием адсорбционного слоя из молекул ПАВ на поверхности частиц в пленке Au–AOT, а также шероховатостью исходной подложки.
Исследование классическими методами. Для оценки возможности использования описанных выше методик АСМ и СЭМ при исследовании дисперсности частиц Au в исходных органозолях они были проверены тремя независимыми методами: ФКС, ПЭМ и спектрофотометрией.
Согласно теории Ми, имеет место зависимость длины волны максимума поверхностного плазмонного резонансного поглощения (λmax) от диаметра наночастиц металлов. В обзоре [28] на основе анализа литературных данных [29, 30, 43–49] предложена эмпирическая зависимость, связывающая λmax сферических наночастиц Au, с их размером при λmax < 523 нм:
(3)
${{d}_{{{\text{Au}}}}} = 3 + 7.5 \times {{10}^{{--5}}}{{({{\lambda }_{{{\text{max}}}}}--500)}^{4}}.$По результатам спектрофотометрии (рис. 1a) максимум плазмонного резонансного поглощения полученными органозолями золота составляет 520 нм (т.е. λmax < 523 нм). Согласно расчету по уравнению (3), диаметр наночастиц Au равен 15 нм (табл. 2).
На рис. 5 приведены результаты измерения гидродинамического диаметра наночастиц. Согласно представленным данным, образец характеризуется узкой функцией распределения, Dz частиц варьируется от 12 до 17 нм. При этом среднее и наиболее вероятное значения Dz совпадают и составляют 13 ± 1 нм (табл. 1).
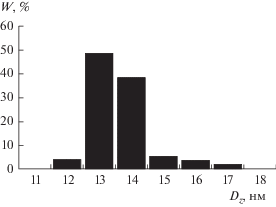
На рис. 6 представлены результаты ПЭМ. Частицы Au имеют круглую форму. Их диаметр варьируется от 3 до 9 нм. Узкая функция распределения частиц по диаметру свидетельствует о достаточно высокой степени монодисперсности исследуемой системы. Среднее и наиболее вероятное значения диаметра совпадают и составляют 7 ± 2 нм (табл. 1).
Результаты измерения диаметра наночастиц Au с помощью СЭМ и АСМ по описанным выше методикам коррелируют с данными ФКС, ПЭМ и теории Ми (табл. 2).
Наименьшее значение диаметра наночастиц получено с помощью ПЭМ, а наибольшее значение – из данных спектрофотометрии по теории Ми. Различие достигает 8 нм. Завышение результатов связано с использованием эмпирической математической модели, позволяющей получить только оценочные значения диаметра наночастиц.
Диаметр наночастиц, определенный с помощью ФКС, также превышает значения диаметра, полученные методами ПЭМ и СЭМ, на 6 и 2 нм соответственно. Завышение результатов связано в этом случае с наличием адсорбционного слоя ПАВ на наночастицах Au в органозолях. Толщина адсорбционного слоя (l) может быть найдена по следующей формуле:
(4)
$l = {{({{D}_{{\text{z}}}}--{{d}_{{\text{i}}}})} \mathord{\left/ {\vphantom {{({{D}_{{\text{z}}}}--{{d}_{{\text{i}}}})} 2}} \right. \kern-0em} 2},$Размер частиц, определенный с помощью АСМ, отличается от их размера по данным СЭМ и ПЭМ на 1 и 5 нм соответственно. Различие в значениях обусловлено наличием адсорбционного слоя ПАВ на поверхности пленки. Значение ее толщины составило 2.5 нм (по данным ПЭМ) и 0.5 нм (по результатам СЭМ). Уменьшение толщины адсорбционного слоя в случае пленки по сравнению с его толщиной для исходного золя является следствием частичного разложения стабилизатора и десорбции образующихся продуктов с поверхности композита Au–AOT при термической обработке. Различие в линейных размерах частиц, определенных с помощью АСМ и ПЭМ, находится в пределах статистической погрешности. Следовательно, использованная нами методика АСМ может быть задействована при исследовании дисперсности исходных органозолей наночастиц Au.
Значения диаметра наночастиц, полученные методами СЭМ и ПЭМ, различаются на 4 нм. Данное различие находится в пределах статистической погрешности и обусловлено спеканием частиц Au с подложкой в процессе термообработки композитной пленки Au–AOT. Таким образом, степень дисперсности исходной системы искажена незначительно, и предложенный способ исследования размера частиц с помощью СЭМ может быть использован при определении дисперсности исходных органозолей золота.
ЗАКЛЮЧЕНИЕ
Высокие концентрации ПАВ при синтезе наночастиц металлов способствуют образованию стабильных органозолей, содержащих практически монодисперсные наночастицы, однако зачастую препятствуют применению традиционных методов определения их характеристик in situ.
В представленной работе предложен вариант решения этой проблемы посредством термического “запекания” – фиксации наночастиц в твердофазном композите Au–AOT с целью сохранения их исходной дисперсности. Концентрация наночастиц золота при этом должна быть оптимальной, так как высокие концентрации приводят к коагуляции и срастанию наночастиц и, как следствие, к уменьшению степени дисперсности. Низкие концентрации частиц не дают возможность провести корректную статистическую обработку результатов измерений. В перспективе разработанные нами подходы, основанные на использовании СЭМ и АСМ, могут быть адаптированы для анализа дисперсности наночастиц непосредственно в полученных в результате синтеза системах без предварительного их концентрирования и отделения избытка ПАВ.
Список литературы
Novembre C., Guerin D., Lmimouni K., Gamrat C., Vuillaume D. // Appl. Phys. Lett. 2008. V. 92. 103 314.
Homberger M., Simon U. // Phil. Trans. R. Soc. A. 2010. V. 368. P. 1405.
Grisel R., Weststrate K.-J., Gluhoi A., Nieuwenhuys B.E. // Gold Bull. 2002. V. 35. № 2. P. 39.
Li G., Jin R. // Nanotechnol. Rev. 2013. V. 2. P. 529.
Lahtinen E., Kukkonen E., Kinnunen V., Lahtinen M., Kinnunen K., Suvanto S., Vaisanen A., Haukka M. // ACS Omega. 2019. V. 4. P. 16891.
Suchomel P., Kvitek L., Prucek R., Panacek A., Halder A., Vajda S., Zboril R. // Sci. Rep. 2018. V. 8. P. 4589.
Dykman L.A., Khlebtsov N.G. // Acta Naturae. 2011. V. 3. № 2. P. 34.
Kumar P., Roy. I. // Int. J. Pharm. Pharm. Sci. 2016. V. 8. № 7. P. 9.
Peixoto de Almeida M., Pereira E., Baptista P., Gomes I., Figueiredo S., Soares L., Franco R. // Compr. Anal. Chem. 2014. V. 66. P. 529.
Saha K., Agasti S.S., Kim C., Li X., Rotello V.M. // Chem. Rev. 2012. V. 112. P. 2739.
Zhang Y., Chu W., Foroushani A.D., Wang H., Li D., Liu J., Barrow C.J., Wang X., Yang W. // Materials. 2014. V. 7. P. 5169.
Zeng M., Zhang Y. // J. Mater. Chem. A. 2019. V. 7. P. 23 301.
Hussain K., Hussain T. // South Indian J. Biol. Sci. 2015. V. 1. P. 127.
Voliani V., Signore G., Nifosi R., Ricci F., Luin S., Beltram F. // Recent Pat. Nanomed. 2012. V. 2. P. 34.
Vines J.B., Yoon J.-H., Ryu N.-E., Lim D.-J., Park H. // Front. Chem. 2019. V. 7. P. 167.
Kim H.S., Lee D.Y. // Polymers. 2018. V. 10. P. 961.
Kongsuwan C., Warisnoicharoen W. // Thai J. Pharm. Sci. 2013. V. 38. P. 229.
Bac L.H., Kim J.S., Kim. Size J.C. // Rev. Adv. Mater. Sci. 2011. V. 28. P. 117.
Trbojevich R., Pellegri N., Frattini A., de Sanctis O. // J. Mater. Res. 2002. V. 17. P. 1973.
Faoucher E., Nativo P., Black K., Claridge J.B., Gass M., Romani S., Bleloch A.L., Brust M. // Chem. Commun. 2009. P. 6661.
Abdelrasoul G.N., Farkas B., Romano I., Diaspro A., Beke S. // Mater. Sci. Eng. C. 2015. V. 56. P. 305.
Corbierre M.K., Cameron N.S., Sutton M., Laaziri K., Lennox R.B. // Langmuir. 2005. V. 21. P. 6063.
Corbierre M.K., Cameron N.S., Sutton M., Mochrie S.G.J., Lurio L.B., Ruhm A., Lennox R.B. // J. Am. Chem. Soc. 2001. V. 123. P. 10411.
Smetana A.B., Wang J.S., Boeckl J., Brown G.J., Wai C.M. // Langmuir. 2007. V. 23. P. 10429.
Herrera A.P., Resto O., Briano J.G., Rinaldi C. // Nano-technology. 2005. V. 16. P. 618.
Bulavchenko A.I., Popovetskiy P.S. // Langmuir. 2014. V. 30. P. 12729.
Kolodin A.N., Tatarchuk V.V., Bulavchenko A.I., Poleeva E.V. // Langmuir. 2017. V. 33. P. 8147.
Khlebtsov N.G. // Anal. Chem. 2008. V. 80. P. 6620.
Haiss W., Thanh N.T.K., Aveard J., Fernig D.G. // Anal. Chem. 2007. V. 79. P. 4215.
Njoki P.N., Lim, I.-I.S., Mott D., Park H.-Y., Khan B., Mishra S., Sujakumar R., Luo J., Zhong C.-J. // J. Phys. Chem. B. 2007. V. 111. P. 14664.
Sobczak-Kupiec A., Malina D., Zimowska M., Wzorek Z. // Dig. J. Nanomater. Biostructures. 2011. V. 6. P. 803.
Georgiev P., Bojinova A., Kostova B., Momekova D., Bjornholm T., Balashev K. // Colloids Surf. A. 2013. V. 434. P. 154.
Pletikapić G., Žutić V., Vinković Vrček I., Svetličić V. // J. Mol. Recognit. 2012. V. 25. P. 309.
Шапаренко Н.О., Арымбаева А.Т., Демидова М.Г., Плюснин П.Е., Колодин А.Н., Максимовский Е.А., Корольков И.В., Булавченко А.И. // Коллоид. журн. 2019. Т. 81. С. 532.
Sergievskaya A.P., Tatarchuk V.V., Makotchenko E.V., Mironov I.V. // J. Mater. Res. 2015. V. 30. P. 1925.
ГОСТ 25 142-82. Межгосударственный стандарт “Шероховатость поверхности”: Термины и определения. М.: ИУС 7-2017, 2018.
ГОСТ 4287. Геометрические характеристики изделий (GPS): Структура поверхности. Профильный метод. Термины, определения и параметры структуры поверхности. Москва: Стандартинформ, 2015.
Surface Texture (Surface Roughness, Waviness, and Lay). New York: The American Society of Mechanical Engeneers, 2003.
Циркунова Н.Г., Кухаренко Л.В., Чижик С.А., Борисенко В.Е. // Доклады БГУИР. 2008. Т. 3. № 33. С. 71.
Шевкина А.Ю., Соснов Е.А., Малыгин А.А. // Коллоид. журн. 2012. Т. 74. С. 408.
Brahma R., Krishma M.G. // Bull. Mater. Sci. 2012. V. 35. № 4. P. 551.
Arman A., Talu S., Luna C., Ahmadpourian A., Naseri M., Molamohammadi M. // J. Mater. Sci.: Mater Electron. 2015. V. 26. P. 9630.
Brown K.R., Walter D.G., Natan M.J. // Chem. Mater. 2000. V. 12. P. 306.
Brown K.R., Natan M.J. // Langmuir. 1998. V. 14. P. 726.
Chithrani B.D., Ghazani A.A., Chan W.C.W. // Nano Lett. 2006. V. 6. P. 662.
Andreescu D., Sau T.K., Goia D.V. // J. Colloid Interface Sci. 2006. V. 298. P. 742.
Horisberger M. // In Techniques in Immunocytochemistry / Ed. by Bullock G.R., Petrusz P. London: Academic Press, 1985. P. 155.
Slouf M., Kuzel R., Matej Z.Z. // Kristallogr. Suppl. 2006. V. 23. P. 319.
Алексеева А.В., Богатырев В.А., Хлебцов Б.Н., Мельников А.Г., Дыкман Л.А., Хлебцов Н.Г. // Коллоид. журн. 2006. Т. 68. С. 725.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал