

Координационная химия, 2019, T. 45, № 1, стр. 38-42
Влияние условий синтеза на молекулярную и кристаллическую структуру гетерометаллических 1D-полимерных ацетатных комплексов с мотивом {Dy2Co}nА. В. Гавриков, А. Б. Илюхин, Н. Н. Ефимов
А. В. Гавриков 1, *, А. Б. Илюхин 1, Н. Н. Ефимов 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
, Москва, Россия
* E-mail: penguin1990@yandex.ru
Поступила в редакцию 22.08.2018
После доработки 28.08.2018
Принята к публикации 24.08.2018
Аннотация
Синтезированы и исследованы новые гетерометаллические 1D-полимерные ацетатные комплексы с мотивом металлоостова {Dy2Co}n – [Dy2Co(CH3COOO)8(H2O)4]n · 6nH2O (I) и [Dy2Co(CH3COOO)8(H2O)2]n · 2nCH3COOH (II). Молекулярные структуры полученных соединений (CCDC № 1861619 (I), 1861620 (II)) различаются качественным составом координационного окружения Dy и структурными функциями ацетат-анионов, что существенно влияет на величины соответствующих расстояний Dy···Dy и Dy···Co в цепочке. Указанные различия, а также разный сольватный состав I и II определяются условиями синтеза комплексов.
В настоящее время разработка методов направленного синтеза и комплексное исследование новых представителей 3d-4f-гетерометаллических координационных соединений (КС) – одна из важнейших междисциплинарных задач [1–3]. Подобный интерес обусловлен возможностью проявления этими соединениями уникальных и практически важных свойств, например магнитных [4–6] и люминесцентных [7], и, следовательно, возможностью их использования в качестве действующих компонентов функциональных материалов [8]. Кроме того, в случае оптимального стехиометрического соотношения атомов гетерометаллов эти комплексы можно использовать в качестве прекурсоров для смешанных оксидов, уже находящих применение в качестве основы функциональных материалов [9–12].
Известно, что определяющее влияние на свойства практически всех соединений (в том числе КС) оказывает их строение. Последнее, в свою очередь, во многом определяется условиями синтеза – природой реагентов, их стехиометрическим соотношением и т.д. В случае КС управление их свойствами, в принципе, возможно на уровне отдельных структурных единиц [13, 14] и кристаллической структуры. Последнее достигается во многом за счет варьирования сольватных молекул [15, 16]. Очевидно, что для направленной модификации КС и улучшения их ценных характеристик (магнитных, люминесцентных и т.д.), в принципе, могут успешно применяться оба указанных метода. Однако этому должно предшествовать тщательное исследование комплексообразования в каждой конкретной системе.
В настоящей работе исследовано влияние условий синтеза на молекулярное и кристаллическое строение гетерометаллических ацетатных комплексов, образующихся в системе Co(Acac)2 · · 2H2O–Dy(Acac)3 · 3H2O–CH3COOH–C2H5OH.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для синтеза новых соединений использовали коммерчески доступные реактивы и растворители: CoCl2 · 6H2O (“х. ч.”, Лабтех) DyCl3 · 6H2O (>99%, ЛАНХИТ), ацетилацетон HAcac (99%, Acros Organics), концентрированный водный раствор аммиака (“х. ч.”, Лабтех), ледяную уксусную кислоту (“х. ч.”, Химмед) и этанол (96%). Все реактивы использовали без предварительной очистки.
Ацетилацетонаты Co(Acac)2 · 2H2O, Dy(Acac)3 · · 3H2O синтезировали по обменным реакциям в водных растворах соответствующих хлоридов с ацетилацетонатом аммония, образующимся in situ [17].
РСА. Экспериментальные данные для соединений [Dy2Co(CH3COOO)8(H2O)4]n · 6nH2O (I) и [Dy2Co(CH3COOO)8(H2O)2]n · 2nCH3COOH (II) собраны на дифрактометре Bruker SMART APEX2 (λ(MoKα), графитовый монохроматор) [18] (табл. 1). Поглощение учтено полуэмпирическим методом по эквивалентам (SADABS) [19]. Структуры, определенные комбинацией прямого метода и Фурье-синтезов, уточнены полноматричным анизотропно-изотропным МНК. Атомы водорода метильных фрагментов рассчитаны из геометрических соображений и уточнены в модели наездника. Атомы H, связанные с атомами O, локализованы из разностных Фурье-синтезов и уточнены в изотропном приближении. Все расчеты выполнены по программам SHELXS и SHELXL [20].
Таблица 1.
Основные структурные данные и результаты уточнения соединений I и II
| Параметр | Значение | |
|---|---|---|
| Соединение | I | II |
| T, K | 173(2) | 120(2) |
| Сингония | Триклинная | Триклинная |
| Пр. гр. | P1 | P1 |
| a, Å | 8.1587(2) | 9.0696(2) |
| b, Å | 10.2000(2) | 9.2538(2) |
| c, Å | 11.2955(2) | 10.5208(2) |
| α, град | 111.4820(10) | 79.8531(4) |
| β, град | 108.3450(10) | 75.2262(4) |
| γ, град | 92.8960(10) | 77.4092(4) |
| V, Å3 | 815.49(3) | 826.44(3) |
| Z | 1 | 1 |
| ρ (выч.), г/см3 | 2.110 | 2.034 |
| μ, мм–1 | 5.134 | 5.055 |
| F(000) | 507 | 491 |
| Размер кристалла, мм | 0.2 × 0.12 × 0.1 | 0.2 × 0.16 × 0.12 |
| Область θ, град | 2.076–31.563 | 2.274–31.522 |
| Интервал индексов | –12 ≤ h ≤ 12 –15 ≤ k ≤ 15 –16 ≤ l ≤ 16 |
–12 ≤ h ≤ 10 –13 ≤ k ≤ 12 –15 ≤ l ≤ 15 |
| Собранных отражений | 12 194 | 9703 |
| Независимых отражений, Rint | 5359, 0.0238 | 5002, 0.0224 |
| Полнота до θ = 25.242°, % | 100 | 100 |
| Max, min пропускание | 0.7462, 0.4142 | 0.7462, 0.496 |
| Уточняемых параметров | 249 | 222 |
| GOOF | 1.007 | 1.054 |
| R1, wR2 (I > 2σ(I)) | 0.0198, 0.0500 | 0.0206, 0.0480 |
| R1, wR2 (весь массив) | 0.0215, 0.0508 | 0.0224, 0.0488 |
| Δρmax/Δρmin, е Å–3 | 1.280, –0.898 | 1.041, –0.855 |
Экспериментальные данные для структур I и II депонированы в Кембриджском банке структурных данных (CCDC № 1861619, 1861620 соответственно); deposit@ccdc.cam.ac.uk или http:// www.ccdc.cam.ac.uk).
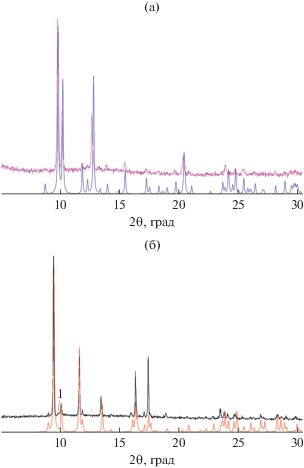
Рентгенофазовый анализ (РФА) проведен на дифрактометре Bruker D8 Advance (CuKα, Ni-фильтр, LYNXEYE детектор, геометрия на отражение).
Элементный анализ выполнен на автоматическом C,H,N-анализаторе EUROEA 3000 (Carlo Erba).
Синтез I. К раствору Co(Acac)2 · 2H2O (0.171 г, 0.58 ммоль) в 10 мл EtOH прибавляли при перемешивании раствор Dy(Acac)3 · 3H2O (0.3 г, 0.58 ммоль) в 10 мл EtOH, затем приливали смесь 10 мл ледяной уксусной кислоты и 30 мл этанола. Полученный малиново-красный раствор упаривали на водяной бане при пониженном давлении (водоструйный насос) и 76°C и охлаждали до комнатной температуры. Образовавшиеся через несколько часов малиново-красные кристаллы I отделяли от маточного раствора и промывали холодным этанолом. Выход 0.21 г (70%).
По данным РФА, соединение однофазно (pис. 1а).
Синтез II. К смеси Co(Acac)2 · 2H2O (0.15 г, 0.51 ммоль) и Dy(Acac)3 · 3H2O (0.526 г, 1.02 ммоль) приливали 20 мл ледяной уксусной кислоты и перемешивали до полного растворения. К полученному малиново-красному раствору добавляли 15 мл этанола и упаривали на водяной бане при пониженном давлении (водоструйный насос) и температуре 90°C. При охлаждении до комнатной температуры в течение несколько часов сформировались малиново-красные кристаллы II, которые отделяли от маточного раствора и промывали холодным этанолом. Выход 0.33 г (68%).
По данным РФА, продукт содержит следовые количества соединения I (пик 1, рис. 1б).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Метод синтеза 3d-4f-гетерометаллических карбоксилатных комплексов, использованный в данном исследовании и основанный на реакции взаимодействия трис-ацетилацетонатов металлов с карбоновыми кислотами, мы применяли ранее для синтеза гомометаллических карбоксилатных комплексов лантанидов [12, 13, 21, 22]. Движущей силой соответствующих реакций, очевидно, является существенное различие кислотных свойств ацетилацетона и карбоновых кислот. В случае реакций образования комплексов I и II, Ka(CH3COOH) ≈ 2 × 10–5 [23], что на четыре порядка превышает соответствующую величину для ацетилацетона (Ka ≈ 1.5 × 10–9 [24]). Таким образом, можно утверждать, что образование I и II термодинамически выгодно. Более полному протеканию подобных реакций способствует также частичное удаление из реакционной среды побочного продукта реакции – молекулярного ацетилацетона – в результате постепенного упаривания маточного раствора при нагревании.
Совместное действие вышеуказанных факторов приводит к существенному увеличению выхода I и II по сравнению с выходами ранее исследованных изоморфных комплексов других Ln, полученных в результате обменных реакций [25].
Структуры I и II образованы полимерными цепочками (рис. 2) и сольватными молекулами H2O (I) или CH3COOH (II). Координационное окружение атомов Co и Dy в I и II одинаковое – атомы Co расположены в центрах инверсии, координационный полиэдр – октаэдр. КЧ атомов Dy равно 9, полиэдр – одношапочная квадратная антипризма.
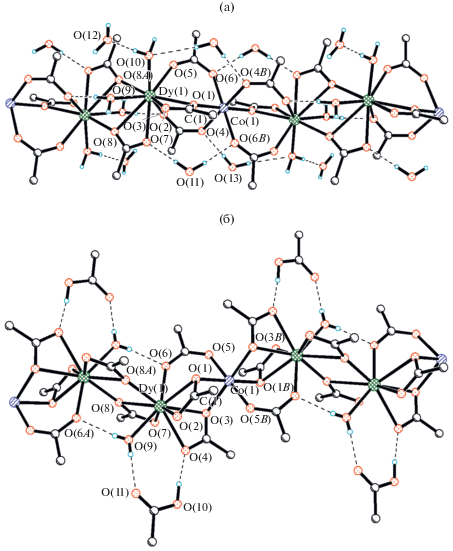
В структуре I два μ2-η2:η1-CH3COO лиганда объединяют два атома Dy в центросимметричный димер, а два μ2-CH3COO и один μ2-η2:η1-CH3COO связывают атомы Dy и Co. Два атома О молекул H2O достраивают координационное окружение Dy.
В структуре II два μ2-η2:η1-CH3COO лиганда объединяют два атома Dy в центросимметричный димер, один μ2-CH3COO и два μ2-η2:η1-CH3COO связывают атомы Dy и Co. Атом О молекулы H2O достраиваeт координационное окружение Dy.
Такое различие в строении цепочек приводит к существенной разнице в расстояниях Dy···Dy и Dy···Co в цепочке – 3.974 и 4.309 Å в I, 3.578 и 4.050 Å в II. В связи со столь значительными различиями указанных величин несомненный интерес представляет сравнительное исследование важнейших физико-химических свойств I и II, например магнитных свойств, термического поведения и др. Исследования в указанных направлениях будут продолжены.
Кристаллы соединения I изоструктурны кристаллам CoNd [26], CoGd [25], CoTb [27], MnNd [27], MnGd [25, 28]; II – аналогам CoEu, CoGd, CoTb, CoHo, CoEr, CoTm, CoYb, CoLu, MnEu, MnGd, MnTb, MnHo, MnEr, MnTm, MnYb, MnLu [27].
В [29] приводятся данные о синтезе Na2[Nd2Mn(CH3COOO)8(H2O)4]n(OH)2n · 2nH2O (a = 8.255, b = 10.394, c = 11.550 Å, α = 111.48°, β = = 107.86°, γ = 93.51°, пр. гр. $P\bar {1}$). Значение Uэкв атома Na в этой структуре 0.094 Å2, в то время как аналогичные значения атомов O (H2O, OH) 0.037–0.062 Å2. КЧ атома Na равно 4, расстояния Na···O 2.74–2.99 Å. Расчет баланса валентных усилий по [30] приводит для Na к нереальному значению 0.25. Все это однозначно доказывает, что кристаллы соединения, синтезированного в [29], изоструктурны кристаллам I и ранее описаны в [27].
Можно утверждать, что различное молекулярное строение (количество молекул воды, координированных атомами Dy) и сольватный состав комплексов I и II определяются условиями синтеза этих соединений. Действительно, комплекс I, в структуре которого содержится большее количество молекул H2O, получили из системы, содержащей избыток воды за счет использования неабсолютированного (содержащего до 4 об. % воды) этанола. Напротив, синтез соединения II проводится в присутствии большого избытка ледяной уксусной кислоты, а избыток воды существенно ниже, чем при образовании I (за счет использования меньшего количества этанола). Избыток CH3COOH в данном случае обеспечивает не только “вытеснение” сольватных молекул воды, но и удержание молекул воды в растворе (за счет образования прочной системы водородных связей) и их последующее удаление при упаривании.
Таким образом, в рамках данного исследования показано определяющее влияние условий синтеза (состава сольвосистемы, температуры) на молекулярную и кристаллическую структуру гетерометаллических 1D-полимерных ацетатных комплексов, образующихся в системе Co(Acac)2 · 2H2O–Dy(Acac)3 · 3H2O–CH3COOH–C2H5OH. Изученные комплексы, хотя и характеризуются одинаковым мотивом металлоостова – {Dy2Co}n, различаются качественным составом координационного окружения атомов Dy (за счет количества координированных молекул H2O). Указанное отличие, в свою очередь, приводит к существенному изменению расстояний Dy···Dy и Dy···Co в цепочках исследованных комплексов, что определяет интерес к дальнейшему сравнительному исследованию полученных соединений (в частности, их магнитных свойств).
Исследования проводили с использованием оборудования ЦКП ФМИ ИОНХ РАН.
Работа выполнена при финансовой поддержке Российского научного фонда (грант № 16-13-10407).
Список литературы
Winpenny R.E.P. // Chem. Soc. Rev. 1998. V. 27. № 6. P. 447.
Sakamoto M., Manseki K., Ōkawa H. // Coord. Chem. Rev. 2001. V. 219–221. P. 379.
Andruh M., Costes J.-P., Diaz C., Gao S. // Inorg. Chem. 2009. V. 48. № 8. P. 3342.
Benelli C., Gatteschi D. // Chem. Rev. 2002. V. 102. № 6. P. 2369.
Huang Y.-G., Wang X.-T., Jiang F.-L. et al. // Chem. Eur. J. 2008. V. 14. № 33. P. 10340.
Zaleski C.M., Depperman E.C., Kampf J.W. et al. // Angew. Chem. Int. Ed. 2004. V. 43. № 30. P. 3912.
Huang Y.-G., Jiang F.-L., Hong M.-C. // Coord. Chem. Rev. 2009. V. 253. № 23. P. 2814.
He R., Liang Q., Song H.-H., Wei Z. // Struct. Chem. 2010. V. 21. № 5. P. 923.
Dobrohotova Z.V., Sidorov A.A., Kiskin M.A. et al. // J. Solid State Chem. 2010. V. 183. № 10. P. 2475.
Zauzolkova N., Dobrokhotova Z., Lermontov A. et al. // J. Solid State Chem. 2013. V. 197. P. 379.
Gavrikov A., Koroteev P., Ilyukhin A. et al. // Polyhedron. 2017. V. 122. P. 184.
Gavrikov A.V., Koroteev P.S., Dobrokhotova Z.V. et al. // Polyhedron. 2015. V. 102. P. 48.
Gavrikov A.V., Koroteev P.S., Efimov N.N. et al. // Dalton Trans. 2017. V. 46. № 10. P. 3369.
Koroteev P.S., Ilyukhin A.B., Efimov N.N. et al. // Polyhedron. 2018. V. 154. P. 54.
Sun W.-B., Yan B., Jia L.-H. et al. // Dalton Trans. 2016. V. 45. № 21. P. 8790.
Zhang S., Ke H., Sun L. et al. // Inorg. Chem. 2016. V. 55. № 8. P. 3865.
Binnemans K. // Handbook on the Physics and Chemistry of Rare Earths. V. 35 / Eds. Gschneidner K.A., Bünzli J.-C.G., Pecharsky V.K. Elsevier, 2005. P. 107.
APEX II and SAINT. Madison (W, USA): Bruker AXS Inc., 2007.
Sheldrick G.M. SADABS. Göttingen (Germany): Univ. of Göttingen, 1997.
Sheldrick G. // Acta Crystallogr. C. 2015. V. 71. № 1. P. 3.
Gavrikov A.V., Efimov N.N., Dobrokhotova Z.V. et al. // Dalton Trans. 2017. V. 46. № 35. P. 11806.
Gavrikov A.V., Efimov N.N., Ilyukhin A.B. et al. // Dalton Trans. 2018. V. 47. № 17. P. 6199.
Pham H.H., Taylor C.D., Henson N.J. // Chem. Phys. Lett. 2014. V. 610–611. P. 141.
Bunting J.W., Kanter J.P., Nelander R., Wu Z. // Can. J. Chem. 1995. V. 73. № 8. P. 1305.
Pan Y.-Y., Yang Y., Long L.-S. et al. // Inorg. Chem. Front. 2014. V. 1. № 8. P. 649.
Gonzalez A., Beltran A., Le Bail A. // Acta Crystallogr. C. 1991. V. 47. № 8. P. 1624.
Bierke T. Thesis. Koln (Deutschland): Department fur Chemie. Univ. zu Koln, 2012.
Zhang C., Zhang D., Ma H. et al. // J. Mol. Struct. 2013. V. 1054–1055. P. 53.
Zhang C., Chen Y., Ma H. et al. // New J. Chem. 2013. V. 37. № 5. P. 1364.
Brese N.E., O’Keeffe M. // Acta Crystallogr. B. 1991. V. 47. № 2. P. 192.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия