

Коллоидный журнал, 2020, T. 82, № 6, стр. 749-760
Особенности формирования адсорбционных слоев продуктов механохимической модификации гуминовых кислот на границе раздела жидкость–газ
С. Л. Хилько 1, *, М. И. Рогатко 1, Р. А. Макарова 1, Р. Г. Семенова 1
1 Институт физико-органической химии и углехимии им. Л.М. Литвиненко
283114 Донецк-114, ул. Р. Люксембург, 70, Украина
* E-mail: sv-hilko@yandex.ru
Поступила в редакцию 26.03.2020
После доработки 15.05.2020
Принята к публикации 23.05.2020
Аннотация
Получены этокси- и аминопроизводные природных полимеров – гуминовых кислот – методом механохимического синтеза в вибрационном аппарате. Строение и физико-химические свойства их макромолекул изучены методами ИК- и УФ-спектроскопии. Определена их средняя молекулярная масса. Методами формы висячей капли и осциллирующей висячей капли исследованы тензиометрические и дилатационные реологические характеристики поверхностных слоев растворов модифицированных производных гуминовых кислот на границе с воздухом. Установлено, что формирование адсорбционных слоев солей гуминовых кислот согласуется с моделью адсорбции белков, учитывающей возможность существования макромолекул в n состояниях и их способность к агрегации в поверхностных слоях, при использовании соотношений модели в полуэмпирическом виде. Для этоксилированных производных гуминовых кислот показано увеличение модуля вязкоупругости поверхностных слоев по сравнению с немодифицированной формой. Аминопроизводные гуминовых кислот проявляют бóльшую устойчивость в кислой среде за счет наличия аминогрупп.
ВВЕДЕНИЕ
Главными и наиболее реакционноспособными компонентами гуминовых веществ являются гуминовые кислоты (ГК). ГК представляют собой один из наиболее обширных классов природных соединений. Это ароматические полиоксиполикарбоновые кислоты, способные вступать в ионные и донорно-акцепторные взаимодействия, образовывать водородные связи, участвовать в окислительно-восстановительных и сорбционных процессах [1–3], что позволяет использовать их в качестве эффективных и дешевых ПАВ, антиоксидантов, ингибиторов коррозии металлов, редокс-полимеров, сорбентов, основы для получения новых лекарственных препаратов и т.д. [1, 4–9].
Спецификой молекул ГК является их стохастическое строение, которое обусловлено естественным отбором наиболее устойчивых к биоразложению структур. Поскольку гуминовые вещества являются биогеополимерами, их состав определяется набором сходных молекул, который отражает характерное поведение полимеров нерегулярного строения [10]. Поэтому для ГК понятие “молекула” трансформируется в понятие “молекулярного ансамбля” [1, 3, 11]. Соли ГК являются природными полиэлектролитами, которые характеризуются невыраженной дифильностью структуры, то есть нерегулярным расположением гидрофильных и гидрофобных фрагментов в структуре макромолекул, подобно белковым макромолекулам.
Актуальной задачей современной химии является разработка методов направленной химической модификации природных соединений и исследование их физико-химических свойств с целью создания новых типов эффективных продуктов с заданными свойствами [12, 13]. В этой связи перспективной является разработка методов структурного модифицирования гуминовых веществ путем введения в состав их макромолекул новых функциональных фрагментов.
В настоящее время механохимические методы активации твердофазных реакций широко применяются в различных областях химии, химической технологии и материаловедения [14, 15]. Интерес к твердофазным механохимическим реакциям связан с возможностью их использования в технике, особенно при создании новых, так называемых “сухих” технологических процессов, которые более экологически безопасны и экономически выгодны по сравнению с традиционными [16, 17]. В процессах механосинтеза под действием механических сил вещества изменяют свой состав и строение. В результате чисто физические процессы измельчения и трения становятся причиной химических реакций. Тонкое измельчение твердых материалов сопровождается интенсивной электризацией, эмиссией электронов, выделением тепла и другими эффектами [18, 19].
При интенсивном механическом воздействии на органические вещества углей, торфа и продуктов их переработки (гуминовые вещества) наблюдается изменение их химического состава. Возрастает атомное соотношение Н/С, уменьшается содержание кислорода в гетероциклах, эфирных мостиках, альдегидных и лактонных группах. Увеличивается число активных функциональных групп: спиртовых, фенольных, карбоксильных, хиноидных. Уменьшается доля углерода, приходящегося на СН2-группы, и увеличивается доля СН3-групп. Изменения химического состава природных органических материалов связано с разрушением связей С–С (алифатические мостики, связи между ароматическими кольцами, алифатические боковые цепи) [20–23]. Образование спиртовых групп при тонком диспергировании обусловлено деструкцией наиболее слабых связей C–O в кислородных мостиках [20, 23].
Механохимические процессы деструкции органических полимеров связаны, прежде всего, с радикальным механизмом реакций [22–25]. Возможные реакции механохимического преобразования связей полимерного угольного вещества и полиэтиленгликоля приведены на рис. 1 [20–22].
Поверхностную активность природных (нативных) гуминовых соединений можно увеличить путем введения в состав их макромолекул дополнительных функциональных групп. Введение в состав гуминовых кислот оксиэтиленовых групп (–CH2–CH2–O–CH2–CH2–) позволит получить новые ПАВ, совмещающие анионактивные, за счет присутствующих в макромолекулах ГК групп СООН и ОН, и неионогенные свойства – за счет оксиэтиленовых групп. Введение аминогрупп позволит получить новые ПАВ, совмещающие анионактивные и катионактивные свойства.
Целью работы было проведение реакций этоксилирования и аминирования ГК в вибрационном аппарате и исследование тензиометрических и реологических свойств адсорбционных слоев продуктов этих реакций на границе фаз водный раствор–газ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Гуминовые кислоты выделяли из образцов бурого угля Александрийского месторождения однократной экстракцией при соотношении твердой и жидкой фаз 1 : 8 при температуре 100°C (ГК100). Затем из “сырого” экстракта получали нерастворимые в воде ГК осаждением 5%-ным раствором HCl, который добавляли при постоянном перемешивании до достижения pH 1–2. Выпавший осадок ГК отделяли от надосадочной жидкости центрифугированием и промывали дистиллированной водой до нейтральной реакции среды (pH 6–7). Промытые ГК сушили при 80°C до постоянной массы. Растворы гуматов натрия (ГН) для исследования получали растворением промытых и сухих ГК в 0.1 н. растворе NaOH.
Механохимическую твердофазную реакцию этоксилирования ГК проводили в присутствии полиэтиленгликоля (ПЭГ) с молекулярной массой 6000, а реакцию аминирования ГК – в присутствии мочевины, гуанидина или гидроперита в вибрационном аппарате 75Т-ДрМ при частоте вибрации ν = 50 Гц и амплитуде A = 3.0–5.0 мм. Величина виброускорения (I = 4π2Аν2) составляла 296–493 м/с2; сила F, действующая на слой материала со стороны рабочего тела массой MT = 1.13 кг, составляла 334–557 Н (F = MTI).
После обработки смеси компонентов в механореакторе продукты реакции многократно промывали водой. При этом отделяли избыток водорастворимых непрореагировавших компонентов – ПЭГ-6000, мочевины или гуанидина. Нерастворимые в воде производные ГК отделяли центрифугированием.
ИК-фурье-спектры образцов ГК записывали на спектрометре Bruker Tensor 37 в таблетках KBr (содержание ГК в таблетке составляло 0.5%).
Электронные спектры поглощения солей ГК регистрировали с помощью спектрофотометра Genesys 10S UV-Vis в растворах NaOH.
Количество активных кислых групп (–COOH и –OH) определяли методом потенциометрического титрования. Значения pH растворов измеряли на прецизионном pH-метре (Metrohm 744 pH Meter, Швейцария). Растворы натриевых солей ГК (0.05%) титровали 0.1 н. раствором HCl. Точки эквивалентности определяли как максимумы на дифференциальных кривых ΔpH/ΔV = f(VHCl), где VHCl – объем добавленного раствора HCl.
Величины средней молекулярной массы $(\bar {M})$ фракций ГК измеряли вискозиметрически с использованием методики определения $\bar {M}$ для гуминовых соединений [26–28].
Динамическое и равновесное поверхностное натяжение (γ, мН/м) водных растворов Na-солей ГК при их фиксированной концентрации в широком диапазоне времени жизни межфазной поверхности (t = 1–105 с) измеряли методом висячей капли с помощью тензиометра PAT-2P (Sinterface Technologies, Германия). Ошибка при измерении γ не превышала ±0.1 мН/м.
Дилатационные реологические характеристики поверхностных слоев на границе жидкость–газ изучали методом осциллирующей капли также с помощью тензиометра PAT-2P [29, 30]. В этом методе капля заданного объема формируется на конце капилляра. После достижения адсорбционного равновесия поверхность капли подвергается периодической синусоидальной деформации (осцилляции) малой амплитуды (относительные изменения площади поверхности A составляют ΔA/A = ± 7–8%) с частотой f в диапазоне 0.005–0.5 Гц. Результаты экспериментов с гармоническими осцилляциями поверхности капли были проанализированы с использованием преобразования Фурье [29, 30]:
где A0 – начальная площадь поверхности капли, Δγ – изменение поверхностного натяжения раствора ПАВ при изменении площади поверхности капли при ее осцилляции.Дилатационный модуль $\left| Е \right|$ характеризует вязкоупругие свойства поверхностных слоев ПАВ и учитывает все релаксационные процессы, влияющие на поверхностное натяжение γ. При малой амплитуде ΔA гармонических осцилляций поверхности с угловой частотой Ω = 2π f, $\Delta A = \Delta \bar {A}\exp (i\Omega t),$ имеет место следующее выражение для дилатационного модуля вязкоупругости [31, 32]:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
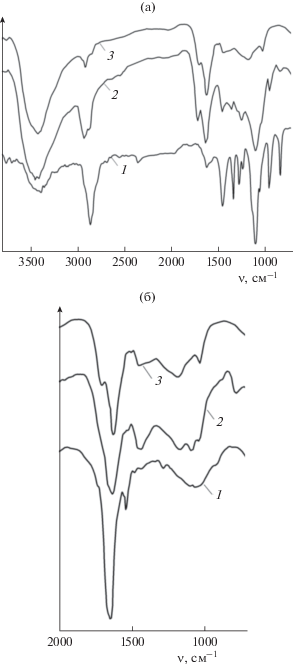
Как следует из рис. 2а, в ИК-спектре продукта механохимической реакции ГК с ПЭГ-6000 появляется полоса поглощения в области 1100 см–1, которая соответствует валентным колебаниям νС–О эфирной группы (–СН2–О–СН2–). Эта интенсивная полоса поглощения наблюдается в ИК спектре ПЭГ-6000, но отсутствует в спектре исходного образца ГК (ГКисх). По сравнению с образцом ГКисх увеличивается интенсивность полосы поглощения в области 2950 см–1, соответствующей колебаниям алифатических групп, что может указывать на увеличение доли алифатической составляющей в макромолекуле после присоединения этоксигрупп.
Рис. 2.
ИК-спектры ГК и модифицирующих добавок: (а) 1 – ПЭГ-6000, 2 – ГКэтокси, 3 – ГКм/а; (б) 1 – гуанидин, 2 – ГКамино, 3 – ГКм/а.
Макромолекулы гуминовых соединений содержат конденсированные ароматические фрагменты и алифатические цепи разной длины и степени разветвленности. Для анализа молекулярных свойств гуминовых веществ широко используется электронная спектроскопия поглощения. Соотношение между ароматической и алифатической частями в макромолекулах гуминовых соединений принято определять как отношение значений оптической плотности при 465 и 665 нм, E465/E665 [33–35]. Низкие значения этого отношения указывают на превалирование конденсированных ароматических фрагментов, а его высокие значения означают преобладание алифатических фрагментов в структуре макромолекул гуминовых веществ.
Как следует из данных, приведенных в табл. 1, для раствора соли этоксилированной гуминовой кислоты (ГНэтокси) величина E465/E665 выше, чем для растворов солей ГНисх и ГНм/а, где ГНм/а – Na-соль механоактивированной исходной ГК. Это также указывает на увеличение доли алифатических фрагментов в структуре макромолекул. Кроме того, возрастает средняя молекулярная масса $\bar {M}$ этоксилированного образца ГН.
Таблица 1.
Характеристики образцов натриевых солей ГК
| Образец ГН | [OH], мг-экв/г |
[COOH], мг-экв/г |
E465/E665 | $\bar {M}$ |
|---|---|---|---|---|
| ГНисх | 3.2 | 3.2 | 5.05 | 14 000 |
| ГНм/а | 3.4 | 3.4 | 5.47 | 13 000 |
| ГНэтокси | 3.2 | 2.4 | 6.94 | 15 500 |
| ГНамино | 1.6 | 4.8 | 6.04 | 12 500 |
| ГН(ГК + мочевина) | 1.6 | 4.8 | 5.80 | 12 500 |
| ГН(ГК + гидроперит) | 2.0 | 5.8 | 6.78 | 12 000 |
На основании приведенных данных можно полагать, что в результате механохимической реакции происходит включение в структуру макромолекулы ГК оксиэтиленовых групп (–CH2–CH2–O–CH2–CH2–).
Присоединение оксиэтиленовых групп может происходить по фрагментам макромолекулы ГК, содержащим “кислый” водород: группам OH, СООН, NН2 [36, 37]. По данным потенциометрического титрования растворов в этоксилированном образце ГН уменьшается количество групп COOH (табл. 1), что может указывать на участие этих групп в механосинтезе. С другой стороны, механохимические превращения полимеров связаны с разрывом макромолекулярных цепей, в результате чего реакции могут идти и по другим путям [14–17, 38]. При механическом воздействии на ПЭГ возможна деструкция его молекул [24, 39]. Разрыв слабых с механохимической точки зрения связей в ГК с одновременным разрывом связей в макромолекулах ПЭГ (рис. 1) может приводить к образованию сополимеров – этоксигуминовых кислот.
В ИК-спектрах аминопроизводных ГК наблюдаются изменения по сравнению с исходными реагентами (рис. 2б). Так, в спектре продукта взаимодействия ГК с гуанидином регистрируется широкая полоса поглощения в области 1600–1700 см–1 в результате наложения полос поглощения C=O гуминового фрагмента и валентных колебаний C=N и деформационных колебаний NH2 гуанидиновой части. Кроме того, в области 1000–1200 см–1 наблюдается изменение по сравнению с исходной гуминовой кислотой, также свидетельствующее о механохимическом взаимодействии ГК с гуанидином.
Как следует из табл. 1, для растворов Na-солей аминопроизводных ГК (ГНамино) отношение E465/E665 выше, а величина $\bar {M}$ ниже, чем для немодифицированных форм. Возможно, при механохимических реакциях с азотсодержащими органическими веществами деструкция связей в макромолекулах ГК более выражена, чем при механохимической активации ГКисх, и может приводить к увеличению доли алифатических фрагментов, что особенно заметно в случае реакции с гидроперитом. Результаты потенциометрического титрования показали существенные различия между немодифицированными и аминированными образцами солей ГК. В солях аминогуминовых кислот уменьшается количество активных гидроксильных групп и увеличивается количество карбоксильных групп (табл. 1).
Известно, что мочевина способна реагировать с соединениями, содержащими активные OH-группы, спиртами и фенолами, с образованием уретанов и аммиака или заменой OH-группы на группу NН2 [40]. Уменьшение количества гидроксильных групп в макромолекулах ГК после механохимических реакций с мочевиной, гуанидином или гидроперитом указывает на возможность такого взаимодействия, а не только реакций, связанных с разрывом макромолекулярных цепей. Возрастание количества карбоксильных групп в продуктах аминирования ГК может быть обусловлено окислением фрагментов макромолекул при механохимическом воздействии. Наиболее сильно это выражено в реакции ГК с гидроперитом, содержащим активную перекись водорода, которая выделяется при распаде этого клатрата в результате механохимического процесса.
Включение этоксильных и аминогрупп в структуру макромолекул ГК может оказывать влияние на процесс формирования адсорбционных слоев таких ПАВ на межфазных границах.
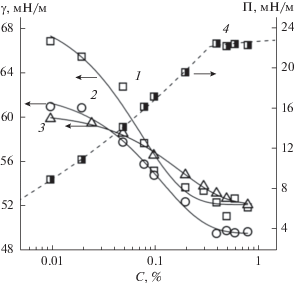
В результате введения этоксигрупп в структуру макромолекул ГК снижается динамическое поверхностное натяжение водных растворов соответствующей Na-соли (ГНэтокси) по сравнению с растворами ГНисх и ГНм/а (рис. 3). В то же время, введение аминогрупп в состав ГК слабо влияет на динамику снижения поверхностного натяжения водного раствора соответствующего гумата. Как следует из рис. 4, равновесное поверхностное натяжение на границе фаз жидкость–газ в случае растворов ГНэтокси также ниже, чем для растворов ГНисх и ГНм/а. В области низких концентраций для этоксилированной и аминосодержащей форм ГН наблюдается существенное снижение величины γ по сравнению с ГНисх.
Рис. 3.
Зависимости динамического поверхностного натяжения от времени для растворов Na-солей ГК. C, %: 0.1 (1, 3, 5) и 0.8 (2, 4, 6). 1, 2 – ГНисх; 3, 4 – ГНамино; 5, 6 – ГНэтокси.
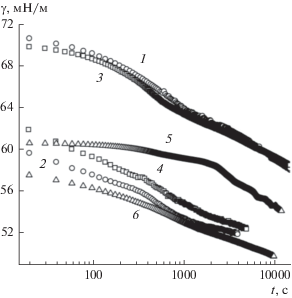
Рис. 4.
Зависимости равновесного поверхностного натяжения (1–3) и поверхностного давления (4) от концентрации растворов ГНисх (1), ГНэтокси (2, 4) и ГНамино (3). Пунктирная кривая – расчет для ГНэтокси по теоретической модели (пояснения в тексте).
Для обработки экспериментальных зависимостей была использована модель адсорбции белков, как природных полиэлектролитов, с учетом возможности существования макромолекул в n состояниях и их способности к агрегации в адсорбционных слоях при полимолекулярной адсорбции.
Как известно [41], макромолекулам белков свойственна упорядоченная пространственная структура, в которой полипептидные цепи свернуты в компактные, жесткие и неоднородные образования, различающиеся для разных видов белков. С биологической точки зрения макромолекулы белков являются высокоорганизованной системой, которая обеспечивает выполнениe специфических биологических функций в организме.
В отличие от белков, ГК, в силу специфики их строения, не способны к образованию высокоупорядоченных пространственных структур. Однако способность гуминовых макромолекул к сильным внутри- и межмолекулярным гидрофобным взаимодействиям и образованию большого числа водородных связей может приводить к формированию статистических клубков с различной плотностью упаковки.
В этой связи можно попытаться использовать соотношения теоретической модели, разработанной для описания поверхностных свойств растворов белков [42], в качестве полуэмпирических применительно к растворам гуминовых веществ.
В этой модели предполагается, что на границе фаз адсорбция может происходить с образованием нескольких слоев, причем первый слой оказывает доминирующее влияние на параметры образования второго и последующих слоев. Модель характеризуется следующими характеристиками: ω0 – молярной площадью в расчете на один сегмент макромолекулы в первом адсорбционном слое (инкремент молярной площади), ωmin – минимальной молярной площадью в первом адсорбционном слое, ωmax – максимальной молярной площадью в этом слое. Взаимосвязь между этими величинами определяется соотношением:
(1)
$\begin{gathered} {{\omega }_{0}} = {{({{\omega }_{{\text{m}}}}_{{{\text{ax}}}}--{{\omega }_{{{\text{min}}}}})} \mathord{\left/ {\vphantom {{({{\omega }_{{\text{m}}}}_{{{\text{ax}}}}--{{\omega }_{{{\text{min}}}}})} {(n - {\text{1}})}}} \right. \kern-0em} {(n - {\text{1}})}}~ \\ {\text{или }}n = \frac{{{{\omega }_{{\max }}} - {{\omega }_{{\min }}}}}{{{{\omega }_{0}}}} + 1, \\ \end{gathered} $В модели [42] предполагается также, что общая адсорбция макромолекул ПАВ в первом слое во всех n состояниях равна
Средняя молярная площадь (ω) связана со степенью заполнения поверхности (θ) в первом адсорбционном слое макромолекулами полиэлектролита во всех n состояниях соотношением
(3)
$\theta = \omega {{\Gamma }^{{(1)}}} = \sum\limits_{i = 1}^n {{{\omega }_{i}}\Gamma _{i}^{{(1)}}} .$Величина полимолекулярной адсорбции полиэлектролита рассчитывается как
где b(X) – константа адсорбционного равновесия для каждого из L “внешних” адсорбционных слоев, c – концентрация ПАВ; величина Г(1)(с) рассчитывается по формуле (2).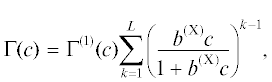
При полимолекулярной адсорбции возможна агрегация макромолекул полиэлектролитов в поверхностном слое (посткритическое состояние). Поверхностный слой, в котором нет агрегатов макромолекул ПАВ, определяется как докритическое состояние. Переход в посткритическое состояние происходит при определенной приповерхностной концентрации ПАВ, c *, при которой поверхностное давление равно Π *, а величина адсорбции – Γ *. При этом предполагается, что уравнение состояния (зависимость поверхностного давления от приповерхностной концентрации), выраженное через степень заполнения поверхности θ, имеет одинаковую форму как для докритической, так и для посткритической области, а именно:
(5)
$ - \frac{{\Pi {{\omega }_{0}}}}{{RT}} = \ln \left( {1 - {{\theta }_{{\text{P}}}}} \right) + {{\theta }_{{\text{P}}}}\left( {1 - {{{{\omega }_{0}}} \mathord{\left/ {\vphantom {{{{\omega }_{0}}} \omega }} \right. \kern-0em} \omega }} \right) + {{a}_{{\text{P}}}}\theta _{{\text{P}}}^{2},$В посткритической области агрегация макромолекул при полимолекулярной адсорбции приводит к изменению выражения для полной адсорбции (Г):
(6)
$\Gamma = \Gamma {\kern 1pt} {\kern 1pt} {\text{*}} + \frac{1}{{{{n}_{{\text{a}}}}}}\left( {{{\Gamma }_{{\text{P}}}} - \Gamma {\kern 1pt} {\text{*}}} \right),$Степень заполнения поверхности в агрегированном слое рассчитывается как
Степень заполнения поверхности и величина адсорбции в первом слое (Г(1)) с учетом агрегации макромолекул полиэлектролита могут быть рассчитаны также следующим образом:(8)
${{\theta }_{{\text{P}}}} = \theta {\kern 1pt} * + \frac{1}{{{{n}_{{\text{a}}}}}}\left( {\theta - \theta {\kern 1pt} *} \right),\,\,\,\,{{\Gamma }^{{\left( {\text{1}} \right)}}} = {{{{\theta }_{{\text{Р}}}}} \mathord{\left/ {\vphantom {{{{\theta }_{{\text{Р}}}}} \omega }} \right. \kern-0em} \omega }.$Величины θP и Г(1) используются для расчета поверхностного давления и величины общей адсорбции в посткритической области по соотношениям (5) и (4).
Для описания адсорбционного поведения гуминовых соединений в рамках приведенной выше модели были использованы параметры ωmax и ωmin, которые находили по результатам определения толщины δ адсорбционного слоя: $\delta \cong {V \mathord{\left/ {\vphantom {V {{{\omega }_{{\max \left( {\min } \right)}}}}}} \right. \kern-0em} {{{\omega }_{{\max \left( {\min } \right)}}}}},$ где V – молярный объем полиэлектролита. Согласно данным [43, 44], толщина адсорбционного слоя для различных образцов солей гуминовых веществ может изменяться в пределах 3–20 нм и достигать 50 нм. Если принять, что величина молярного объема примерно равна средней молекулярной массе гуминового вещества, V $ \cong $ $\bar {M}$ (при плотности гуминового вещества, близкой к единице), то величины ωmin и ωmах могут находиться в диапазоне от 0.5 × 106 до 2.0 × × 105 м2/моль. При аппроксимации экспериментальных зависимостей γ = f(CПАВ) значения ωmin и ωmах выбирали близкими к величинам из этого диапазона. Параметры ω0, aP, b(X), na находили фитированием экспериментальной зависимости γ = f(CГК). Величины n рассчитывали по формуле (1). При обсуждении результатов мы, однако, не учитывали значения aPи b(X), которые достаточно сложно точно оценить, ограничившись значениями параметров ω0 и nа, найденными фитированием, и величинами n, оцененными приближенно.
Как было показано в работах [7, 45], для солей ГК наиболее вероятна бимолекулярная адсорбция. Используя возможности приведенной выше модели, мы рассчитали зависимости Π = f(C) для Na-солей ГК. В качестве примера на рис. 4 приведены экспериментальная и рассчитанная (пунктирная кривая 4) зависимости для ГНэтокси. Видно их хорошее согласие.
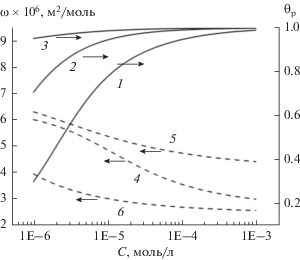
Для солей ГК были получены концентрационные зависимости величин молярной площади, ω, и степени заполнения межфазной поверхности, θР. Теоретические кривые рассчитаны с помощью программ ProteinR и ProteinG [46]. С ростом концентрации растворов солей ГК уменьшаются значения ω и возрастает степень заполнения поверхностного слоя (рис. 5). Величина θP для всех образцов солей ГК закономерно возрастает и в исследуемом диапазоне концентрации практически достигает своего максимума. При этом для образца ГНамино значение θP уже при минимальной концентрации раствора равно 0.96 и заметно отличается от соответствующих значений для исходного и этоксилированного ГН, что может быть связано с меньшей средней молекулярной массой образца ГНамино и большим количеством карбоксильных групп, определяющим заряд макромолекулы (табл. 1).
Рис. 5.
Расчетные зависимости степени заполнения поверхностного слоя θР (1–3) и средней молярной площади ω (4–6) при бимолекулярной адсорбции Na-солей ГК от концентрации их растворов: 1, 4 – ГНисх, 2, 5 – ГНэтокси, 3, 6 – ГНамино.
Рассчитанные значения параметров модели адсорбции полиэлектролитов для солей ГК приведены в табл. 2. Отношение ωmax/ωmin для образца ГНэтокси близко к величинам такого отношения для глобулярных белков [47]. Возможное количество состояний n, которое может принимать макромолекула ГНэтокси, меньше, чем для других образцов, а способность к агрегации в поверхностном слое (значение na) максимальна в этом ряду ПАВ.
Таблица 2.
Параметры модели полимолекулярной адсорбции полиэлектролитов для натриевых солей ГК
| Образец ГН | ω0 × 105, м2/моль | ωmin × 106, м2/моль | ωmах × 106, м2/моль | ωmax/ωmin | n | na |
|---|---|---|---|---|---|---|
| ГНисх | 4.0 | 3.0 | 15.0 | 5.0 | 31 | 6 |
| ГНм/а | 3.0 | 2.5 | 10.0 | 4.0 | 26 | 7 |
| ГНэтокси | 5.0 | 4.0 | 10.0 | 2.5 | 13 | 23 |
| ГНамино | 7.7 | 2.5 | 14.0 | 5.6 | 16 | 10 |
Дилатационные реологические характеристики межфазной поверхности позволяют получать новую важную информацию о процессах формирования и структуре адсорбционных слоев высокомолекулярных полиэлектролитов.
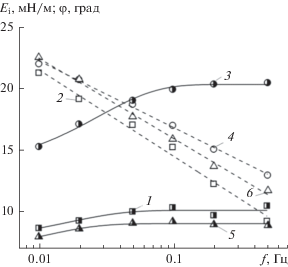
Согласно данным, приведенным на рис. 6, величины модулей вязкоупругости и упругости (реальная компонента величины $\left| Е \right|$) возрастают при увеличении частоты осцилляции капли. Значения $\left| Е \right|$ и Еr значительно выше для этоксилированного образца по сравнению с немодифицированной и аминированной формами ГН. При этом для ГНамино величины $\left| Е \right|$ и Еr слабо отличаются от соответствующих величин для ГНисх. Следует отметить близкие значения $\left| Е \right|$ и Еr как для ГНисх, так и для его модифицированных производных. Модуль Еi (мнимая компонента $\left| Е \right|$) незначительно возрастает, а величина фазового угла уменьшается с ростом частоты осцилляции капли для всех образцов ГН (рис. 7). Такое поведение (рис. 6 и 7) характерно для упругих поверхностных слоев ПАВ [47–51].
Рис. 6.
Зависимости модуля вязкоупругости $\left| Е \right|$ (1, 3, 5) и действительной части динамической поверхностной упругости Еr (2, 4, 6) для адсорбционных слоев ГНисх (1, 2), ГНэтокси (3, 4) и ГНамино (5, 6) от частоты осцилляции площади поверхности капли f. C = 0.4%.
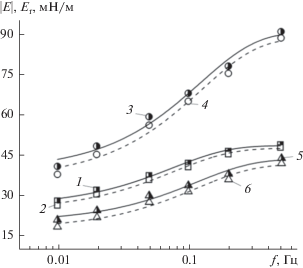
Рис. 7.
Зависимости мнимой части динамической поверхностной упругости Еi (1, 3, 5) и фазового угла ${\varphi }$ (2, 4, 6) для адсорбционных слоев ГНисх (1, 2), ГНэтокси (3, 4) и ГНамино (5, 6) от частоты осцилляции площади поверхности капли f. C = 0.4%.
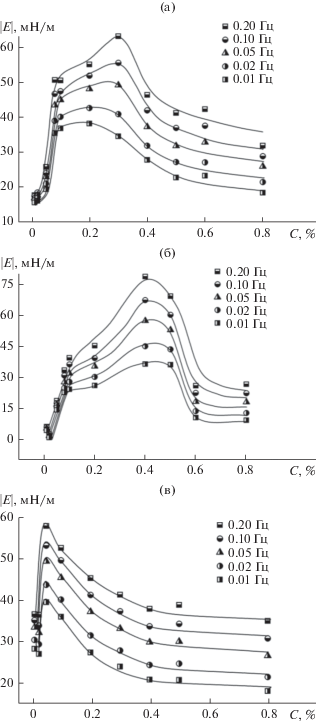
Как следует из рис. 8, зависимости $\left| Е \right|$ = f(C) для образца ГНисх и его модифицированных производных имеют выраженный максимум. Для образца ГНисх максимальные значения модуля дилатационной вязкоупругости $\left| Е \right|$ наблюдаются при концентрации раствора C ≈ 0.2–0.3%, для образца ГНэтокси – при C ≈ 0.4–0.5%, а для ГНамино – при C ≈ 0.05%.
Рис. 8.
Зависимости модуля поверхностной вязкоупругости от концентрации Na-солей ГК при разных частотах осцилляции капли: (а) ГНисх, (б) ГНэтокси, (в) ГНамино.
Низкие экспериментальные значения фазового угла и мнимой части динамической поверхностной упругости (Ei) при частоте осцилляции капли f = 0.1 Гц позволяют сравнивать значения модуля вязкоупругости $\left| {\rm E} \right|$ с величинами предельного (высокочастотного) модуля упругости ${{E}_{0}} = {{ - d\gamma } \mathord{\left/ {\vphantom {{ - d\gamma } {d\ln {{\Gamma }_{\Sigma }}}}} \right. \kern-0em} {d\ln {{\Gamma }_{\Sigma }}}}$ при этой частоте [47, 48]. Экспериментальные зависимости модуля поверхностной вязкоупругости растворов ГНэтокси и глобулярных белков от поверхностного давления при бимолекулярной адсорбции представлены на рис. 9. Там же приведены рассчитанные зависимости предельного (высокочастотного) модуля упругости E0 для этих веществ. В работах [49, 51] показано, что величина $\left| {\rm E} \right|$ для глобулярных белков монотонно увеличивается с ростом поверхностного давления до некоторого предельного значения. В области высоких значений поверхностного давления, выше 12–15 мН/м, модуль дилатационной вязкоупругости адсорбционных слоев глобулярных белков не является чисто упругим и зависит от механизмов релаксации.
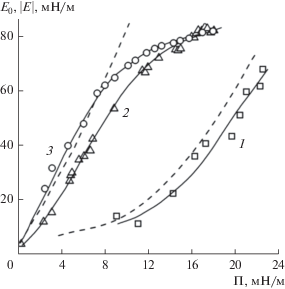
Для раствора ГНэтокси величина $\left| {\rm E} \right|$ монотонно возрастает с ростом поверхностного давления. Зависимость $\left| {\rm E} \right|$ = f(Π) в области Π ≤ 22 мН/м практически совпадает с расчетной зависимостью ${{E}_{0}} = {{ - d\gamma } \mathord{\left/ {\vphantom {{ - d\gamma } {d\ln {{\Gamma }_{\Sigma }}}}} \right. \kern-0em} {d\ln {{\Gamma }_{\Sigma }}}}.$ Отсюда следует, что при увеличении поверхностного давления вплоть до 22 мН/м, модуль вязкоупругости адсорбционного слоя ГНэтокси является упругим.
Для растворов ГНисх и ГНамино модель бимолекулярной адсорбции отражает экстремальный ход зависимости модуля поверхностной вязкоупругости от поверхностного давления (рис. 10). Такой характер этой зависимости является следствием уменьшения молярной площади полиэлектролита в поверхностном слое с ростом адсорбции. Подобное поведение свойственно белкам с гибкими цепями [48, 50, 58], макромолекулы которых способны изменять молярную площадь на границе фаз в большей степени по сравнению с макромолекулами глобулярных белков. Значение отношения ωmax/ωmin для образцов исходного и аминированного ГН выше, чем для этоксилированного ГН (табл. 2), и занимают промежуточное положение относительно значений этого отношения для глобулярного β-лактоглобулина (ωmax/ωmin = 2) и гибкоцепного β-казеина (ωmax/ωmin = 10) [47].
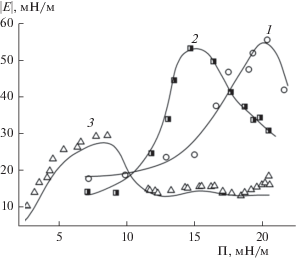
Механизм, определяющий различия в реологическом поведении глобулярных и гибкоцепных белков, обсуждался в работах [47, 48].
Заметные отличия в реологическом поведении макромолекул ГНэтокси по сравнению с макромолекулами других образцов ГН могут быть связаны с изменением конформационных характеристик ГНэтокси и усилением его способности к агрегации в поверхностном слое (табл. 2), которые обусловлены сополимеризацией с ПЭГ.
В табл. 3 представлены максимальные значения модуля вязкоупругости, полученные экспериментально для солей ГК в нашей работе, и, для сравнения, величины $\left| {\rm E} \right|$max для некоторых известных этоксилированных ПАВ и белков, взятые из литературных источников [48–58]. Параметр $\left| {\rm E} \right|$mах для ГНэтокси превышает значения этой величины для приведенных в табл. 3 этоксилированных ПАВ и глобулярных белков.
Таблица 3.
Экспериментальные значения максимального модуля вязкоупругости для адсорбционных слоев различных ПАВ на границе фаз раствор–газ
| ПАВ | |E|max, мН/м |
|---|---|
| ГНисх | 63 |
| ГНэтокси | 90 |
| ГНамино | 65 |
| Полиэтиленгликоль (ПЭГ-6000) | 9 |
| C14Н29(ОСН2СН2)8, | 60 [52, 53] |
| C12Н25(ОСН2СН2)8, | 40 [52, 53] |
| С10Н21(ОСН2СН2)8 | 20 [52, 53] |
| Тритон Х-100 | 12 [54, 55] |
| Тритон X-45 (этоксилаты октилфенола) | 13 [54, 55] |
| Твин 20 (полисорбат) | 26 [56] |
| Альбумин | 70 [57] |
| β-Казеин | 30 [48, 50, 58] |
| β-Лактоглобулин | 80 [49, 51] |
| Овальбумин | 80 [49, 51] |
Известно, что соли ГК могут существовать в щелочных водных растворах (с pH 10–13) в виде развернутых цепей и в виде плотных статистических клубков – в кислых растворах, при pH 1–2 [59, 60]. ГК в протонированной форме нерастворимы в воде [59, 61]. Конформационные превращения растворов солей ГК при снижении pH раствора вызывают агрегацию их макромолекул вплоть до полного выпадения в осадок при критических значениях pH. Изменения конформации и агрегация макромолекул ГК в водных растворах связаны со степенью диссоциации их функциональных групп (–COOH и –OH) [59–63].
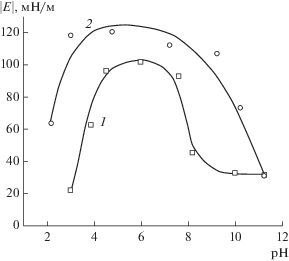
Как следует из рис. 11, снижение pH среды приводит к возрастанию модуля поверхностной вязкоупругости растворов солей ГК. В области pH ≤ 3 значения $\left| Е \right|$ уменьшаются. При переходе от щелочной к нейтральной и слабокислой среде зависимость $\left| Е \right|$ = f(pH) для ГНамино значительно отличается от таковой для образца ГНисх. Более высокие значения $\left| Е \right|$ для поверхностных слоев растворов ГНамино могут быть связаны с бóльшим количеством карбоксильных групп в структуре его макромолекул (табл. 1). Снижение pH среды способствует уменьшению заряда поверхностного слоя за счет протонирования карбоксильных и гидроксильных групп. Уменьшение заряда поверхностного слоя приводит к уменьшению электростатической составляющей адсорбционного барьера и позволяет выйти на поверхность дополнительному количеству ПАВ, что и приводит к заметному росту модуля поверхностной вязкоупругости. При pH < 3 величина $\left| Е \right|$ резко снижается, причем в значительно большей степени для немодифицированного образца ГН. Частичное осаждение ГК приводит к уменьшению их концентрации в поверхностном слое и снижению величины модуля дилатационной вязкоупругости. Как следует из рис. 11, ГНамино проявляет бóльшую устойчивость при критических значениях pH, величина $\left| Е \right|$ остается высокой даже при pH 2.0 (примерно такой же, как при pH 10.0). Такой характер зависимости $\left| Е \right|$ = f(pH), вероятно, связан с введением аминогрупп в структуру макромолекул ГК, поскольку заряд ${\text{NH}}_{3}^{ + }$-групп в кислой области pH способен увеличивать их растворимость при наличии протонированных карбоксильных и гидроксильных группах.
Рис. 11.
Зависимость модуля поверхностной вязкоупругости от pH для растворов Na-солей ГК: ГНисх (1), ГНамино (2). C = 0.8%, f = 0.1 Гц.
Различия в поведении солей ГК на границе фаз водный раствор–воздух обусловлены различиями в структуре макромолекул исходного и модифицированных образцов. Особенности поведения модифицированных образцов ГК обусловлены протеканием механохимических реакций, приводящих к появлению в структуре макромолекул новых функциональных групп. Их отличия от немодифицированного образца определяются перестройкой макромолекул ГК, связанной с преобразованием конденсированного ядра и линейных фрагментов, изменением гидрофильно-гидрофобного баланса, конформационной динамикой полимерных цепей в поверхностном слое и/или в объеме раствора.
ЗАКЛЮЧЕНИЕ
Механохимическим методом в вибрационном аппарате синтезированы новые ПАВ – этоксилированные и аминированные производные гуминовых кислот. Введение функциональных групп в структуру макромолекул ГК подтверждено методами спектроскопии, вискозиметрии и потенциометрического титрования.
Методом формы висячей капли исследовано влияние строения производных ГК на динамическое и равновесное поверхностное натяжение водных растворов их натриевых солей. Характер изотерм равновесного поверхностного натяжения растворов изученных соединений соответствуeт модели реального двумерного раствора, разработанной для белков, с учетом возможности существования макромолекул в n состояниях и способности к агрегации в поверхностных слоях при полимолекулярной адсорбции. В рамках этой модели соли этоксилированных производных ГК обладают высокой способностью к агрегации в поверхностном слое и малой конформационной изменчивостью, подобно глобулярным белкам. Соли аминопроизводных ГК, согласно модели, проявляют свойства гибкоцепных белков.
Методом формы осциллирующей капли исследованы дилатационные реологические характеристики поверхностных слоев растворов солей модифицированных ГК. Экспериментальная зависимость модуля дилатационной вязкоупругости от поверхностного давления для раствора этоксигумата натрия удовлетворительно согласуется с зависимостью предельного (высокочастотного) модуля упругости E0 = f(П), рассчитанной по той же модели, что и равновесное поверхностное натяжение. При значениях поверхностного давления до 22 мН/м модуль вязкоупругости адсорбционных слоев этоксилированного гумата натрия остается упругим. Образец ГНэтокси характеризуется высоким значением модуля дилатационной вязкоупругости $({{\left| {\rm E} \right|}_{{\max }}}$ = 90 мН/м), превышающим величины ${{\left| {\rm E} \right|}_{{\max }}},$ характерные для некоторых этоксилированных ПАВ и глобулярных белков. Для исходного образца ГН и его аминированной формы модель бимолекулярной адсорбции отражает экстремальный ход зависимости модуля дилатационной вязкоупругости от поверхностного давления, что характерно для гибкоцепных белков.
Соли аминогуминовых кислот более устойчивы в кислой области pH по сравнению с исходным образцом ГН, что проявляется в более высоких значениях модуля поверхностной вязкоупругости и может быть связано с протонированием аминогрупп в структуре макромолекул.
Список литературы
Попов А.И. Гуминовые вещества: свойства, строение, образование / Под ред. Ермакова Е.И. СПб.: Изд-во СПбГУ, 2004.
Орлов Д.С. // Соросовский образоват. журн. 1997. № 2. С. 56.
Piccolo A. // Soil Sci. 2001. V. 166. P. 810.
Мальцева Е.В., Шеховцова Н.С., Шиляева Л.П., Юдина Н.В. // Журн. физ. химии. 2017. Т. 91. С. 1174.
Уразова Т.С., Бычков А.Л., Ломовский О.И. // Журн. прикл. химии. 2014. Т. 87. С. 664.
Skripkina T.S., Bychkov A.L., Tikhova V.D., Smolyakov B.S., Lomovsky O.I. // Environ. Technol. Innov. 2018. V. 11. P. 74.
Хилько С.Л., Ковтун А.И., Файнерман В.Б. // Коллоид. журн. 2010. Т. 72. С. 851.
Хилько С.Л., Ковтун А.И., Файнерман В.Б. // Коллоид. журн. 2011. Т. 73. С. 97.
Беркович А.М. Применение гуминовых и гуминоподобных препаратов в ветеринарии и медицине // URL: http://www.humipharm.ru/research/prim.pdf. 2007. 29 с.
Cook R.L., Langford C.H. Understanding Humic Substances. Advanced Methods, Properties and Applications. Cambridge: Royal Soc. Chem., 1999.
Перминова И.В. Анализ, классификация и прогноз свойств гумусовых кислот: Дис. … докт. хим. наук. М.: МГУ, 2000.
Овчинников Ю.А. Биоорганическая химия. М.: Просвещение, 1987.
Смирнова И.Г., Трифонова Ж.П., Катруха Г.С. // Вестник МГУ. Сер. 2. Химия. 2006. Т. 47. С. 149.
Фундаментальные основы механической активации, механосинтеза и механохимических технологий / Отв. ред. Авакумов Е.Г. Новосибирск: Изд-во СО РАН, 2009.
Baláž P. // Chem. Soc. Rev. 2013. V. 42. P. 7571.
Болдырев В.В. // Успехи химии. 2006. Т. 75. С. 203.
James S.L., Friscic T. // Chem. Commun. 2013. V. 49. P. 5349.
Хрусталев Ю.А., Хренкова Т.М., Лебедев В.В. // До-кл. АН СССР. 1981. Т. 257. С. 418.
Хрусталев Ю.А., Хренкова Т.М., Лебедев В.В., Топоров Ю.П. // Химия тверд. топлива. 1983. № 4. С. 64.
Хренкова Т.М. Механохимическая активация углей. М.: Недра, 1993.
Хренкова Т.М., Кирда В.С. // Химия тверд. топлива. 1994. № 6. С. 36.
Иванов А.А., Юдина Н.В., Ломовский О.И. // Известия Томского политехнического университета. 2006. Т. 309. № 5. С. 73.
Лебедев В.В., Никанорова Л.П. // Химия тверд. топлива. 1985. № 2. С. 35.
Радциг В.А. // Хим. физика. 2004. Т. 23. № 10. С. 70.
Бутягин П.Ю. // Успехи химии. 1971. Т. 40. С. 1935.
Сивакова Л.Г., Лесникова Н.П., Ким Н.М., Ротова Г.М. // Химия тверд. топлива. 2011. № 1. С. 3.
Kawahigashi M., Sumida H., Yamamoto K. // J. Colloid Interface Sci. 2005. V. 284. P. 463.
Visser S.A. // Plant Soil. 1985. V. 87. P. 209.
Loglio G., Pandolfini P., Miller R., Makievski A.V., Ravera F. Liggieri L. // Studies in Interface Science / Ed. by Möbius D., Miller R. Amsterdam: Elsevier, 2001. V. 11. P. 439.
Zholob S.A., Makievski A.V., Miller R., Fainerman V.B. // Adv. Colloid Interface Sci. 2007. V. 134–135. P. 322.
Ravera F., Liggieri L., Loglio G. // Progress in Colloid and Interface Science / Ed. by Miller R., Liggieri L. Leiden: Brill, 2009. V. 1. P. 137.
Zholob S.A., Kovalchuk V.I., Makievski A.V., Kragel J., Fainerman V.B., Miller R. // Progress in Colloid and Interface Science / Ed. by Miller R., Liggieri L. Leiden: Brill, 2009. V. 1. P. 77.
Naidja A., Huang P.M., Anderson D.W., Kessel C.V. // Appl. Spectrosc. 2002. V. 56. P. 318.
Chen J., Gu B., LeBoeuf E.J., Pan H., Dai S. // Chemosphere. 2002. V. 48. P. 59.
Silva R.R., Lucena G.N., Machado A.F., Freitas G.A., Matos A.T., Abrahao W.A.P. // Comun. Sci. 2018. V. 9. P. 264.
Umbach W., Stein W. // J. Am. Oil Chem. Soc. 1971. V. 48. P. 394.
Sallaya P., Farkas L., Szlovák Z., Fogassy G. // J. Surfactants Deterg. 2002. V. 5. P. 353.
Барамбойм Н.К. Механохимия высокомолекулярных соединений. М.: Химия, 1978.
Oprea C., Vasiliu-Oprea C., Dan F. Macromolecular Mechanochemistry: Polymer Mechanochemistry. Cambridge: Cambridge Int. Sci. Publ., 2003.
Резников В.А. Химия азотсодержащих органических соединений. Новосибирск: НГУ, 2006.
Ленинджер А. Основы биохимии. М.: Мир, 1985.
Fainerman V.B., Aksenenko E.V., Makievski A.V., Trukhin D.V., Yeganehzad S., Gochev G., Miller R. // Food Hydrocoll. 2020. V. 106. P. 1.
Pranzas P.K., Willumeit R., Gehrke R., Thieme J., Knöchel A. // Anal. Bioanal. Chem. 2003. V. 376. P. 618.
Shang C., Rice J.A. // J. Colloid Interface Sci. 2007. V. 305. P. 57.
Хилько С.Л., Рогатко М.И., Макарова Р.А., Семенова Р.Г. // Коллоид. журн. 2020. Т. 82. С. 119.
Программы и примеры расчетов: http://www.sinterface.com. Разработчик программ Аксененко Е.В. (Eugene_Aksenenko@ukr.net).
Lucassen-Reynders E.H., Fainerman V.B., Miller R. // J. Phys. Chem. B. 2004. V. 108. P. 9173.
Файнерман В.Б., Миллер Р. // Коллоид. журн. 2005. Т. 67. С. 437.
Benjamins J., Lyklema J., Lucassen-Reynders E.H. // Langmuir. 2006. V. 22. P. 6181.
Noskov B.A., Latnikova A.V., Lin S.-Y., Loglio G., Miller R. // J. Phys. Chem. C. 2007. V. 111. P. 16895.
Maldonado-Valderrama J., Miller R., Fainerman V.B., Wilde P.J., Morris V.J. // Langmuir. 2010. V. 26. P. 15 901.
Fainerman V.B., Aksenenko E.V., Krägel J., Miller R. // Langmuir. 2013. V. 29. P. 6964.
Aksenenko E.V., Kairaliyeva T., Makievski A.V., Fainerman V.B., Miller R. // Colloids Surf. A. 2018. V. 547. P. 95.
Fainerman V.B., Lylyk S.V., Aksenenko E.V., Makievski A.V., Petkov J.T. Yorke J., Miller R. // Colloids Surf. A. 2009. V. 334. P. 1.
Fainerman V.B., Mys A.V., Aksenenko E.V., Makievski A.V., Petkov J.T., Yorke J., Miller R. // Colloids Surf. A. 2009. V. 334. P. 22.
Girardet J.M., Humbert G., Creusot N., Chardot V., Campagna S., Courthaudon J.L., Gaillard J.L. // J. Colloid Interface Sci. 2001. V. 243. P. 515.
Pezennec S., Gauthier F., Alonso C., Graner F., Croguennec T., Brule G., Renault A. // Food Hydrocoll. 2000. V. 14. P. 463.
Fainerman V.B., Kovalchuk V.I., Aksenenko E.V., Zinkovych I.I., Makievski A.V., Nikolenko M.V., Miller R. // Langmuir. 2018. V. 34. P. 6678.
Yates L.M., Wandruszka R. // Soil Sci. Soc. Am. J. 1999. V. 63. P. 1645.
Leyton P., Lizama-Vergara P.A., Campos-Vallette M.M., Becker M.I., Clavijo E., Córdova-Reyes I., Vera M., Jerez C.A. // J. Chil. Chem. Soc. 2005. V. 50. P. 725.
Terashima M., Fukushima M., Tanaka S. // Colloids Surf. A. 2004. V. 247. P. 77.
Prado A.G.S., Pertusatti J., Nunes A.R. // J. Braz. Chem. Soc. 2011. V. 22. P. 1478.
Smejkalova D., Piccolo A. // Environ. Sci. Technol. 2008. V. 42. P. 699.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал