

Коллоидный журнал, 2020, T. 82, № 6, стр. 707-714
Нейросистемное моделирование зернограничных нанофаз, локализованных в простых симметричных границах наклона Σ3 (111) и Σ5 (210)
В. В. Королев 1, *, А. А. Митрофанов 1, Ю. М. Неволин 1, В. В. Кротов 1, Д. К. Ульянов 1, П. В. Проценко 1
1 Московский государственный университет им. М.В. Ломоносова,
химический факультет
119991 Москва, Ленинские горы, д. 1, стр. 3, Россия
* E-mail: korolev@colloid.chem.msu.ru
Поступила в редакцию 27.04.2020
После доработки 07.05.2020
Принята к публикации 11.05.2020
Аннотация
Предложен метод нейросистемного анализа существования и устойчивости зернограничных фаз, образующихся на высокосимметричных границах наклона Σ3 (111) И Σ5 (210) в поликристаллическом твердом растворе Ni(Bi). Данный метод основан на использовании реперных потенциалов межчастичного взаимодействия, построенных в рамках теории функционала плотности, в сочетании с конструкционными возможностями искусственной двухуровневой самообучающейся нейронной сети. Значение абсолютной погрешности при определении потенциальной энергии в рамках метода нейросистемного анализа составило 0.012 эВ/атом. Значения энтальпии образования зернограничных фаз для границ зерен типа Σ3 и Σ5 достаточно хорошо согласуются с известными из литературы результатами моделирования указанной системы и экспериментальными данными.
ВВЕДЕНИЕ
Границы зерен (ГЗ) – внутренние границы раздела в поликристаллах, оказывают существенное влияние на свойства материалов. В последние годы все большее распространение приобретает концепция направленного управления свойствами поликристаллов посредством изменения свойств ГЗ (grain boundary engineering [1, 2]). Так, например, длительный отжиг латуни совместно с механической обработкой позволяет повысить долю высокосимметричных ГЗ, и, как следствие, повысить прочность, так как механические свойства поликристалла напрямую зависят от плотности/структуры трехмерной сети ГЗ [3, 4].
Возможности для направленного управления свойствами ГЗ принципиально расширяются для многокомпонентных систем [5–8], то есть в тех случаях, когда возможна адсорбция допирующих добавок или одного из компонентов сплава на внутренних границах раздела – сегрегация. Адсорбировавшиеся атомы образуют квазидвумерные термодинамически устойчивые нанофазы, в англоязычной литературе получившие название grain boundary complexions [9–12]. В противоположность “классическим”, с относительно большой толщиной (порядка микрометров), прослойкам, разделяющим кристаллы при смачивании ГЗ, зернограничные нанофазы демонстрируют разнообразное фазовое поведение, и переход между ними сопровождается скачкообразным изменением физико-химических свойств [2, 9, 10, 13]. В свою очередь, эти фазовые переходы влияют на свойства макроскопического объекта – поликристалла.
Однако помимо положительного влияния на свойства поликристаллов, образование зернограничных фаз в многокомпонентных системах может также привести и к катастрофическим последствиям. Так, например, пластичные металлы, такие как медь, алюминий, никель, в поликристаллическом виде при контакте с некоторыми расплавами склонны к зернограничному охрупчиванию и разрушению при относительно небольших механических нагрузках, что, по сути, является примером проявления эффекта Ребиндера. Практическая значимость проблемы обусловлена, например, использованием жидких металлов в качестве теплоносителей в атомных реакторах нового поколения, т.е. в области, где безопасность и износостойкость материалов играет критически важную роль.
Одна из предложенных классификаций зернограничных фаз предполагает их деление на основе толщины: “чистые” ГЗ, нанофазы – монослои, бислои, трислои, полислои) и микрофазы – слои микрометровой толщины (предельный случай, аналогичный ранее упомянутым трехмерным пленкам). То есть толщина большинства типов зернограничных фаз составляет порядка одного нанометра, и, следовательно, для их экспериментального изучения необходимы методы с атомным разрешением. Так, просвечивающая электронная микроскопия высокого разрешения (ПЭМВР) позволила обнаружить и исследовать зернограничные фазы в ряде систем. Однако в силу ряда причин данный метод удается применить только для отдельных ГЗ (в лучшем случае – порядка десяти типов [8]), что не позволяет получить представление о строении зернограничных фаз для всего разнообразия ГЗ, присутствующих в реальном поликристалле. Кроме того, ПЭМВР, несмотря на высокую разрешающую способность, позволяет определить лишь толщину зернограничных фаз, но не позволяет исследовать их локальное строение. Помимо толщины, зернограничные фазы также могут различаться степенью упорядоченности и плотностью атомной упаковки вблизи ГЗ [14–16]. Кроме того, экспериментальные данные не позволяют судить об эволюции зернограничных фаз во времени, их влиянии на скорость скольжения отдельных зерен, или влиянии на скорость формирования кристаллов из многокомпонентных расплавов.
В качестве альтернативного подхода для исследования атомной структуры и свойств зернограничных фаз были применены разнообразные методы компьютерного моделирования. Метод функционала плотности (density functional theory, DFT), в настоящее время является стандартом качества в области моделирования широкого класса свойств твердого тела. Однако в силу значительных вычислительных затрат (∼N 3), данный метод позволяет моделировать структуры относительного небольшого масштаба в течение ограниченного времени.
Детальные исследования кристаллографии ГЗ показали, что они часто имеют периодическую структуру, период которой варьируется в зависимости от типа ГЗ. К настоящему времени большая часть работ по моделированию зернограничных структур посвящена ГЗ с низким значением Σ в терминах решетки совпадающих узлов, т.е. высокосимметричным ГЗ с относительно небольшой элементарной ячейкой [17–21]. Однако за пределами рассмотрения остаются ГЗ общего типа, широко представленные в реальных поликристаллах, которые имеют достаточно большие элементарные ячейки, что препятствует их моделированию с применением метода функционала плотности. Такие низкосимметричные ГЗ, как правило, характеризующиеся большим избыточным свободным объемом, отрицательно сказываются на прочности поликристаллов.
Так, относительно недавно, благодаря использованию метода функционала плотности, было показано разнообразное фазовое поведение для двух подробно изученных экспериментально ГЗ в меди — Σ3 и Σ5 [18]. Однако обобщение этих результатов на случай ГЗ с низкой симметрией проблематично в силу вышеуказанных причин. Значительная часть ГЗ в реальных поликристаллах относится к низкосимметричным, размер ячеек для их преставления в виде периодической структуры на настоящий момент исключает возможность использования метода функционала плотности. Так, например, ГЗ Σ43 содержит 41 264 атома в ячейке [22].
Основную альтернативу методу функционала плотности составляют эмпирические потенциалы, которые предполагают параметризацию поверхности потенциальной энергии. Аналитическая форма модели подбирается из соображений простоты практического использования, а также на основании общих физико-химических представлений о структуре поверхности потенциальной энергии прототипа.
Параметры потенциала подбираются таким образом, чтобы отдельные физико-химические свойства, будь то макроскопические величины или параметры, характеризующие систему на микроскопическом уровне (значения которых известны из экспериментальных данных или рассчитаны с использованием первопринципных методов), как можно лучше воспроизводились с использованием данного потенциала. Очевидный недостаток параметрических потенциалов состоит в том, что их относительно простая аналитическая форма недостаточно вариативна для удовлетворительного описания широкого класса атомных конфигураций/моделируемых свойств. Увеличение же количества параметров требует большего количества опорных данных, и, кроме того, снижает интерпретируемость и физическую обоснованность модели.
Таким образом, метод функционала плотности и эмпирические потенциалы – две основные альтернативы при выборе между требованиями точности и скейлинговой инвариантности (масштабируемости) при моделировании свойств твердых тел, в частности – свойств зернограничных фаз. Отсюда очевидное стремление совместить достоинства обоих подходов. Это может быть реализовано с применением методов машинного обучения для построения поверхности потенциальной энергии [23] с сохранением точности DFT-расчетов и скейлинговой инвариантности эмпирических потенциалов.
Наибольший интерес с точки зрения моделирования твердого тела представляет воссоздание с максимально возможной точностью поверхности потенциальной энергии, которая позволяет судить об устойчивости произвольных конфигураций атомов в заданной химической системе. Общая идеология подхода с использованием моделей машинного обучения заключается в построении функции, аппроксимирующей поверхность потенциальной энергии без использования функциональных зависимостей, традиционно используемых для описания межчастичных взаимодействий. Успешность данного подхода определяется тремя факторами.
1. Наличие набора атомных конфигураций, достаточно плотно (полно) покрывающего конфигурационное пространство. Качество работы алгоритмов машинного обучения, в особенности – глубоких нейронных сетей, напрямую зависит от объема данных в обучающей выборке. Кроме того, нейронные сети – наиболее распространенный алгоритм машинного обучения для построения потенциалов взаимодействия – являются универсальными функциональными аппроксиматорами [24], т.е. имеют весьма ограниченную предсказательную способность для конфигураций, существенно отличающихся от тех, которые представлены в обучающей выборке.
2. Релевантный способ представления атомных конфигураций. Для преобразования трехмерной структуры в такое представление, которое пригодно в качестве входных данных для планируемой функционально адекватной модели, было предложено несколько подходов. Основное требование – инвариантность относительно операций симметрии, присущих заданным конфигурациям (параллельные сдвиги, вращение, перестановки эквивалентных атомов и т.д.).
3. Эффективный обучающий (итерационный) алгоритм. Революция в области глубоких нейронных сетей позволила существенно продвинуться в распознавания изображений, человеческой речи и т.п. Адаптация методов машинного обучения для решения задач материаловедения связана, прежде всего, с двумя указанными выше пунктами. Архитектуры нейронных сетей общего назначения при должном подборе набора конфигураций и способа их представления можно использовать практически в исходном виде.
Потенциалы взаимодействия, созданные с использованием нейронных сетей, позволили существенно ускорить моделирование для ряда систем, с сохранением точности опорного метода (основанного на теории функционала плотности). Так, среди прочего, такие потенциалы использовались для моделирования фазовых диаграмм аморфных тел [25], а также анализа процессов в системах, содержащих как свободные поверхности [26–28], так и дефекты упаковки [29]. Как следует из приведенного списка, такой подход достаточно универсален с точки зрения химического и структурного разнообразия, что позволяет надеяться на его состоятельность при моделировании структуры и свойств внутренних границ раздела.
Основным результатом данного исследования является разработка методологии, позволяющей определять энергетические характеристики межзеренных структур с высокой точностью при небольших затратах вычислительных ресурсов. Нами получена модель потенциала взаимодействия, основанная на аппроксимации поверхности потенциальной энергии с использованием нейронной сети в качестве аппроксимирующей функции. Данная модель пригодна для прогнозирования устойчивости зернограничных фаз в системе Ni–Bi. Высокая точность предложенного подхода подтверждена на примере двух высокосимметричных ГЗ, для которых в более ранних исследованиях было проведено полноценное моделирование с использованием теории функционала плотности.
МЕТОДЫ
Квантово-механические расчеты проводились в рамках теории функционала плотности с использованием базиса плоских волн, PAW-формализма и обменно-корреляционного функционала PBE (Perdew–Burke–Ernzerhof) [30, 31]. Вычисления выполнены в пакете VASP [32–34].
Для аппроксимации поверхности потенциальной энергии мы использовали двухслойную искусственную нейронную сеть, каждый из скрытых слоев содержал 10 нейронов. Для оптимизации весов модели использовался алгоритм Бройдена–Флетчера–Гольдфарба–Шанно. В качестве целевых функций для предсказания использовались значения потенциальной энергии, приходящейся на элементарную ячейку, характеризующую заданную конфигурацию как единое целое, а также значения сил, действующих на отдельные атомы.
Расчет атомных дескрипторов, использовавшихся для представления атомных конфигураций, и обучение искусственной нейронной сети проводились с использованием пакета amp-atomistics [35].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
1. В силу вышеупомянутых особенностей искусственных нейронных сетей потенциалы взаимодействия, созданные с использованием их в качестве аппроксимирующего алгоритма, имеют ограниченную область адекватного применения даже в пределах заданной химической системы (за редким исключением [36]). Поэтому для создания нейросетевого потенциала взаимодействия (НПВ), пригодного для оценки устойчивости зернограничных фаз необходимо, чтобы набор опорных данных (в данном случае – конфигурации атомов с известными значениями энергии), соответствовал той области конфигурационного пространства, в которой находятся данные структуры.
Для генерации конфигураций, локальное окружение атомов в которых близко по структуре окружению атомов вблизи ГЗ, мы применили следующие структуры:
1) ячейки, содержащие атомы никеля или висмута, с идеальной кристаллической решеткой (гранецентрированной кубической, гексагональной плотнейшей, объемно-центрированной кубической) и с измененными параметрами решетки (±10%),
2) аналогичные конфигурации с произвольной заменой части атомов на атомы другого элемента (Bi → Ni, Ni → Bi),
3) регулярные моно- и бислои висмута, заключенные между двумя кристаллами никеля,
4) все вышеприведенные конфигурации + случайное отклонение атомов от их исходного положения (до 0.1 Å).
Методом DFT были рассчитаны энергии для 562 конфигураций, содержащих от 1 до 16 атомов в элементарной ячейке. Примеры конфигураций приведены на рис. 1.
Рис. 1.
Примеры атомных конфигураций, сгенерированных согласно методологии, приведенной в пункте 1. Атомы никеля отображены зеленым цветом, висмута – фиолетовым.
2. Для расчета энергии многоатомной системы нейронная сеть формальным образом аддитивно суммирует энергетические вклады отдельных атомов. Для преобразования исходного координатного представления периодических структур в форму, пригодную для дальнейшего обучения генерируемой модели, были использованы следующие функции, отличающиеся универсальностью и учитывающие все основные типы симметрии, присущие периодическим структурам [23, 37]:
Стоит отдельно отметить, что для каждого из двух рассмотренных химических элементов использовалась отдельно обученная нейронная сеть.
3. Ключевым фактором, определяющим успешность применения обученной модели нейронной сети, является возможность с ее помощью прогнозировать релевантные значения целевой переменной для объектов, не использовавшихся непосредственно для обучения модели. Таким образом, для валидации нашей модели мы использовали распространенный подход, предполагающий разбиение исходных данных на тренировочный и тестовый наборы.
На рис. 2 представлены значения потенциальной энергии для структур из тестового набора, рассчитанные с использованием теории функционала плотности и полученные в рамках разрабатываемой модели. Из рис. 2 следует, что модель позволяет определять потенциальную энергию тестовой структуры с высокой точностью (0.012 эВ/атом для энергии, 0.078 эВ/Å для силы), достаточной для практического использования.
Рис. 2.
Значения (а) энергии, приходящейся на один атом в ячейке, и (в) силы, действующей на отдельные атомы, рассчитанные с помощью теории функционала плотности и в рамках разрабатываемой модели, использующей DFT-расчеты в качестве опорных данных. Значения абсолютной ошибки расчета (б) энергии, приходящейся на атом, и (г) силы относительно соответствующих значений, полученных методом DFT.
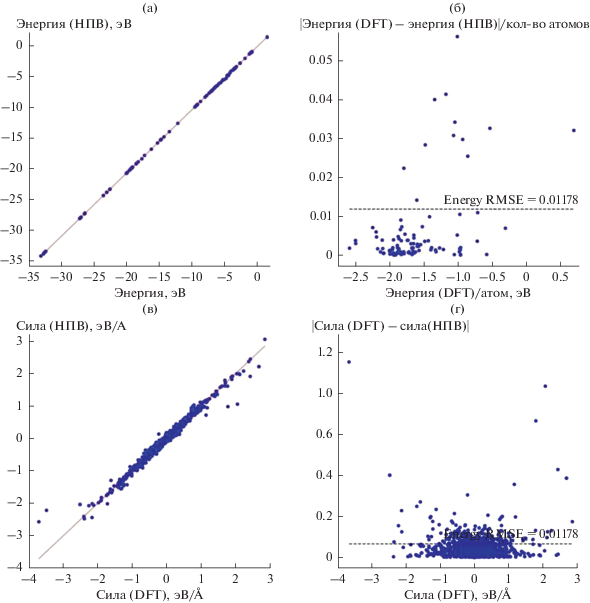
Низкие значения средней квадратичной погрешности для потенциальной энергии и сил позволяют судить о высокой предсказательной способности представленной модели.
4. Моделирование зернограничных фаз с помощью метода функционала плотности сопряжено со значительными сложностями, поэтому в литературе представлено ограниченное число работ, посвященных данной тематике. Так, в работе [18] представлены результаты моделирования длязернограничных фаз в системе никель–висмут.
Постоянная элементарной ячейки, содержащей бикристалл никеля, разделенный ГЗ заданного строения, в общем случае отличается от периода решетки бислоев висмута (для моделирования была выбрана наиболее устойчивая, согласно имеющимся данным, зернограничная фаза). Поэтому для моделирования мы использовали суперячейку, построенную таким образом, чтобы отклонение от истинного значения для постоянной решетки висмута не превышало 3%. Модельные ячейки представлены на рис. 3. В ходе моделирования в качестве варьируемого параметра также рассматривалось расстояние между двумя кристаллами никеля.
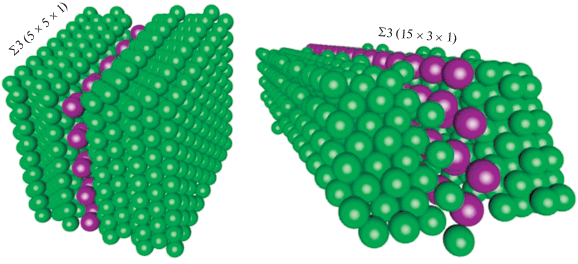
Для оценки устойчивости отдельных зернограничных фаз в системе Ni–Bi использовалась удельная энтальпия образования, приходящаяся на единицу площади:
Таблица 1.
Результаты расчетов энтальпии образования зернограничных фаз для бислоев висмута на ГЗ Σ3 и Σ5 (выделено жирным шрифтом)
| Тип ГЗ | Параметр | DFT-расчет [18] | Расчет с помощью НПВ | |||
|---|---|---|---|---|---|---|
| Σ3 (111) | Плотность слоя, атом/Å2 | 0.084 | 0.167 | 0.251 | 0.293 | 0.115 |
| Энтальпия образования, эВ/Å2 | 70.8 | 44.1 | 76.3 | 61.9 | 56.91 | |
| Σ5 (210) | Плотность слоя, атом/Å2 | 0.074 | 0.146 | 0.219 | 0.292 | 0.101 |
| Энтальпия образования, эВ/Å2 | –21.2 | –53.8 | –15.3 | –36.0 | –17.39 | |
ЗАКЛЮЧЕНИЕ
В данной работе представлена методология построения потенциала межчастичного взаимодействия, основанная на использовании глубоких нейронных сетей в качестве аппроксимирующей функции для поверхности потенциальной энергии. В качестве модельной выборки рассмотрены биметаллические периодические структуры Ni–Bi. Благодаря релевантному выбору опорных данных и способа представления атомных конфигураций, полученные значения энергии с достаточно высокой точностью воспроизводят значения, рассчитанные с применением теории функционала плотности.
Представленный потенциал позволяет прогнозировать устойчивость и термодинамические характеристики зернограничных фаз в системе Ni–Bi. Значения энтальпии образования нанофаз, рассчитанные в рамках нейросистемного анализа, укладываются в диапазон значений, полученных с применением метода DFT. Разработанная на основе нейросистемного анализа модель предсказывает отрицательную энтальпию образования для бислоя висмута на границе Σ5 (210) (–17.39 эВ/A2) и положительную (56.91 эВ/A2) на границе Σ3 (111).
Список литературы
Randle V. // Scr. Mater. 2006. V. 54. P. 1011.
Watanabe T. // J. Mater. Sci. 2011. V. 46. P. 4095.
Kim C.S., Hu Y., Rohrer G.S., Randle V. // Scr. Mater. 2005. V. 52. P. 633.
Rohrer G.S., Randle V., Kim C.S., Hu Y. // Acta Mater. 2006. V. 54. P. 4489.
Duscher G., Chisholm M.F., Alber U., Rühle M. // Nat. Mater. 2004. V. 3. P. 621.
Yamaguchi M., Motoyuki S., Hideo K. // Science. 2005. V. 307. P. 393.
Luo J., Cheng H., Asl K.M., Kiely C.J., Harmer M.P. // Science. 2011. V. 333. P. 1730.
Yu Z., Cantwell P.R., Gao Q., Yin D., Zhang Y., Zhou N., Rohrer G.S., Widom M., Luo J., Harmer M.P. // Science. 2017. V. 358. P. 97.
Dillon S.J., Tang M., Carter W.C., Harmer M.P. // Acta Mater. 2007. V. 55. P. 6208.
Cantwell P.R., Tang M., Dillon S.J., Luo J., Rohrer G.S., Harmer M.P. // Acta Mater. 2014. V. 62. P. 1.
Zhou N., Hu T., Luo J. // Curr. Opin. Solid State Mater. Sci. 2016. V. 20. P. 268.
Gao Q., Widom M. // Curr. Opin. Solid State Mater. Sci. 2016. V. 20. P. 240.
Kaplan W.D., Chatain D., Wynblatt P., Carter W.C. // J. Mater. Sci. 2013. V. 48. P. 5681.
Frolov T., Olmsted D.L., Asta M., Mishin Y. // Nat. Commun. 2013. V. 4. P. 1897.
Frolov T., Divinski S.V., Asta M., Mishin Y. // Phys. Rev. Lett. 2013. V. 110. P. 1.
Frolov T., Asta M., Mishin Y. // Curr. Opin. Solid State Mater. Sci. 2016. V. 20. P. 308.
Kang J., Glatzmaier G.C., Wei S.H. // Phys. Rev. Lett. 2013. V. 111. P. 1.
Gao Q., Widom M. // Phys. Rev. B. 2014. V. 90. P. 1.
Luo G.-N., You Y.-W., Liu C.S., Wang Z., Kong X.-S., Wu X., Chen J.-L., Lu G.-H. // Acta Mater. 2016. V. 120. P. 315.
Wu X., Kong X.S., You Y.W., Liu W., Liu C.S., Chen J.L., Luo G.N. // J. Appl. Phys. 2016. V. 120. 095901.
Li Y., Korzhavyi P.A., Sandström R., Lilja C. // Phys. Rev. Mater. 2017. V. 1. P. 1.
Hu C., Luo J. // Scr. Mater. 2019. V. 158. P. 11.
Behler J., Parrinello M. // Phys. Rev. Lett. 2007. V. 98. P. 1.
Cybenko G. // Math. Control. Signals Syst. 1989. V. 2. P. 303.
Artrith N., Urban A., Ceder G. // J. Chem. Phys. 2018. V. 148. 241711.
Jindal S., Chiriki S., Bulusu S.S. // J. Chem. Phys. 2017. V. 146. 204301.
Kondati Natarajan S., Behler J. // J. Phys. Chem. C. 2017. V. 121. P. 4368.
Shakouri K., Behler J., Meyer J., Kroes G.J. // J. Phys. Chem. Lett. 2017. V. 8. P. 2131.
Sanville E., Bholoa A., Smith R.,Kenny S.D. // J. Phys. Condens. Matter. 2008. V. 20. 285219.
Blöchl P.E. // Phys. Rev. B. 1994. V. 50. P. 17953.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Kresse G., Hafner J. // Phys. Rev. B. 1993. V. 47. P. 558.
Kresse G., Furthmüller J. // Phys. Rev. B. 1996. V. 54. P. 11 169.
Kresse G., Furthmüller J. // Comput. Mater. Sci. 2004. V. 6. P. 15.
Khorshidi A., Peterson A.A. // Comput. Phys. Commun. 2016. V. 207. P. 310.
Bartók A.P., Kermode J., Bernstein N., Csányi G. // Phys. Rev. X. 2018. V. 8. 041048.
Behler J. // J. Chem. Phys. 2011. V. 134. 074106.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал