

Координационная химия, 2019, T. 45, № 1, стр. 24-31
Псевдополимерный мopфолиндитиокарбамат ртути(II), [Hg{S2CN(CH2)4O}2]n: супрамолекулярная структура (роль вторичных связей Hg···S), MAS ЯМР (13C, 15N) и термическое поведениеО. В. Лосева, Т. А. Родина, А. В. Иванов
О. В. Лосева 1, Т. А. Родина 2, А. В. Иванов 1, *
1 Институт геологии и природопользования ДВО РАН
, Благовещенск, Россия
2 Амурский государственный университет
, Благовещенск, Россия
* E-mail: alexander.v.ivanov@chemist.com
Поступила в редакцию 28.02.2018
После доработки 15.06.2018
Принята к публикации 19.04.2018
Аннотация
Препаративно выделен новый представитель дитиокарбаматных комплексов ртути(II) – кристаллическая бис-(морфолиндитиокарбамато-S,S')ртуть(II) с псевдо-1D-полимерной структурой, охарактеризованная методами MAS ЯМР (13С, 15N) спектроскопии и РСА (CIF file CCDC № 1821609). Пары симметричных вторичных связей Hg⋅⋅⋅S 3.400 Å объединяют моноядерные молекулы [Hg{S2CN(CH2)4O}2], включающие плоские полигоны [HgS4], в линейную псевдополимерную цепочку. Исследование термического поведения показало, что двухстадийный процесс потери массы, регистрируемый кривой ТГ, обусловлен термической деструкцией комплекса с образованием HgS и его последующей сублимацией.
Дитиокарбаматные комплексы ртути(II) – удобные прекурсоры при получении пленочных и нанокристаллических сульфидов ртути в различного рода одностадийных термохимических процессах [1–3]. Последние, благодаря малой ширине запрещенной зоны, входят в число перспективных материалов для полупроводниковой промышленности при создании ультразвуковых датчиков, фотоэлектрических преобразователей, инфракрасных детекторов, катализаторов и др. Кроме того, дитиокарбаматы ртути(II) обнаруживают способность к люминесценции, как в кристаллическом состоянии, так и в растворах [4, 5], а также эффективному хемосорбционному связыванию золота(III) из растворов [6, 7]. В связи с высоким химическим сродством ртути к сере ряд дитиореагентов предложен для эффективного связывания токсичной элементной ртути, которое включает окисление и формирование устойчивых комплексов Hg(II) в качестве индивидуальных форм ее фиксации [8].
С диалкил(арил)дитиокарбаматными (симметрично и несимметрично замещенными) и алкилендитиокарбаматными лигандами ртуть(II) в основном формирует два типа молекулярных комплексов: моноядерные [Hg(S2CNR2)2] и биядерные [Hg2(S2CNR2)4]. Первые включают по два S,S′-бидентатно-хелатных лиганда и характеризуются квадратным (R = CH3 [9], C2H5 [10, 11], CH2CH2OH [12]) или искаженно-тетраэдрическим (R = изо-C3H7 [13, 14], изо-C4H9 [15], C9H10 [2], цикло-C6H11 [16]; R2 = изо-C3H7, цикло-C6H11 [15], C2H5, C6H5 [17], CH2–C4H3N–CH3, CH2–C6H5 [4], CH2–C4H3N–CH3, CH2–C5H4N [5]) строением координационного узла [HgS4]. В составе вторых имеется два бидентатно-хелатных и два тридентатно-мостиковых лиганда (R = C2H5 [10], изо-С3Н7 [7, 14], C4H9 [15], R2 = (CH2)4 [18], (CH2)6 [19], CH3, C6H5 [20], С2Н5, цикло-C6H11 [15], изо-C3H7, CH2CH2OH [21]). Формально биядерные молекулы можно было бы рассматривать как сдвоенные, за счет двух дополнительных связей Hg–S, моноядерные фрагменты. Однако в последних у мостиковых лигандов одна из связей Hg–S существенно ослаблена, тогда как прочность связи, образуемой с атомом ртути в соседнем моноядерном фрагменте, сопоставима с таковой для концевых лигандов. Центральный восьмичленный цикл [Hg2S4C2] в биядерных молекулах стабилизирован в конформации кресло при единственном известном исключении – [Hg2{S2CN(C4H9)2}4] (конформация ванны) [15]. Проявление полиморфизма в обсуждаемых комплексах ртути (например, для ди-изо-пропилдитиокарбаматного комплекса описано три модификации: α [13], β [14] и γ [7]) обусловлено именно участием в построении кристаллической решетки моноядерных или/и биядерных молекул.
Среди биядерных молекулярных форм дитиокарбаматов ртути(II) комплекс состава [Hg2(S2CNR2)4] (R2 = CH2–C10H7, CH2–C5H4N) выделяeтся особым способом димеризации – за счет двух симметричных связей Hg–N при участии гетероциклических атомов азота в периферической части лигандов [5]. Кроме моноядерных и биядерных форм, для ртути(II) известен единственный трехъядерный комплекс [Hg3{S2CN(C9H10)}6] · N(C5H5), включающий 1,2,3,4-тетрагидрохинолиндитиокарбаматный лиганд и внешнесферную молекулу пиридина [2], а также полиядерные катионные комплексы, например [Hg3{S2CN(C2H5)2}4]2+ и [Hg5{S2CN(C2H5)2}8]2+ [22].
Проявление в кристаллических дитиокарбаматах ртути(II) вторичных взаимодействий невалентного типа (Hg⋅⋅⋅S, C–H⋅⋅⋅S, O–H⋅⋅⋅O, C–H⋅⋅⋅π) приводит к формированию различного рода супрамолекулярных структур, систематика которых подробно обсуждается в [12, 21].
В настоящей работе получена и детально оха-рактеризована кристаллическая бис-(морфолиндитиокарбамато-S,S′)ртуть(II), [Hg{S2CN-(CH2)4O}2] (I) – новый представитель дитиокарбаматов ртути(II) с редкой псевдо-1D-полимерной структурой, формирование которой обеспечивают парные вторичные взаимодействия Hg···S между соседними молекулами. Спектральные характеристики, структурная организация и термическое поведение комплекса I установлены методами MAS ЯМР (13С, 15N) РСА и синхронного термического анализа (СТА).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Морфолиндитиокарбамат натрия, полученный взаимодействием сероуглерода (Merck) и морфолина (Aldrich) в щелочной среде [23], охарактеризован по данным MAS ЯМР 13С, 15N (δ, м.д.): Na{S2CN(CH2)4O} · 2H2O: 204.8 (−S2CN=), 67.6, 67.2 (−OCH2−), 54.6, 53.9, 53.5 (=NCH2−); 130.8 (=N−) [24].
Синтез комплекса I. Ионы Hg2+ осаждали из водной фазы морфолиндитиокарбаматом натрия, взятым в стехиометрическом отношении. К раствору, содержащему 0.0651 г (0.190 ммоль) Hg(NO3)2 · H2O (Fluka) в 10 мл воды, приливали раствор, содержащий 0.0841 г (0.380 ммоль) Na{S2CN(CH2)4O} · 2H2O в 10 мл воды. Для подавления гидролиза раствор нитрата ртути(II) подкисляли азотной кислотой до pH 2. Образовавшийся мелкий творожистый осадок белого цвета с желтоватым оттенком многократно промыли дистиллированной водой и высушили на фильтре. Выход 99.3%. Для дифрактометрического эксперимента порошок комплекса растворяли в N,N-диметилформамиде при нагревании. Остывание раствора сопровождалось выделением мелких игольчатых желтоватых кристаллов I.
По данным MAS ЯМР 13С, 15N (δ, м.д.): [Hg{S2CN(CH2)4O}2]n: 200.4 (48)* (−S2CN=), 65.8, 65.3 (1 : 1, −OCH2−), 54.4, 53.1 (1 : 1, =NCH2−); 121.9 (60)** (=N−) (* асимметричный дублет 13C–14N, в Гц; ** константа спин-спинового взаимодействия 3J(15N–199Hg), в Гц).
Спектры CP-MAS ЯМР 13C/15N регистрировали на спектрометре Ascend Aeon (Bruker) с рабочей частотой 100.64/40.56 МГц, сверхпроводящим магнитом (В0 = 9.4 Тл) с замкнутым циклом конденсации гелия через внешний компрессор и Фурье-преобразованием. Применяли кросс-поляризацию (CP) c протонов, а для подавления взаимодействий 13C−1H и 15N−1H эффект декаплинга, при использовании радиочастотного поля на резонансной частоте протонов [25]. Образец комплекса массой ~60 мг помещали в 4.0 мм керамический ротор из ZrO2. При измерениях MAS ЯМР 13C/15N использовали вращение образцов под магическим углом на частоте 10 000(1) Гц; число накоплений 656/20 360; длительность протонных π/2-импульсов 2.7/2.5 мкс; контактное время 1H−13C/1H−15N 3.0/3.0 мс; интервал между импульсами 3.0/3.0 с. Изотропные хим. сдвиги ядер 13C/15N (м.д.) даны относительно одной из компонент внешнего стандарта – кристаллического адамантана (δ = 38.48 м.д., относительно (CH3)4Si)/кристаллического NH4Cl (δ = = 0.0 м.д., –341 м.д. в абсолютной шкале [26]) с поправкой на дрейф напряженности магнитного поля, частотный эквивалент которого составил 0.025/0.09 Гц/ч.
РСА выполнен с игольчатых монокристаллов I на дифрактометреBruker-Nonius X8 Apex CCD (MoKα-излучение, λ = 0.71073 Å, графитовый монохроматор) при 296(2) K. Сбор данных проведен по стандартной методике: φ и ω сканирование узких фреймов. Поглощение учтено эмпирически по программе SADABS [27]. Структура определена прямым методом и уточнена методом наименьших квадратов (по F 2) в полноматричном анизотропном приближении неводородных атомов. Положения атомов водорода рассчитаны геометрически и включены в уточнение в модели “наездника”. Расчеты по определению и уточнению структуры выполнены по комплексу программ SHELXTL [27]. Основные кристаллографические данные и результаты уточнения структуры I приведены в табл. 1, длины связей и валентные углы − в табл. 2.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структуры I
| Параметр | Значение |
|---|---|
| Брутто-формула | C10H16N2O2S4Hg |
| М | 525.08 |
| Сингония | Моноклинная |
| Пр. гр. | P21/n |
| a, Å | 4.2660(7) |
| b, Å | 11.616(2) |
| c, Å | 14.999(3) |
| α, град | 90.00 |
| β, град | 94.067(5) |
| γ, град | 90.00 |
| V, Å3 | 741.4(2) |
| Z | 2 |
| ρ(выч.), г/см3 | 2.352 |
| μ, мм–1 | 10.941 |
| F(000) | 500 |
| Размер кристалла, мм | 0.12 × 0.02 × 0.02 |
| Область сбора данных по θ, град | 2.22–27.61 |
| Интервалы индексов отражений | –5 ≤ h ≤ 5, –15 ≤ k ≤ 15, –1 ≤ l ≤ 19 |
| Измерено отражений | 5366 |
| Независимых отражений (Rint) | 1668 (0.0489) |
| Отражений с I > 2σ(I) | 1229 |
| Переменных уточнения | 89 |
| GOOF | 1.012 |
| R-факторы по F 2 > 2σ(F 2) | R1 = 0.0347, wR2 = 0.0666 |
| R-факторы по всем отражениям | R1 = 0.0590, wR2 = 0.0718 |
| Остаточная электронная плотность (min/max), e/Å3 | –1.065/1.320 |
Таблица 2.
Основные длины связей (d), валентные (ω) и торсионные (φ) углы в структуре I*
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| Hg(1)–S(1) | 2.4028(16) | N(1)–C(2) | 1.470(8) |
| Hg(1)–S(2) | 2.9041(18) | N(1)–C(5) | 1.462(7) |
| Hg(1)⋅⋅⋅S(1)b | 3.400(2) | O(1)–C(3) | 1.425(8) |
| S(1)–C(1) | 1.763(6) | O(1)–C(4) | 1.417(8) |
| S(2)–C(1) | 1.681(7) | C(2)–C(3) | 1.512(8) |
| N(1)–C(1) | 1.335(7) | C(4)–C(5) | 1.521(7) |
| Угол | ω, град | Угол | ω, град |
| S(1)Hg(1)S(2) | 67.45(5) | Hg(1)S(2)C(1) | 78.9(2) |
| S(1)Hg(1)S(2)a | 112.55(5) | S(1)C(1)S(2) | 119.5(3) |
| S(1)аHg(1)S(1)b | 86.96(5) | S(1)C(1)N(1) | 116.8(5) |
| S(1)Hg(1)S(1)b | 93.04(5) | S(2)C(1)N(1) | 123.7(5) |
| S(2)Hg(1)S(1)b | 94.77(5) | C(1)N(1)C(2) | 124.0(5) |
| S(2)aHg(1)S(1)b | 85.23(5) | C(1)N(1)C(5) | 121.8(5) |
| Hg(1)S(1)C(1) | 93.4(2) | C(2)N(1)C(5) | 113.3(5) |
| Угол | φ, град | Угол | φ, град |
| Hg(1)S(1)S(2)C(1) | 170.2(4) | S(1)C(1)N(1)C(5) | 171.9(4) |
| S(1)Hg(1)C(1)S(2) | 171.8(4) | S(2)C(1)N(1)C(2) | –176.7(5) |
| S(1)C(1)N(1)C(2) | 3.3(8) | S(2)C(1)N(1)C(5) | –8.1(8) |
Координаты атомов, длины связей и углы депонированы в Кембриджском банке структурных данных (№ 1821609; deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk).
Термическое поведение I изучали методом СТА на приборе STA 449C Jupiter (NETZSCH) в корундовых тиглях под крышкой с отверстием, обеспечивающим давление паров при термическом разложении образца в 1 атм. Скорость нагрева составляла 5°С/мин до 600°С в атмосфере аргона. Масса навесок 2.972–7.295 мг. Точность измерения температуры ±0.7°С, изменения массы ±1 × 10–4 мг. При съемке кривых ТГ и ДСК использовали файл коррекции, а также калибровки по температуре и чувствительности для заданной температурной программы и скорости нагрева. Независимое определение температуры плавления I проводили на приборе ПТП(М) (ОАО Химлаборприбор).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
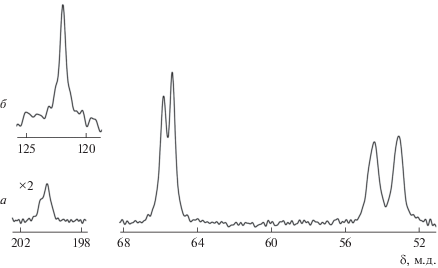
Резонансные сигналы 13C в MAS ЯМР спектре поликристаллического образца I отнесены к группам =NC(S)S–, −OCH2− и =NCH2− (рис. 1a). Пáры сигналов равной интенсивности от каждой из двух последних групп указывают на их структурную неэквивалентность в циклическом фрагменте ‒N(CH2–CH2)2O лиганда. В дитиокарбаматной группе атом углерода напрямую связан с азотом короткой связью N–C(S)S, характеризующейся значительным вкладом двоесвязанности. Поэтому группа =NC(S)S– представлена в спектре асимметричным дублетом с соотношением интенсивностей 1 : 2 (рис. 1а), вследствие диполь-дипольного взаимодействия ядра 13C с квадрупольным ядром 14N (I = 1) [28, 29]. Единственный резонансный сигнал 15N в спектре MAS ЯМР (рис. 1б) указывает на эквивалентность MfDtc-лигандов (с унифицированной структурной функцией) в комплексе I, что согласуется с данными ЯМР 13C. Обсуждаемый сигнал характеризуется уширением с двух сторон за счет перекрывания с двумя сателлитными линиями, симметрично расположенными относительно основного сигнала 15N, с соотношением интенсивностей близким к 1 : 10 : 1 (рис. 1б). Это довольно типично для дитиокарбаматов ртути(II) [19], поскольку природная ртуть включает нуклид 199Hg (μ = 0.5058852 μN, I = = 1/2). Поэтому мультиплетная структура резонансного сигнала 15N проявляется за счет спин-спинового взаимодействия между ядрами 15N и 199Hg с константой 3J(15N–199Hg) = 60 Гц. Природное содержание нуклида 199Hg (16.87 ат. %) определяет вклад эквидистантных сателлитных сигналов в общую интенсивность сигнала 15N.
Рис. 1.
Спектры MAS ЯМР 13С (а) и 15N (б) комплекса I. Частота вращения образца 10 кГц; число накоплений: 656 (а) и 20360 (б).
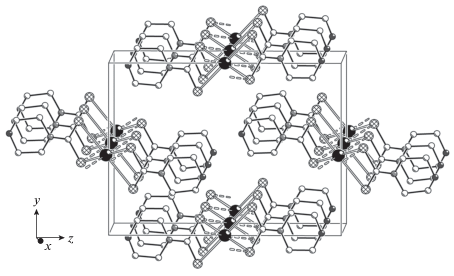
Структурная организация комплекса I установлена методом РСА. В состав элементарной ячейки входят две формульные единицы (упаковку структурных единиц см. на рис. 2). Структурная единица I – центросимметричная молекула состава [Hg{S2CN(CH2)4O}2] (рис. 3), в которой ртуть(II) S,S'-анизобидентатно координирует два MfDtc-лиганда, формируя прямоугольный хелатный узел [HgS4]. Внутрилигандные расстояния S(1)⋅⋅⋅S(2) 2.976 Å намного короче межлигандных, S(1)⋅⋅⋅S(2)a 4.423 Å. Строго плоская конфигурация последнего (диагональные валентные углы S(1)Hg(1)S(1)a и S(2)Hg(1)S(2)a равны 180°) обусловлена внешнеорбитальным sp2d-гибридным состоянием комплексообразователя. Связи Hg–S существенно неэквивалентны: Hg(1)–S(1) 2.4028, Hg(1)–S(2) 2.9041 Å (табл. 2). Однако, если первая из связей согласуется с суммой ковалентных/эмпирических ковалентных радиусов атомов серы и ртути (2.37/2.51 Å [30]), длина второй намного превосходит эти значения. Тем не менее это межатомное расстояние значительно меньше суммы ван-дер-ваальсовых радиусов (3.35 Å [30]), что позволяет считать вторую связь также ковалентной, хотя и существенно ослабленной. Диагональное расположение однородных связей Hg–S определяет длинную (S(2)–S(2)a 5.808 Å) и короткую ось (S(1)–S(1)a 4.806 Å) ромбического искажения узла [HgS4], а также отклонение углов, образуемых атомами серы, от прямого угла: S(1)S(2)S(1)a 78.34°, S(2)S(1)S(2)a 101.66°.
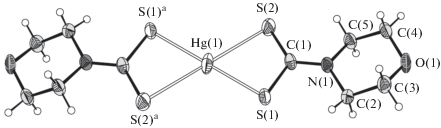
Бидентатная координация MfDtc-лигандов приводит к образованию двух металлоциклов [HgS2C] (объединяемых общим атомом ртути), малые размеры которых отражают межатомные расстояния Hg–C 3.064 и S–S 2.976 Å. Взаимное расположение атомов в циклах несколько отклоняется от копланарного, на что указывают значения торсионных углов Hg(1)S(1)S(2)C(1) 170.2° и S(1)Hg(1)C(1)S(2) 171.8° (табл. 2). Прочность связей N–C(S)S заметно выше, чем N–CН2 (табл. 2). Валентные углы при атомах углерода и азота близки к 120°, что отражает вклад двоесвязанности в формально одинарную связь N–C(S)S (за счет примешивания sp2- к sp3-гибридному состоянию атомов азота и углерода). Небольшие отклонения (на 3.3° и 8.1°) торсионных углов SCNC от 0° или 180° указывают на преимущественно плоскую геометрию группировок C2NC(S)S в MfDtc-лигандах. (табл. 2). Шестичленные гетероциклы −N(CH2)4О стабилизированы в конформации кресла и располагаются по разные стороны плоскости хелатного узла [HgS4] (транс-ориентация).
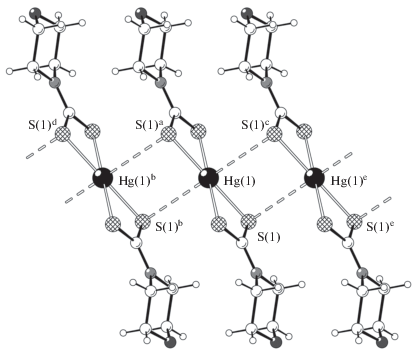
Формирование супрамолекулярной структуры I осуществляется за счет парных вторичных взаимодействий невалентного типа, в которых принимают участие атомы Hg(1) и диагонально ориентированные менее прочно связанные с комплексообразователем атомы S(1) (рис. 4). Поэтому каждая молекула [Hg{S2CN(CH2)4O}2] образует две пары вторичных связей с ближайшими соседями, в результате чего формируются линейные псевдополимерные цепочки [Hg{S2CN(CH2)4O}2]n (межатомное расстояние Hg–Hg 4.266 Å), ориентированные вдоль кристаллографической оси x. Длина парных вторичных связей Hg(1)⋅⋅⋅S(1)b и Hg(1)⋅⋅⋅S(1)с 3.400 Å близка сумме ван-дер-ваальсовых радиусов атомов ртути и серы: 3.35 Å [30]. За счет дополнительных вторичных связей Hg⋅⋅⋅S в аксиальных положениях атом ртути достраивает свой полиэдр до искаженного октаэдра [HgS6] (угол S(1)bHg(1)S(1)c 180°). Таким образом, среди множества известных к настоящему времени дитиокарбаматов ртути(II) для моноядерного соединения I характерно сочетание двух структурных особенностей: плоский полигон [HgS4] и самоорганизация псевдополимерной цепочки, что установлено еще только для двух комплексов общего состава [Hg(S2CNR2)2]n (R = C2H5 [10, 11] и CH2CH2OH [12]).
Рис. 4.
Фрагмент псевдополимерной цепочки комплекса I, ориентированной вдоль кристаллографической оси x; вто-ричные взаимодействия Hg···S показаны пунктиром. Симметрические преобразования: a 1 – x, 1 – y, –z; bx – 1, y, z; c 2 – x, 1 – y, –z; d –x, 1 – y, –z; e 1 + x, y, z.
Термическое поведение I изучено методом синхронного термического анализа с одновременной регистрацией кривых термогравиметрии (ТГ) и дифференциальной сканирующей калориметрии (ДСК). Соединение термически устойчиво до ~210°C. На кривой ТГ две основные стадии потери массы определяются диапазонами 210–315°C и 315–405°C (рис. 5а). При этом первый, круто падающий участок, делится на две части точкой перегиба при 291.0°C. Потеря массы до точки перегиба (26.76%) дает основание предположить, что начальный этап термолиза комплекса I связан с отщеплением алкоксильных фрагментов лигандов (=CHCH2)2O (расч. 26.70%), чему может соответствовать образование промежуточного соединения [Hg(S2CNH2)2]. Дальнейшее повышение температуры сопровождается возрастанием скорости термолиза, результатом которого является высвобождение HgS [7, 20]. Здесь важно отметить, что потеря массы после точки перегиба, составляющая 47.32%, значительно превышает расчетную величину 28.99%.
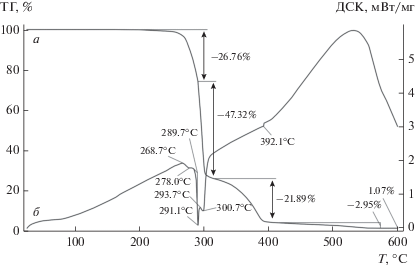
Вторая, более пологая, ступень кривой ТГ (315–405°C) связана с сублимацией HgS [7, 20]. Однако потеря массы, регистрируемая на этой ступени (21.89%), занижена более чем вдвое по сравнению с расчетным значением для HgS (44.31%). Принимая во внимание завышение потери массы на первой ступени кривой ТГ (после 291°С), можно сделать вывод, о том, что значительная часть HgS (18.33%) сублимирует еще до начала второй ступени, накладываясь на первый этап термолиза. Ранее исследование термического поведения компактного порошка HgS позволило установить начало его сублимации уже при 225°C [7]. С 405°C и до стабилизации массы тигля при 570°C на кривой ТГ представлен участок финальной десорбции (2.95%) летучих продуктов термолиза. По окончании термолиза при 600°C на дне тигля обнаружены следы светло-серого налета (1.07%), что может быть связано с выделением элементного углерода.
Участок кривой ДСК, проецируемый на первую ступень кривой ТГ, включает три тепловых эффекта (рис. 5б). Первый, слабовыраженный эндоэффект с экстремумом при 278.0°C (экстраполированная температура 268.7°C) отражает начальный этап потери массы. Второй эндоэффект при 291.1°С следует отнести к плавлению вещества в процессе разложения (экстраполированная температура 289.7°C). Независимым определением в стеклянном капилляре плавление образца с разложением установлено при 289–290°C. Третий эндоэффект при 300.7°C с экстраполированной температурой 293.7°C отражает термолиз вещества после точки перегиба. Завершению сублимации HgS (вторая ступень потери массы) на кривой ДСК соответствует слабо выраженный тепловой эффект при 392.1°C.
Авторы выражают признательность проф. О.Н. Анцуткину и докт. В. Говда (Университет технологий г. Лулео, Швеция) за предоставленную возможность и помощь в съемке спектров MAS ЯМР 13С, 15N.
Список литературы
Dar S.H., Thirumaran S., Selvanayagam S. // Polyhedron. 2015. V. 96. P. 16.
Srinivasan N., Thirumaran S., Ciattini S. // RSC Adv. 2014. V. 4. № 44. P. 22971.
Onwudiwe D.C., Ajibade P.A. // Mat. Lett. 2011. V. 65. № 21–22. P. 3258.
Yadav M.K., Rajput G., Gupta A.N. et al. // Inorg. Chim. Acta. 2014. V. 421. P. 210.
Rajput G., Yadav M.K., Thakur T.S. et al. // Polyhedron. 2014. V. 69. P. 225.
Лосева О.В., Родина Т.А., Смоленцев А.И., Иванов А.В. // Коорд. химия. 2016. Т. 42. № 11. С. 683. (Loseva O.V., Rodina T.A., Smolentsev A.I., Ivanov A.V. // Russ. J. Coord. Chem. 2016. V. 42. № 11. P. 719. doi 10.1134/ S1070328416110063).
Loseva O.V., Rodina T.A., Smolentsev A.I., Ivanov A.V. // Polyhedron. 2017. V. 134. P. 238.
Mercuri M.L., Serpe A., Marchiò L. et al. // Inorg. Chem. Comm. 2014. V. 39. P. 47.
Cox M.J., Tiekink E.R.T. // Z. Kristallogr. 1997. V. 212. № 7. P. 542.
Iwasaki H. // Acta Crystallogr. B. 1973. V. 29. № 10. P. 2115.
Healy P.C., White A.H. // Dalton Trans. 1973. № 3. P. 284.
Howie R.A., Tiekink E.R.T., Wardell J.L., Wardell S.M.S.V. // J. Chem. Crystallogr. 2009. V. 39. № 4. P. 293.
Ito M., Iwasaki H. // Acta Crystallogr. B. 1979. V. 35. № 11. P. 2720.
Iwasaki H., Ito M., Kobayashi K. // Chem. Lett. 1978. V. 7. P. 1399.
Cox M.J., Tiekink E.R.T. // Z. Kristallogr. 1999. V. 214. № 9. P. 571.
Cox M.J., Tiekink E.R.T. // Main Group Met. Chem. 2000. V. 23. № 12. P. 793.
Onwudiwe D.C., Ajibade P.A. // J. Chem. Crystallogr. 2011. V. 41. № 7. P. 980.
Altaf M., Stoeckli-Evans H., Batool S.S. et al. // J. Coord. Chem. 2010. V. 63. № 7. P. 1176.
Иванов А.В., Корнеева Е.В., БуквецкийБ.В. и др. // Коорд. xимия. 2008. Т. 34. № 1. С. 61 (Ivanov A.V., Korneeva E.V., Bukvetskii B.V. et al. // Russ. J. Coord. Chem. 2008. V. 34. № 1. P. 59. doi 10.1134/ S1070328408010107).
Onwudiwe D.C., Ajibade P.A. // Int. J. Mol. Sci. 2011. V. 12. № 3. P. 1964.
Jotani M.M., Tan Y.S., Tiekink E.R.T. // Z. Kristallogr. 2016. V. 231. № 7. P. 403.
Bond A.M., Colton R., Hollenkamp A.F. et al. // J. Am. Chem. Soc. 1987. V. 109. № 7. P. 1969.
Бырько В.М. Дитиокарбаматы. М.: Наука, 1984. 341 с.
Иванов А.В., Ивахненко Е.В., Герасименко А.В., Форшлинг В. // Журн. неорган. химии. 2003. Т. 48. № 1. С. 52 (Ivanov A.V., Ivakhnenko E.V., Gerasimen-ko A.V., Forsling W. // Russ. J. Inorg. Chem. 2003. V. 48. № 1. P. 45).
Pines A., Gibby M.G., Waugh J.S. // J. Chem. Phys. 1972. V. 56. № 4. P. 1776.
Ratcliffe C.I., Ripmeester J.A., Tse J.S. // Chem. Phys. Lett. 1983. V. 99. № 2. P. 177.
APEX2 (version 1.08), SAINT (version 7.03), SADABS (version 2.11), SHELXTL (version 6.12). Madison (WI, USA): Bruker AXS Inc., 2004.
Hexem J.G., Frey M.H., Opella S.J. // J. Chem. Phys. 1982. V. 77. № 7. P. 3847.
Harris R.K., Jonsen P., Packer K.J. // Magn. Res. Chem. 1985. V. 23. № 7. P. 565.
Winter M. Web Elements Periodic Table of the Elements, http://www.webelements.com (accessed January 2010).
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия