

Коллоидный журнал, 2022, T. 84, № 1, стр. 3-14
Моделирование коллоидной кристаллизации селенида кадмия
С. М. Асадов 1, 2, *, М. А. Анисимов 2, К. И. Кельбалиев 1, В. Ф. Лукичев 3
1 Институт катализа и неорганической химии Национальной
академии наук Азербайджана
Az1143 Баку, Азербайджан
2 Институт физических наук и технологий,
Университет Мэриленда, Колледж-Парк
20742 Мэриленд, США
3 Физико-технологический институт им. К.А. Валиева РАН
117218 Москва,
Нахимовский просп., 36, корп. 1, Россия
* E-mail: salim7777@gmail.com
Поступила в редакцию 11.06.2021
После доработки 01.08.2021
Принята к публикации 04.08.2021
- EDN: LJUTJQ
- DOI: 10.31857/S0023291222010025
Аннотация
Статья посвящена моделированию методом Монте-Карло кинетики коллоидной кристаллизации наночастиц (НЧ) селенида кадмия CdSe, т.е. роста наночастиц из отдельных молекул CdSe. С учетом экспериментальных данных модифицирована модель кристаллизации НЧ CdSe, включающая константы скорости реакции кристаллизации в коллоидном растворе. Принимается, что управление размером, распределением по размерам и выходом НЧ играет ключевую роль в улучшении свойств НЧ. В приближении динамического равновесия определены константы модели зародышеобразования, роста и последующей кристаллизации. Ключевыми параметрами процесса формирования НЧ являются частоты переходов присоединения и отрыва комплекса мономер–НЧ в зависимости от размеров и концентрации НЧ. Превращения частиц предшественника в мономер, формирование эффективного мономера и зародышеобразование переходят в стадию роста НЧ CdSe. Методом Монте-Карло аппроксимирована кинетическая модель процессов зарождения и рост НЧ CdSe с учетом отдельных вкладов (концентрации сформировавшихся мономеров и коэффициента диффузии мономеров) в константы скорости реакции формирования НЧ. Смоделированы кинетические зависимости среднего радиуса и концентрации НЧ CdSe, а также распределение НЧ по размерам при различных значениях кинетических констант процесса коллоидной кристаллизации.
ВВЕДЕНИЕ
Коллоидная кристаллизация с получением полупроводниковых нанокристаллов (НК) является важной задачей. В этом плане исследований актуальными являются бинарные сульфиды (селениды) группы AIIBVI [1] и AIIIBVI [2]. Такие материалы, например, как CdSe в виде коллоидных нанокристаллов и квантовых точек имеют высокие оптоэлектронные свойства даже при комнатной температуре [3, 4]. Отметим, что под словосочетанием “коллоидная кристаллизация” можно понимать два различных процесса: агрегация отдельных наночастиц в большой нанокластер, как это описано, например, в статье [5], или же рост одной наночастицы из отдельных молекул вещества. НК CdSe перспективны для применения в наноэлектронике, промышленности солнечных элементов, детекторах излучения, фотодетекторах, фотопроводниках [6]. В частности, из-за высоких светочувствительных [7] и люминесцентных [8] свойств CdSe можно использовать в технологии тонкопленочных транзисторов, детекторов гамма-излучения и элементов памяти. CdSe обычно имеет p-тип проводимости и прямую запрещенную зону ∼1.72 эВ, что подходит для преобразования видимого света. С учетом сказанного важным является также изучение кинетики коллоидной кристаллизации CdSe в виде НК.
Существуют методы коллоидного формирования CdSe в виде НК из различных растворов [9–11]. Нелинейная кристаллизация НК из коллоидного раствора – сложный процесс, и его физико-химические закономерности до конца не выяснены [12, 13]. Несмотря на множество экспериментальных данных по формированию наночастиц (НЧ), эффективная теория кинетики кристаллизации НК из коллоидных растворов не существует. Вместе с тем экспериментальные результаты по кинетике коллоидной кристаллизации НК не всегда можно интерпретировать в рамках классической теории кристаллизации [14, 15]. Поэтому определение кинетических закономерностей нанокристаллизации требует развития существующих моделей коллоидной кристаллизации НК.
Коллоидная кристаллизация НК происходит в пространственно неоднородной среде, т.е. концентрация формирующихся НК распределяется неоднородно в пространстве. Такое распределение НК возможно, в частности, в приповерхностной зоне контакта между коллоидным раствором и твердой фазой за счет влияния диффузии, тепло- и массопереноса [16]. Кроме того, на рост НК могут влиять и другие факторы, в частности размеры частиц и фазовые переходы.
Известны различные способы коллоидного синтеза НК CdSe. В зависимости от условий формирования НК CdSe могут быть получены модификации сфалерита (цинковой обманки c-CdSe; a = = 6.05 Å) с кубической сингонией [17] и вюрцита с гексагональной сингонией (w-CdSe; a = 4.300 Å, c = 7.011 Å). По данным разных авторов фазы со структурами вюрцита и сфалерита обнаружены для всех халькогенидов кадмия и цинка. В этих соединениях при высокой температуре стабильной является фаза со структурой вюрцита, а при низкой – со структурой сфалерита.
Среди коллоидных нанокристаллов перспективными являются двумерные (2D) нанопластинки c-CdSe со структурой цинковой обманки как важный класс частиц с высокими оптическими свойствами [17]. Оптические свойства макроскопических кристаллов не зависят от размера, а зависят в основном от химического состава. При уменьшении размеров кристалла до наномасштабов движение электронов лимитируется физическими размерами области, в которой они могут находиться. В кристалле влияние электростатических сил становится более выраженным, и электроны ограничиваются потенциальным барьером, т.е. электроны оказываются в потенциальной яме.
Цель настоящей работы – моделирование методом Монте-Карло (МК) кинетических зависимостей коллоидной нанокристаллизации (зародышеобразование–рост) CdSe с кубической сингонией с учетом имеющихся экспериментальных данных.
Кинетика кристаллизации НК CdSe нами изучена методом МК [18–22]. В руководстве [18] рассмотрены основные аспекты метода, приведены примеры и алгоритмы моделирования сложных физических систем методом МК. Показано, что использование моделирования сложных молекул методом МК расширяет обсуждение, дополняет эксперимент и аналитическую теорию.
Интерес к методу МК связан с его эффективностью в управлении свойствами получающихся частиц, включая размер, гранулометрический состав и форму. Подход, продемонстрированный, например, в [19] для синтеза однородных сферических частиц диоксида кремния размером менее 100 нм, начинается с небольшого числа тестовых экспериментов, а затем направлен на корректировку условий реакции, которые контролируют нежелательные свойства получаемых частиц, оставляя неизменными условия, определяющие желаемые свойства. Применение данной стратегии показало, что синтезированные частицы размером 27–100 нм имели относительные стандартные отклонения размера <13.3%. В работе [20] влияние отношения потоков атомов Ga/As на субмонослойный рост GaAs на поверхности (001) GaAs моделируется методом МК. Рост осуществляется методом молекулярно-лучевой эпитаксии при различных значениях параметров процесса. Рассчитана поверхностная плотность островков в зависимости от отношения потоков атомов GaAs, которая согласуется с экспериментальными данными. Соотношение потоков атомов сильно влияет на плотность островков при пониженной температуре и повышенной скорости роста, когда десорбция мышьяка слабая. Оценена доля атомов мышьяка в растущей пленке при различных условиях процесса. Кинетический метод МК (KMK) использован для моделирования ван-дер-ваальсова роста гетероструктур, включающих однослойные дихалькогениды переходных металлов [21]. Методом KMK исследуются множественные кинетические процессы, связанные с различным поведением роста, наблюдаемым в экспериментах. Метод KMK используется в исследовании механизмов роста, что позволяет дать рекомендации для руководства экспериментальным исследованием. В работе [22] отмечается, что хотя и управление размерами, и распределение по размерам, и выход коллоидных квантовых точек PbS играют ключевую роль в улучшении их свойств, детали механизма образования, влияющие на размер, концентрацию и распределение размеров квантовых точек PbS в сложных условиях реакции остаются плохо изученными. Уравнение кинетики кристаллизации (стадии зародышеобразования и оствальдова созревания) использовалось для моделирования и описания экспериментальных данных по образованию квантовых точек PbS при переменной температуре раствора.
Таким образом, КМК-моделирование позволяет описать случайные изменения свойств и “временнýю зависимость” процесса, в частности кристаллизации. Принимается, что при кристаллизации изменение свойств системы происходит не заранее заданным образом, а стохастически, при этом протекание вероятностных процессов определяется последовательностями случайных чисел, генерируемых во время КМК-моделирования.
Термины “нанокристалл” и “наночастица” в данной работе подразумеваются как синонимы.
1. МЕТОДИКА ЭКСПЕРИМЕНТА И КМК-МОДЕЛИРОВАНИЕ
Синтез наночастиц
Синтез НЧ CdSe проводили по известной методике коллоидной кристаллизации [23–26]. Суть метода коллоидного синтеза заключается в гомогенном зародышеобразовании новой фазы в присутствии стабилизатора. В горячую, непрерывно перемешиваемую дисперсионную среду инжектируется смесь прекурсоров (источников) элементов нужных групп. В результате реакции и образования пересыщенного раствора появляются зародыши твердой фазы, происходит нуклеация. По мере снижения степени пересыщения нуклеация прекращается, а сформировавшиеся зародыши продолжают расти.
В качестве реагента, способствующего образованию наночастиц, использовали ацетат кадмия (Cd(OAc)2). Смесь, содержащую 0.13 г дигидрата ацетата кадмия (Cd(OAc)2 ⋅ 2H2O), 0.08 мл олеиновой кислоты и 10 мл октадецена, нагревали до 230°C в токе аргона. После этого в смесь вводили 100 мкл 1 М раствора селена в триоктилфосфине. В этих условиях начиналась кристаллизация, и в течение 1 ч продолжался рост НК CdSe. Затем реакционную смесь охлаждали до комнатной температуры. При охлаждении в смесь вводили 1 мл олеиновой кислоты и НК CdSe в виде нанопластинок осаждали ацетоном. Редиспергирование осажденных нанопластинок CdSe проводили в гексане; при этом в растворе получаются НЧ с минимальным содержанием примесей.
Кристаллическая структура 2D-нанокристаллов CdSe охарактеризована методом рентгенофазового анализа. Эксперименты проводились с использованием дифрактометра ДРОН-4-07 (производства НПП “Буревестник”) на Kα-линии медного излучения. Экспериментальный образец представлял собой полированную кремниевую подложку, ориентированную в направлении [100], с нанесенными на нее НЧ CdSe методом испаряющейся капли из дисперсии (концентрация дисперсии ∼10–100 мкг/мл). Дифрактограммы получены в диапазоне углов 2θ = 10°–65°. Изучены 2D-нанокристаллы CdSe со структурой сфалерита различной толщины (3, 4 и 5 монослоев CdSe). На дифрактограмме максимумы при 2θ = 25.3°, 41.5°, 49.5° и 60.1° соответствовали положениям рефлексов (111), (220), (311) и (400) для CdSe с кристаллической структурой сфалерита (файл № 19-0191 для кубической сингонии CdSe; рис. 1). Т.е. синтезированные НЧ CdSe имели элементарную ячейку с кубической сингонией (цинковая обманка).
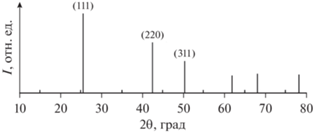
Толщину полученных нанопластинок CdSe изучали в просвечивающем электронном микроскопе. Латеральные размеры нанопластинок CdSe составляли 100–200 нм. На рис. 2 приведена зависимость толщины нанопластинок CdSe от времени синтеза.

Существуют два классификационных подхода к выделению видов полиморфных переходов – структурный и термодинамический. Согласно структурной классификации, основой для выделения различных видов полиморфизма служат степень и характер изменения структуры в процессе перехода. При этом рассматривается, меняются или нет первое координационное число, структурный тип и структурный мотив кристалла. Такой полиморфный переход относят к реконструктивному переходу. Т.е., при переходе от структуры одной модификации к другой необходимы разрыв связей и перестройка структуры. Полиморфный переход CdSe сфалерит ⇄ вюрцит можно отнести к изменению типа укладки координационных полиэдров. При этом в качестве характеристики относительной плотности структуры используют упаковочные коэффициенты. В частности, коэффициент компактности структуры и атомный индекс упаковки определяют долю занятого атомами кристаллического пространства. Для кристаллов, состоящих из атомов разных сортов, упаковочные коэффициенты зависят также от соотношения размеров атомов, например для CdSe ${{r}_{{{\text{Cd}}}}}{\text{/}}{{r}_{{{\text{Se}}~}}}.$
Связь термодинамических функций с полиморфизмом известна из физической химии. Наиболее устойчивой при данных термодинамических условиях является та полиморфная модификация вещества, которая обладает минимальной свободной энергией Гиббса (G). При этом в точке равновесия у двух полиморфных модификаций свободные энергии равны: ${{G}_{1}}\left( {T,P} \right) = {{G}_{2}}\left( {T,P} \right).$ Полиморфное превращение сопровождается изменением энергии Гиббса: $~\Delta G\left( {{{\alpha }} \to {{\beta }}} \right) = \Delta {{H}_{0}}\left( {T - {{T}_{0}}} \right){\text{/}}{{T}_{0}}$, где $\Delta {{H}_{0}}$ – изменение энтальпии при фазовом переходе, T0 – температура равновесия фаз. Полиморфный переход относится к фазовому переходу 1-го рода, где в точке фазового перехода скачок энергии Гиббса $\frac{{dG}}{{dT}} = - S$ и $\frac{{dG}}{{dP}} = V.$
Анализ литературных данных по полученным различными методами кристаллам указывает на то, что при уменьшении размеров частиц кристалла до наноразмерных в них возможны фазовые переходы, изменение фазового состояния, фазовых границ и координации атомов, отличные от превращений в объемных кристаллических образцах. В частности, известные данные по физическим, химическим и технологическим свойствам CdSe показывают, что они могут меняться в широком интервале значений в зависимости от условий синтеза, кинетических и термодинамических параметров, размера и формы НЧ [27].
КМК-моделирование
Процесс коллоидной кристаллизации в системе CdSe рассматривали с применением метода КМК [18]. КМК-моделирование позволяет описать нелинейные процессы, которые включают влияние случайных факторов на кристаллизацию [28–30].
Нелинейные процессы подчиняются вероятностному распределению частиц. В условиях термодинамического равновесия зависимость концентрации молекул, например, идеального газа от их потенциальной энергии при функции распределения Больцмана определяется в каноническом ансамбле в виде: $\left\langle U \right\rangle = \int {U\left( \xi \right){{\rho }}\left( \xi \right)d\xi } .$ $\left\langle U \right\rangle $ – средняя потенциальная энергия, $\xi $ – количество конфигураций частиц, встречающихся с вероятностью $p\left( \xi \right).$
Получение потенциальной энергии по всем возможным конфигурациям $\xi $ системы НЧ CdSe численными методами сложно. Для нахождения значений $\xi $ мы использовали случайный отбор с заданной вероятностью. Т.е. значение $\xi $ находили преобразованием значений другой псевдослучайной величины γ, равномерно распределенной в интервале (0,1). Для случайной величины $\xi $ соответствующая вероятность отвечала уравнению $\int_a^b {p\left( x \right)dx = {{\gamma }}} .$ В процессе КМК-моделирования кинетики коллоидной кристаллизации НЧ CdSe изображения формирующихся НЧ изучали на выбранной координатной сетке. Выбирали двухмерный массив случайных чисел для величины γ. Затем их генерировали вычислительной программой таким образом, чтобы элемент массива, равный единице, соответствовал, например, черной клетке, а равный нулю – белой клетке. При оценке вероятности $p\left( \xi \right)$ события (т.е. формирование конфигураций наночастиц CdSe) результат исхода i-той реализации КМК-моделирования xi равен
При моделировании процесса необходимо заранее знать темпы основных событий. После выбора каждого события (переход в конфигурацию наночастиц) рассчитывали темпы для новых потенциальных событий, и список для КМК-моделирования обновляли. Таким способом изучали динамическую эволюцию коллоидной кристаллизации НЧ CdSe. Затем составили алгоритм, масштабируемый как lg(N), где $N$ – количество возможных переходов от начального состояния к конечному состоянию НЧ CdSe на каждом временном шаге. При этом учитывали адсорбцию, десорбцию, диффузию, зарождение и рост нанокристалла CdSe.
При КМК моделировании коллоидной кристаллизации НЧ CdSe рассматривали возможные случайные события перехода частиц в конфигурацию наночастиц. Стохастическую последовательность этих событий выбирали случайным образом путем включения другого случайного числа для имитации реального времени. Общий алгоритм КМК состоял из следующих шагов.
1. Инициирование: вычисление темпа возможных событий ${{r}_{i}}$ и их суммы $\sum {{r}_{i}}$.
2. Выбор и выполнение события: генерирование случайного числа ${{{{\xi }}}_{1}}$ ∈ [0,1] и выбор для продолжения q-го процесса, для которого выполняется условие $\sum\nolimits_{i = 1}^q {{{r}_{i}} \leqslant {{{{\xi }}}_{1}}} \sum\nolimits_{i = 1}^N {{{r}_{i}} < } \sum\nolimits_{i = 1}^{q + 1} {{{r}_{i}}} .$
3. Изменение времени: выбирается другое случайное число ${{\xi }_{2}}$ ∈ [0,1], и реальное время увеличивается на ${{\Delta }}t = - \frac{{{\text{ln}}{\kern 1pt} {{\xi }_{2}}}}{{\sum {{r}_{i}}}}.$
4. Обновление: пересчет скоростей возможных событий ${{r}_{i}}$ и их суммы $\sum {{r}_{i}}$ в новой конфигурации.
5. Сходимость: повторение шагов 2–4 до достижения кинетически стационарного состояния.
6. Накопление данных: повторение шагов 2–4 и сбор данных.
КМК-моделированием случайных величин в виде наборов чисел находили вероятностно точные значения параметров процессов. Значения коэффициента диффузии (D) для CdSe сравнивали с экспериментальными данными [1, 7, 31–33], а затем использовали для вычисления скорости процесса кристаллизации НЧ CdSe.
Коллоидную кристаллизацию НЧ CdSe моделировали с учетом элементарных процессов. Принимали, что каждый элементарный процесс происходит с заданной вероятностью (W). В таких случаях, величину, равную числу событий в единицу времени, принимали за темп данного события. Если $N$ – возможное число всех событий (a = 1…$N$) в данной конфигурации $C$ системы в момент времени t, а темпы событий ${{R}_{a}}$, то полный темп равен: $Q = Q\left( C \right) = \sum\nolimits_{a = 1}^N {{{R}_{a}}{{V}^{a}}\left( {C \to C{\kern 1pt} '} \right)} $, где ${{V}^{a}}\left( {C \to C{\kern 1pt} '} \right)$ – стохастическая матрица. Такой матрицей определяется вероятность $W$ перехода системы между состояниями $C~$ и $C{\kern 1pt} '$ путем реализации события a. При реализации события $a$ вероятность изменения конфигурации системы равна ${{R}_{a}}{\text{/}}Q\left( C \right).$
КМК-моделирование кристаллизации НЧ CdSe реализовали программой, составленной на языке программирования DELPHI. Использовали модельные квадратные ячейки (координатную сетку) с узлами $N \times N$ базовой плоскости при периодических граничных условиях и $N~$ > 10. Здесь $N$ – количество кубиков по горизонтали модельной решетки НЧ CdSe. При выборе такой координатной сетки подразумевается, что НЧ CdSe могут располагаться только в узлах квадратной сетки, где и происходит кристаллизация. В процессе кристаллизации эти частицы могут либо появляться в новых узлах сетки, либо переходить из одного узла сетки в соседний. При моделировании двумерных НЧ (т.е., нанопластинок) CdSe, учитывая кубическую сингонию кристаллической решетки с-CdSe, шаг модельной решетки принимали равным известному из экспериментальных данных параметру кристаллической решетки с-CdSe. Затем выполняли преобразование формы модели НК CdSe с использованием системы компьютерного конструирования.
Температура при расчетах изменялась от 473 до 573 K с шагом 5 K. При КМК использовали два этапа: начальная инициализация системы и этап моделирования. На первом этапе устанавливали параметры системы (температура, концентрация компонентов, время, размер НЧ), составляли список событий, распределение событий по группам, подсчет суммарного темпа всех событий. На втором этапе выбирали группы реализуемых процессов, проводили реализацию процесса, осуществляли пересчет вероятностей и сбор статистических данных о свойствах НЧ CdSe.
Суть алгоритма КМК-моделирования рассмотрим на примере перемещения частицы (выбранной молекулы CdSe) в кристаллической решетке. На каждом временном шаге для выбранной молекулы случайным образом выбирается действие для перехода на другой узел кристаллической решетки CdSe. В алгоритме вероятность принятия (${{P}_{{{\text{acc}}}}}$) новой конфигурации зависит от изменения энергии $\Delta E$ и температуры $T$ как ${{P}_{{{\text{acc}}}}} = {\text{min}}\left( {1,{\text{exp}}\left[ {\frac{{\Delta E}}{{{{k}_{{\text{B}}}}T}}} \right]} \right)$. После расчета вероятности принятия ${{P}_{{{\text{acc}}}}}$ выбирается случайное число x от 0 до 1, и если ${{P}_{{{\text{acc}}}}} > x$, то переход разрешается, в противном случае частица, т.е. одна молекула CdSe, остается на своем начальном узле кристаллической решетки, и весь процесс расчета повторяется.
Выбранный способ определения сходимости расчета по достижению стационарных скоростей роста позволяет оценить степень релаксации соответствующих параметров и судить о вероятности достижения стационарного состояния формирования НЧ CdSe. КМК-моделирование адсорбции, десорбции и миграции частиц НЧ CdSe выполняли с учетом справочных термодинамических параметров и теплоемкости CdSe. Энергетический барьер в процессах определяли выражением: ${{E}_{{i \to f}}} = {{E}^{0}} + \frac{1}{2}\left( {{{\epsilon }_{f}} - {{\epsilon }_{i}}} \right),$ где ${{E}^{0}}$ – энергетический барьер для прыжков изолированного адатома, ${{\epsilon }_{{i\left( f \right)}}}$ – сумма энергий парного взаимодействия в начальном (и конечном) состоянии.
В халькогенидных полупроводниках при кристаллизации коэффициент диффузии компонентов D на поверхности относительно высокий [34]. Значение D зависит от параметров системы, таких как температура, концентрация распределяемых частиц, форма и размер диффундирующих частиц, их способность к ассоциации и диссоциации, а также от свойств растворителя. Значение D частиц при кристаллизации НЧ CdSe оценивали расчетом среднеквадратического смещения частиц с учетом экспериментальных данных [31]. В изотропном приближении для диффузии частиц в базовой плоскости можем записать $D = {{X}^{2}}{\text{/}}4{{N}_{t}}.$ Здесь ${{X}^{2}}$ – сумма квадратов смещений всех частиц, отнесенная к их числу, ${{N}_{t}}$ – число шагов моделирования. Эффективную энергию активации при диффузии в CdSe аппроксимировали экспоненциальной функцией $D\left( T \right)$ типа Аррениуса вблизи предварительно выбранных значений температуры. Коэффициент самодиффузии в CdSe рассчитывали с помощью соотношения ${{D}_{0}} = {{{{\mu }}}_{{\text{m}}}}{{k}_{{\text{B}}}}T{\text{/}}q,$ где μm – подвижность диффундирующей частицы, q – заряд частицы. Вычисляемые величины коэффициентов диффузии зависят от числа шагов процедуры, сходясь в итоге к своим предельным значениям.
Сходимость вычисляемых величин и константы скорости реакций получали путем вычисления их значений для каждых 5000 шагов. Продолжительность начального периода составляла 15 000–20 000 шагов. Таким способом вычисляли кинетические коэффициенты, которые не изменялись со временем.
2. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
При коллоидной кристаллизации предполагали, что процесс фазообразования НК CdSe включает нелинейные взаимосвязанные и протекающие одновременно явления зародышеобразования, роста и кристаллизации (т.е. коллоидная кристаллизация – это нелинейный процесс). Важным параметром статистической модели служит размер частиц. Эволюцию распределения НЧ CdSe по размерам рассмотрим, исходя из уравнения, включающего кинетические коэффициенты скорости при переменных температурах коллоидного раствора, способствующих образованию НЧ CdSe. Эволюцию распределения характеристик НЧ CdSe моделировали методом КМК со стохастическим алгоритмом [29]. В рамках этого алгоритма совокупность дискретных однокомпонентных кинетических уравнений задается как [30]:
(1)
$\begin{gathered} \frac{{\partial N\left( {m,n;t} \right)}}{{\partial t}} = \frac{1}{2}\mathop \sum \limits_{m' = 0\,\,}^m \mathop \sum \limits_{n' = 0}^n K\left( {m - m{\kern 1pt} ',n - n{\kern 1pt} ';m{\kern 1pt} ',n{\kern 1pt} ';t} \right) \times \\ \times \,\,N\left( {m - m{\kern 1pt} ',n - n{\kern 1pt} ';t} \right)N\left( {m{\kern 1pt} ',n{\kern 1pt} ';t} \right) - \\ - \,\,N\left( {m,n;t} \right)\mathop \sum \limits_{m' = 0\,\,}^m \mathop \sum \limits_{n' = 0}^n K\left( {m,n;m{\kern 1pt} ',n{\kern 1pt} '} \right)N\left( {m{\kern 1pt} ',n{\kern 1pt} ';t} \right), \\ \end{gathered} $(2)
$\begin{gathered} \frac{{\partial N\left( {n,m,t} \right)}}{{\partial t}} = \frac{1}{2}\mathop \smallint \limits_0^m dm{\kern 1pt} '\mathop \smallint \limits_0^n dn{\kern 1pt} 'K\left( {m - m{\kern 1pt} ',n - n{\kern 1pt} ';m{\kern 1pt} ',n{\kern 1pt} '} \right) \times \\ \times \,\,N\left( {m - m{\kern 1pt} ',n - n{\kern 1pt} ';t} \right)N\left( {m{\kern 1pt} ',n{\kern 1pt} ';t} \right) - \\ - \,\,N\left( {m,n;t} \right)\mathop \smallint \limits_0^\infty dm{\kern 1pt} '\mathop \smallint \limits_0^\infty dn{\kern 1pt} 'K\left( {m,n;m{\kern 1pt} ',n{\kern 1pt} '} \right)N\left( {m{\kern 1pt} ',n{\kern 1pt} ';t} \right). \\ \end{gathered} $В уравнениях (1) и (2) $K\left( {m,n;m{\kern 1pt} ',n{\kern 1pt} '} \right)$ – постоянная совокупности ядер, которая зависит от состава коагулирующих частиц. Дискретное уравнение (1) дает временную скорость изменения среднего числа мономера-поликристалла с массой m и мономера-нанокристалла с массой n как разность двух членов, первый член дает выигрыш в количестве частиц, масса поликристалла которых имеет размер m, а масса монокристалла – размер n.
Нами не рассмотрены явления, связанные со вторичным зародышеобразованием, т.е. зародышеобразованием, которое происходит из-за предварительного присутствия кристаллов CdSe c кубической и/или гексагональной кристаллической структурой. Принимали, что формирование НЧ CdSe c кубической структурой состоит из зародышеобразования, контролируемого частицей-предшественником-мономером (одна молекула CdSe), стадии оствальдова созревания и роста. Т.е., конденсация пересыщенной фазы НЧ CdSe происходит за счет мелких коллоидных НЧ CdSe в условиях их растворения.
Предположим, что коллоидные НЧ CdSe с узким диапазоном распределения по размеру получены реакцией контролируемого осаждения. Т.е. рост крупных НЧ CdSe происходит за счет использования затравки из маленьких частиц CdSe. В этом случае происходит формирование НЧ CdSe в виде квазимонодисперсных частиц в разбавленной суспензии. Стабильность образующихся НЧ CdSe обеспечивается динамическим равновесием: ${{\left( {{\text{C}}{{{\text{d}}}^{{2 + }}}} \right)}_{{{\text{solvate}}}}} + {{\left( {{\text{S}}{{{\text{e}}}^{{2 - }}}} \right)}_{{{\text{solvate}}}}} \rightleftarrows {{\left( {{\text{CdSe}}} \right)}_{{{\text{НЧ}}}}}{\kern 1pt} .$ По этой реакции происходит растворение мелких и рост более крупных и стабильных НЧ CdSe (оствальдово созревание). Т.е. в процессе моделирования нами рассматривается рост заданного набора мономеров, из которых могут расти несколько НЧ, которые способны затем подвергаться оствальдовому созреванию.
В соответствии с указанным равновесием формирование НЧ халькогенида металла (${\text{MX}}$) представим следующим образом. Запишем схему образования НЧ, которая состоит из трех этапов: образование мономеров НЧ – процесс зародышеобразования – рост НЧ.
(3)
$\begin{gathered} \left[ {\text{M}} \right] + \left[ {\text{X}} \right]\xrightarrow{{{{k}_{{{\text{f}}1}}}}}\left[ {{{{\left( {{\text{MX}}} \right)}}_{1}}} \right]\xrightarrow{{{{k}_{{{\text{f}}2}}}}} \\ \to \left[ {{{{\left( {{\text{MX}}} \right)}}_{2}}} \right]\xrightarrow{{{{k}_{{\text{f}}}}}}\left[ {{\text{MX}}} \right]{\text{\;}}\left( {{\text{мономер\;НЧ\;}}} \right), \\ \end{gathered} $(4)
$\left[ {{\text{MX}}} \right]\left( {{\text{мономер\;НЧ\;}}} \right)\xrightarrow{{{{k}_{{\text{D}}}}}}~\left[ {{\text{MX}}} \right]\left( {c_{1}^{*}} \right),$(5)
$\left[ {{\text{MX}}} \right]\left( {{{c}_{n}}} \right) + \left[ {{\text{MX}}} \right]\left( {c_{1}^{*}} \right)\mathop \rightleftharpoons \limits_{{{k}_{{d,n + 1}}}}^{{{k}_{{g,n}}}} \left[ {{\text{MX}}} \right]\left( {{{c}_{{n + 1}}}} \right),$С учетом химических уравнений (3)–(5) ниже запишем соответствующие уравнения для прекурсоров ${\text{P}}$ (т.е. вещества P, участвующего в реакции, приводящей к образованию целевого вещества – мономеров и нанокластеров). Поэтому соответствующие $N$ совместных обыкновенных дифференциальных уравнений для прекурсора P, мономеров и нанокластеров, используя закон действующих масс, запишем в виде:
(7)
$\frac{{d\left[ {{\text{MX}}} \right]\left( {{{c}_{1}}} \right)}}{{dt}} = {{k}_{{\text{f}}}}\left[ {\text{P}} \right] - {{k}_{{\text{D}}}}\left[ {{\text{MX}}} \right]\left( {{{c}_{1}} - c_{1}^{*}} \right),$(8)
$\begin{gathered} \frac{{d\left[ {{\text{MX}}} \right]\left( {c_{1}^{*}} \right)}}{{dt}} = {{k}_{{\text{D}}}}\left[ {{\text{MX}}} \right]\left( {{{c}_{1}} - c_{1}^{*}} \right) - \\ - \,\,\mathop \sum \limits_{n = 1}^N \left( {{{k}_{{g,n{\text{\;}}}}}\left[ {{\text{MX}}} \right]~\left( {c_{1}^{*}{{c}_{n}}} \right) - {{k}_{{d,n + 1}}}\left[ {{\text{MX}}} \right]~({{c}_{{n + 1}}}} \right), \\ \end{gathered} $(9)
$\begin{gathered} \frac{{d\left[ {{\text{MX}}} \right]\left( {{{c}_{n}}} \right)}}{{dt}} = {{k}_{{g,n - 1}}}\left[ {{\text{MX}}} \right]\left( {c_{1}^{*}{{c}_{{n - 1}}}} \right) - {{k}_{{d,n}}}\left[ {{\text{MX}}} \right]\left( {{{c}_{n}}} \right) - \\ - \,\,{{k}_{{g,n}}}\left[ {{\text{MX}}} \right]\left( {c_{1}^{*}{{c}_{n}}} \right) + {{k}_{{d,n + 1}}}\left[ {{\text{MX}}} \right]({{c}_{{n + 1}}}), \\ \end{gathered} $Решение вышеперечисленных кинетических уравнений требует знания физических и термодинамических параметров. Необходимые значения параметров для CdSe брали из справочника [32]. Условия формирования нанопластинок CdSe, соответствующие изотермическому и неизотермическому процессам, взяты из работ по синтезу нанопластинок CdSe [23–26].
Кинетические параметры
С учетом теории переходного состояния, константу скорости ${{k}_{{\text{f}}}}$ превращения частиц-предшественников вычисляли из выражения
(10)
${{k}_{{\text{f}}}} = \frac{{{{k}_{{\text{B}}}}T}}{h}\frac{{{{Q}_{{{\text{TS}}}}}}}{{\prod\limits_{i = 1}^m {{{Q}_{{\text{R}}}}} }}{\text{exp}}\left[ { - \frac{{\Delta {{E}_{{\text{a}}}}}}{{{{k}_{{\text{B}}}}T}}} \right],$В процессе коллоидной кристаллизации на поверхности образованной НЧ может развиваться гетерогенный процесс. Согласно кинетике гетерогенных реакций формальная скорость (W) такого процесса оценивается уравнением
(11)
$W = \frac{{dx}}{{dt}} = {{k}_{0}}{\text{exp}}\left[ { - \frac{{\Delta {{E}_{{\text{a}}}}}}{{RT}}} \right]S\mathop \prod \limits_{i = 1}^m c_{i}^{{{{n}_{i}}}},$Однако значение площади S оценить трудно, поскольку S меняется с течением времени. В условиях самопроизвольного процесса гетерогенного образования НЧ можно принять, что поверхность полусферического зародыша изменяется пропорционально объему в степени 2/3.
Коэффициент диффузии ${{k}_{{\text{D}}}}$ мономера НЧ CdSe характеризует взаимодействие между мономером и другими молекулами CdSe в растворе. Предположим, что в результате теплового движения молекул при градиенте концентрации диффузия мономера происходит в приповерхностном слое кристалла CdSe, который содержит растворитель и адсорбционный слой мономера. Градиент концентрации мономера зависит от толщины слоя и изменяется во времени. Если мономер очень медленно перемещается в растворителе, то диффузия мономера конкурирует с седиментацией. Тогда согласно уравнению Лаама диффузия растворенного вещества будет равна ${{k}_{{\text{D}}}} = {\text{s}}{{{{\omega }}}^{2}},$ где$~{{\omega }}$ – угловая скорость вращения, ${\text{s}}$ – коэффициент седиментации мономера в растворе. Коэффициент $s$ включает в себя плотность и вязкость раствора, объем фракции мономера, молярную массу и молярный объем мономера с критическим радиусом.
Коэффициенты ${{k}_{{g,n}}}$, ${{k}_{{d,n + 1}}}$ характеризуют эффективность связывания мономера с другими частицами. Эти кинетические константы реакции мономера могут зависеть от различных факторов. Сложно учесть все факторы, которые вносят вклад в коэффициенты ${{k}_{{g,n}}}$, ${{k}_{{d,n + 1}}}$, которые определяют скорости роста мономера, т.е. скорости реакции между мономером и нанокластерами. Мы рассматривали кинетическую модель, включающую стадии зародышеобразования, оствальдовское созревание за счет рекристаллизации нанокластеров CdSe в стабильные НЧ и рост НЧ. Эффекты агломерации и разрушения НЧ не учитывали.
Предположим, что реакционная система, включающая частицы CdSe, находится в изотермических условиях и пересыщение ее раствора задано в качестве начального условия. Для этого случая примем, что коэффициенты ${{k}_{{g,n}}}$, ${{k}_{{d,n + 1}}}$ слабо зависят от формы и размера мономера на поверхности формируемого нанокластера CdSe. Тогда константы роста и диссоциации НЧ могут быть представлены выражениями [33]
(13)
${{k}_{{d,n + 1}}} \propto {{\alpha }}{{k}_{{\text{a}}}}{{r}^{b}}{{c}_{{1,\infty }}}{\text{exp}}\left[ { - \frac{{{\alpha }}}{{a\sqrt[3]{r}}}} \right],$Превращение исходных реагентов в продукты, как правило, состоит из последовательности элементарных стадий. При коллоидной кристаллизации НЧ CdSe переменные (концентрация, температура, время) кинетической модели на отдельных стадиях процесса являются непрерывными функциями. Непрерывные части кинетической модели удобно описать дифференциальными уравнениями типа Фоккера–Планка и решать численными методами [34]. Если переменные состояния системы не изменяются во времени и отсутствуют переходные процессы, то такое стационарное состояние можно также исследовать аналитически.
Однако при коллоидной кристаллизации число образующихся зародыщей-мономеров зависит от времени, пересыщения раствора, температуры, плотности, теплоемкости и коэффициента теплопередачи раствора, отношения объема к площади сосуда, в котором происходит процесс и т.д. Поскольку стационарность лежит в основе многих статистических процедур, то используемые при моделировании коллоидной кристаллизации нестационарные параметры на отдельных стадиях процесса предполагали стационарными. Так, стационарными полагали пересыщение раствора, температуру, плотность и теплоемкость раствора, в котором происходила коллоидная кристаллизация НЧ CdSe с кубической сингонией. Принимали, что в стационарном стохастическом процессе кристаллизации сформированная НЧ CdSe не меняет свои свойства и характеристики со временем. Т.е., в таком стохастическом процессе вероятность распределения средних значений параметров полагалась неизменной во времени.
Другими словами, для стационарного процесса “зарождение–рост мономеров НЧ CdSe”, принимали осредненные для всего объема реакционного сосуда значения удельной массовой изобарной теплоемкости, площади поверхности теплообмена, температуры насыщения теплопередачи и коэффициента теплопередачи. С учетом этого дифференциальные уравнения кинетики процесса решали методом КМК.
Было проведено компьютерное КМК-моделирование формирования НЧ CdSe. Вычислены как функции времени соответствующие величины, включая средний радиус ${{r}_{{{\text{av}}}}}$ и общую концентрацию НЧ CdSe. На рис. 3–5 показаны вычисленные из кинетической модели зависимости параметров ${{k}_{{\text{f}}}},$ ${{k}_{{\text{D}}}}$, ${{k}_{{\text{a}}}}$ и ${{k}_{{\text{d}}}}$ от среднего размера и концентрации НЧ при изотермическом (Т = 483 К) формировании НЧ CdSe.
Рис. 3.
Влияние кинетических констант скорости на средний радиус НЧ CdSe как функцию времени при температуре синтеза НЧ T = 483 К. Кинетические кривые 1–4 аппроксимированы при следующих значениях параметров: (а) ${{k}_{{\text{f}}}}$ изменяется в интервале от 0.01 до 10 с–1 при ${{k}_{{\text{a}}}}$ = 100 с–1, ${{k}_{{\text{D}}}}$ = 0.1 с–1, ${{k}_{{\text{d}}}}$ = 7 × 10–8 с–1, (б) ${{k}_{{\text{a}}}}$ изменяется в интервале от 102 до 105 с–1 при ${{k}_{{\text{f}}}}$ = 0.01 с–1, ${{k}_{{\text{D}}}}$ = 0.1 с–1, ${{k}_{{\text{d}}}}$ = 7 × 10–8 с–1, (в) ${{k}_{{\text{D}}}}$ изменяется в интервале от 0.01 до 100 с–1 при ${{k}_{{\text{a}}}}$ = = 100 с–1, ${{k}_{{\text{D}}}}$ = 0.1 с–1, ${{k}_{{\text{d}}}}$ = 7 × 10–8 с–1, (г) ${{k}_{{\text{d}}}}$ изменяется в интервале от 7 × 10–8 до 7 × 10–2 с–1 при ${{k}_{{\text{f}}}}$ = 0.01 с–1, ${{k}_{{\text{D}}}}$ = = 0.1 с–1, ${{k}_{{\text{a}}}}$ = 100 с–1. Значения кинетических констант скорости увеличиваются в указанных интервалах (а, б, в, г) в следующем порядке: кривые 1–2–3–4.
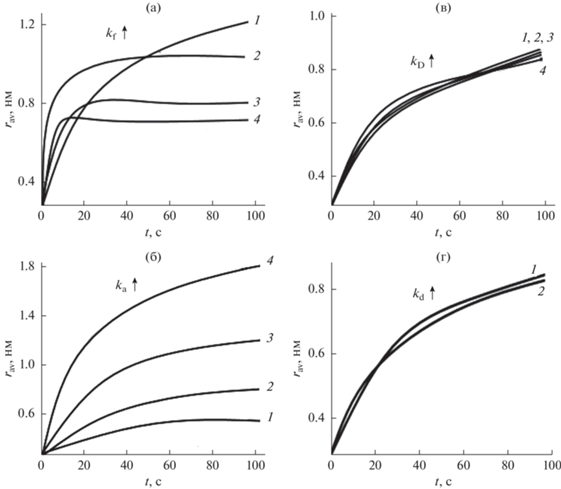
Рис. 4.
Влияние кинетических констант скорости на концентрацию НЧ CdSe как функцию времени. Значения всех параметров такие же, как и на рис. 3.

Рис. 5.
Влияние кинетических констант скорости на функцию распределения НЧ CdSe по размерам через 90 с. Значения всех параметров такие же, как и на рис. 3.

Оптимизацию функции с участием параметров ${{k}_{{\text{f}}}},$ ${{k}_{{\text{a}}}},$ ${{k}_{{\text{D}}}}$ и ${{k}_{{\text{d}}}}$ проводили методом МК. Для достижения максимума (или минимума) функции, которую нужно было оптимизировать в интервале температур от 473 до 573 K, использовали случайные величины на поверхности этой функции, генерированные компьютером. Пределы изменения параметров выбирали на поверхности этой функции с учетом экспериментальных данных коллоидной кристаллизации CdSe. Кинетические кривые аппроксимированы на основе следующих данных: ${{k}_{{\text{f}}}}$ = 10–2–10 с–1 (а), ${{k}_{{\text{a}}}}$ = 102–105 с–1 (б), ${{k}_{{\text{D}}}}$ = 10–1–102 с–1 (в), ${{k}_{{\text{d}}}}$ = 7 × 10–8–7 × 10–2 с–1 (г). Например, при расчете параметра ${{k}_{{\text{f}}}}$ значения остальных зафиксированных параметров для кривых 1–2–3–4 (рис. 3–5) были следующими: ${{k}_{{\text{a}}}}$ = 102 с–1 (а), ${{k}_{{\text{a}}}}$ = = 102.5 с–1 (б), ${{k}_{{\text{D}}}}$ = 10–1 с–1 (в), ${{k}_{{\text{d}}}}$ = 7 × 10–8 с–1 (г), ${{k}_{{\text{a}}}}$ = 102 с–1 (а), ${{k}_{{\text{a}}}}$ = 103 с–1 (б); ${{k}_{{\text{D}}}}$ = 10 с–1 (в), ${{k}_{{\text{d}}}}$ = = 7 × 10–5 с–1 (г); ${{k}_{{\text{a}}}}$ = 102 с–1 (а), ${{k}_{{\text{a}}}}$ = 105 с–1 (б), ${{k}_{{\text{D}}}}$ = 102 с–1 (в), ${{k}_{{\text{d}}}}$ = 10–2 с–1 (г). Вычисленные значения параметра ${{y}_{i}}$ принимали как среднее значение отдельных оценок ${{y}_{{ij}}}.$ Стандартное отклонение рассчитывали по формуле ${{{{\sigma }}}_{{i~}}} = \sqrt {\sum\nolimits_j {{{{{{\left( {{{y}_{{ij}}} - ~{{y}_{i}}} \right)}}^{2}}} \mathord{\left/ {\vphantom {{{{{\left( {{{y}_{{ij}}} - ~{{y}_{i}}} \right)}}^{2}}} {\left( {n - 1} \right)}}} \right. \kern-0em} {\left( {n - 1} \right)}}} } $, где $n$ – общее количество оценок.
Временн${\text{ы}}'$е зависимости характеристик сформированных нанокристаллов CdSe сильно зависят от условий синтеза. Вычисленные нами временные зависимости распределения НЧ CdSe по размерам согласуются с известными экспериментальными данными [35–40].
Увеличение скорости превращения предшественника НЧ CdSe ${{k}_{{\text{f}}}}$ от 10–2 до 10 с–1 приводит к следующим эффектам: а) на начальном этапе средний радиус НЧ быстро увеличивается, а затем происходит насыщение; б) с увеличением времени распределения НЧ по размерам становятся более узкими.
Согласно обсужденной выше кинетической схеме, если большее количество предшественников мономеров превращается в мономеры с концентрацией ${{c}_{1}}$, то все константы скорости, связанные с концентрацией генерируемого мономера, увеличиваются. Таким образом, увеличиваются скорости роста и диссоциации НЧ, так же, как и концентрация НЧ. В случае ${{k}_{{\text{f}}}}$ = 10–1 с–1 рост размера НЧ прекращается через 60 с, что означает, что в коллоидном растворе больше нет свободных мономеров.
Увеличение коэффициента ${{k}_{{\text{a}}}}$ от 102 до 105 с–1 приводит к быстрому росту НЧ CdSe; их концентрация уменьшается и соответствующее концентрационное распределение сдвигается вправо. Когда ускоряется процесс роста (увеличение ${{k}_{{\text{D}}}}$), наблюдается быстрый рост НЧ уже в начальное время реакции. Чем выше эффективная концентрация мономера, тем уже распределение НЧ по размерам и больше концентрация НЧ. Низкое значение ${{k}_{{\text{D}}}}$ приводит к более полидисперсному распределению НЧ, чем более высокое значение ${{k}_{{\text{D}}}}$, и к смещению максимума распределения НЧ по размерам в сторону более мелких частиц.
При увеличении коэффициента ${{k}_{{\text{d}}}}$ путем повышения концентрации от 2 × 10–10 до 2 × 10–4 моль/л (увеличения объемной растворимости) соответствующие временные зависимости радиуса и концентрации НЧ CdSe смещаются влево. Причина такого смещения заключается в том, что по мере увеличения ${{k}_{{\text{d}}}}$ все больше НЧ растворяются и поглощаются более крупными частицами.
3. ЗАКЛЮЧЕНИЕ
Экспериментально исследован процесс кристаллизации нанопластинок CdSe из пересыщенного водного раствора. Определенные в эксперименте параметры процесса (температурный режим, кинетика роста твердой фазы в при различных размерах формирующихся частиц) позволили модифицировать модель кристаллизации. В частности, учтено, что собственные размеры частиц играют важную роль в процессе зарождения нанокристаллической фазы в дисперсных системах, где небольшие изменения среднего размера частиц или концентрации могут приводить к заметным изменениям структуры и состояния дисперсионной среды. Введены понятия частиц-предшественников и эффективных мономеров НЧ, способных сформировать зародыш НЧ. Сформулирована математическая модель, описывающая процессы формирования и роста НЧ при гетерогенном зародышеобразовании. Параметры модели определены в приближении динамического равновесия. При численном моделировании процессов формирования НЧ CdSe учтены константа скорости реакции присоединения мономеров НЧ, эффективная концентрация созданных мономеров, коэффициент диффузии мономера, концентрация нанокластеров, содержащих мономерные звенья, кинетические коэффициенты модели, которые необходимы для оценки того, насколько быстро эффективный мономер связывается с другими частицами (или молекулами CdSe). Физико-химическая задача рассмотрена в условиях нелинейности процессов кристаллизации. Установлено удовлетворительное совпадение полученных при моделировании зависимостей среднего радиуса НЧ CdSe от времени с экспериментальными данными. Тем самым подтверждена адекватность предложенной кинетической модели и определенных на основании эксперимента параметров процесса кристаллизации нанокристаллов селенида кадмия (НК CdSe) с кубической сингонией. В рамках кинетических предположений и уравнений получены зависимости скорости роста НК CdSe от времени зародышеобразования и размера НЧ в изотермических условиях (Т ≈ 483 К). Определенные кинетическим методом Монте-Карло изотермические зависимости кинетических констант скорости роста от среднего радиуса НЧ CdSe как функции времени, концентрации и распределения нанокристаллов по размерам могут быть интерпретированы в терминах, принятых в теории кристаллизации. Построенные кинетические зависимости позволяют оценить влияние параметров процесса кристаллизации на временные зависимости концентрации НЧ CdSe. Эти зависимости важны для интерпретации экспериментальных данных, развития технологии синтеза нанокристаллов, в частности НК CdSe, и для количественного прогнозирования процессов формирования НК с более высоким выходом целевого продукта.
Список литературы
Asadov S.M., Mamedov A.N., Kulieva S.A. // Inorg. Mater. 2016. V. 52. P. 876.
Asadov S.M., Mustafaeva S.N., Mammadov A.N. // J. Therm. Anal. Calorim. 2018. V. 133. P. 1135.
Shornikova E.V., Biadala L., Yakovlev D.R., Sapega V.F., Kusrayev Y.G., Mitioglu A.A., Ballottin M.V., Christianen P.C.M., Belykh V.V., Kochiev M.V., Sibeldin N.N., Golovatenko A.A., Rodina A.V., Gippius N.A., Kuntzmann A., Jiang Y., Nasilowski M., Dubertret B., Bayer M. // Nanoscale. 2018. V. 10. P. 646.
Yu J., Chen R. // InfoMat. 2020. V. 2. P. 905.
Compton O.C., Osterloh F.E. // J. Am. Chem. Soc. 2007. V. 129. P. 7793.
Subila K.B., Kumar G.K., Shivaprasad S.M., Thomas K.G. // J. Phys. Chem. Lett. 2013. V. 4. P. 2774.
Koryakin A.A., Kukushkin S.A., Redkov A.V. // J. Phys.: Conf. Ser. 2018. V. 1124. 022044.
Saidzhonov B.M., Zaytsev V.B., Berekchiian M.V., Vasiliev R.B. // J. Lumin. 2019. V. 209. P. 170.
van Embden J., Mulvaney P. // Langmuir. 2005. V. 21. P. 10226.
Peng Z.A., Peng X. // J. Am. Chem. Soc. 2001. V. 123. P. 183.
Talapin D.V., Rogach A.L., Mekis I., Haubold S., Kornowski A., Haase M., Weller H. // Colloids Surf. A. 2002. V. 202. P. 145.
Rempel J.Y., Bawendi M.G., Jensen K.F. // J. Am. Chem. Soc. 2009. V. 131. P. 4479.
Maceiczyk R.M., Bezinge L., de Mello A.J. // React. Chem. Eng. 2016. V. 1. P. 261.
Wu S., Sun Y. // Nano Res. 2019. V. 12. P. 1339.
Frenette L.C., Krauss T.D. // Nat. Commun. 2017. V. 8. P. 2082.
Gasser U., Weeks E.R., Schopeld A., Pusey P.N. // Science. 2001. V. 292. P. 258. http://www.tinyurl.com/y49gsb4y
Jana S., Davidson P., Abcassis B. // Angew. Chem. 2016. V. 128. P. 9517.
Landau D.P., Binder K. A Guide to Monte Carlo Simulations in Statistical Physics. Third Ed. New York: Cambridge University Press, 2009.
Huang Y., Pemberton J.E. // Colloids Surf. A. 2010. V. 360. P. 175.
Ageev O.A., Solodovnik M.S., Balakirev S.V., Mikhaylin I.A. // Tech. Phys. 2016. V. 61. P. 971.
Nie Y., Liang C., Cha P.C., Luigi W., Robert M., Cho K. // Sci. Rep. 2017. V. 7. 2977.
Wang D., Xing M., Wei Y., Wang L., Wang R., Shen Q. // ACS Omega. 2021. V. 6. P. 3701.
Ithurria S., Bousquet G., Dubertret B. // J. Am. Chem. Soc. 2011. V. 133. P. 3070.
Bouet C., Mahler B., Nadal B., Abecassis B., Tessier M.D., Ithurria S., Xu X., Dubertret B. // Chem. Mater. 2013. V. 25. P. 639.
Ithurria S., Talapin D.V. // J. Am. Chem. Soc. 2012. V. 134.P. 18585.
Peng X. // Nano Res. 2009. V. 2. P. 425.
Wang Z., Wen X.-D., Hoffmann R., Son J.S., Li R., Fang C.-C., Smilgies D.-M., Hyeon T. // PNAS. 2010. V. 107. P. 17119.
Ohno K., Esfarjani K., Kawazoe Y. // Computational Materials Sciences. Berlin: Springer, 2018. Ch. 5. P. 261.
Gillespie D.T. // J. Comput. Phys. 1976. V. 22. P. 403.
Asadov S.M. // Bul. Univ. Karaganda. Chem. Ser. 2021. V. 102. № 2. P. 31. https://doi.org/10.31489/2021Ch2/31-42
Antipov V.V., Kukushkin S.A., Osipov A.V., Rubets V.P. // Phys. Solid State. 2018. V. 60. P. 504.
Semiconductor Nanocrystals and Silicate Nanoparticles. Ed. by Peng X., Mingos D.M.P. Berlin: Springer-Verlag, 2005.
Kashchiev D. Nucleation. Basic Theory with Application. 2nd Ed. Oxford: Butterworth Heinemann, 2003.
Asadov S.M. // Russ. Microelectron. 2021. V. 50. P. 264.
Manna L., Scher E.C., Alivisatos A.P. // J. Am. Chem. Soc. 2000. V. 122. P. 12700.
Peng X., Manna L., Yang W., Wickham J., Scher E., Kadavanich A., Alivisatos A.P. // Nature. 2000. V. 404. P. 59.
Peng X., Mingos D.M.P. Controlled Synthesis of High-Quality Semiconductor Nanocrystals // In: Structure and Bonding. Semiconductor Nanocrystals and Silicate Nanoparticles. Berlin: Springer-Verlag, 2005. V. 118. P. 79.
Kwon S.G., Hyeon T. // Acc. Chem. Res. 2008. V. 41. P. 1696.
Kwon S.G., Hyeon T. // Small. 2011. V. 7. P. 2685.
Асадов С.М., Мустафаева С.Н., Лукичев В.Ф., Келбалиев К.И. // Микроэлектроника. 2021. Т. 49. № 6. С. 11.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал