

Координационная химия, 2019, T. 45, № 12, стр. 756-762
Молекулярные и внутрикомплексные соединения диоксомолибдена(VI) с двузамещенными салицилиденалкогольиминами. Кристаллическая структура сольвата (1 : 1) диоксо(3,5-дибромсалицилиденмоноэтанолиминато)молибдена(VI) с метанолом [MoO2(L1) · MeOH], L1 = C9H7Br2NO2
В. С. Сергиенко 1, 2, *, В. Л. Абраменко 3, Ю. Е. Горбунова 1, А. В. Чураков 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
2 Всероссийский институт научной и технической информации РАН
Москва, Россия
3 Луганский национальный университет им. Владимира Даля
Луганск, Украина
* E-mail: sergienko@igic.ras.ru
Поступила в редакцию 20.02.2019
После доработки 27.05.2019
Принята к публикации 03.06.2019
Аннотация
Синтезированы комплексные соединения диоксомолибдена(VI) молекулярного (МК) МоО2Сl2 · 2H2L и внутрикомплексного (ВКС) типа [MoO2L · Solv] (H2L = азометины, производные двузамещенных R1,R2-салициловых альдегидов (R1,R2 = 3.5-Вr2; R1 = 3-МеО, R2 = 5-Вr) и моноэтаноламина, Solv = молекула метанола, диметилформамида, пиридина, α-пиколина). На основании данных ИК‑спектроскопии сделан вывод о цис-октаэдрическом строении комплексов. В МК лиганды координированы через атом О карбонильной группы хиноидной таутомерной формы H2L, в ВКС – в депротонированной бензоидной форме. Методом РСА (СIF file CCDC № 1898088) установлено строение [MoO2(L1) · МеОН] (I), где L1 – C9H7Br2NO2. В моноядерной молекуле I атом Мо имеет октаэдрическую координацию двумя оксолигандами, двумя атомами кислорода и атомом азота тридентатного бис(хелатного) двухзарядного лиганда (L1)2–, атомом O молекулы метанола. В транс-позициях к лигандам О(оксо) расположены нейтральные атомы N(1), O(1) лигандов L1 и МеОН соответственно. Связи Mo–N(1) 2.265 и Мо–О(1) 2.372 Å существенно удлинены вследствие структурного проявления транс-влияния кратносвязанных оксолигандов. Межмолекулярные водородные связи (МеОН)О–Н···О(оксо) объединяют молекулы в супрамолекулярные 1D-цепочки.
Салицилиденалкогольимины (Н2L) представляют собой потенциально тридентатные двухосновные лиганды и при взаимодействии с солями d-металлов могут образовывать несколько типов внутрикомплексных соединений (ВКС) мономерного [МLn], [М(НL)n], ди- [МLn]2 или олигомерного [МLn]m строения, проявляя при этом би- или тридентатную функцию [1–5]. В ранних работах авторы [6–9] на основании данных ИК- и ЯМР-спектроскопии пришли к выводу о мономерном строении комплексов МоО2L (КЧ 5) и МоО2L · Solv (Solv = молекула растворителя) (КЧ 6). Позже по результатам РСА мы установили [1, 2], что сольватированные комплексы диоксомолибдена(VI) с замещенными салицилиденмоноэтаноламинами (R1,R2 = 3.5-Cl2 и R1 = 3-МеО, R2 = H) имеют цис-октаэдрическое строение с координацией лигандов через азометиновый атом азота и два атома кислорода депротонированных гидроксогрупп альдегидного и спиртового фрагментов. Шестую вершину в октаэдре занимает донорный атом молекулы растворителя (Solv = MeOH).
Молекулы в структурах ВКС попарно объединены в центросимметричные псевдодимеры почти линейными межмолекулярными водородными связями. К аналогичным выводам пришли авторы [10, 11] на основании данных РСА ряда ВКС диоксомолибдена(VI) с монозамещенными салицилиденалкогольиминами.
Менее известны в литературе комплексы молекулярного типа (аддукты) (МК) МХn · mН2L (Х– = анион), в структуре которых лиганды Н2L координированы в нейтральной форме. В развитие представлений о строении и свойствах комплексов диоксомолибдена(VI) с салицилиденалкогольиминами мы осуществили синтез и ИК‑спектроскопическое исследование МК и ВКС диоксомолибдена(VI) с двузамещенными (R1,R2 = 3,5-Вr2 (Н2L1) и R1 = 3-МеО, R2 = 5-Вr (Н2L2)) салицилиденмоноэтанолиминами, имеющие, по данным элементного анализа, состав МоО2Сl2 · 2Н2L1, 2 и [MoO2L1, 2 · Solv] (Solv = молекула метанола, ДМФА, пиридина (Pу), α-пиколина (α-Pіс) соответственно).
Строение комплекса [MoO2(L1) · МеОН] (I) (L1 = C9H7Br2NO2) определено методом РСА.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходный диоксид молибдена получали по общеизвестной методике восстановлением триоксида молибдена квалификации “х. ч.” водородом при 400°С. В работе использовали реактивы и растворители зарубежных фирм Lancaster, Mark, Fluka.
Синтез МоО2Сl2 выполняли хлорированием порошка МоО2 при 350°С [12]. Образующийся в ходе реакции МоО2Сl2 уносился из горячей зоны и конденсировался в холодной части реактора в виде легких бледно-желтых чешуек. Избыток хлора удаляли продуванием диоксодихлорида молибдена сухим аргоном при слабом подогреве (40–50°С) до отрицательной реакции на свободный хлор. Ввиду очень низкого давления паров исходного диоксида молибдена, образующийся МоО2Сl2 не требовал дополнительной очистки.
Ацетилацетонат молибденила МоО2(Aсас)2 получали по методике [13].
Синтез Н2L1 и Н2L2 осуществляли конденсацией 3,5-дибром- и 3-метокси-5-бромсалицилового альдегида и моноэтаноламина в кипящем этаноле. После перекристаллизации из этанола они представляют собой иглы лимонного (HL1) или желтого (HL2) цвета, Tпл = 147–148 и 124–125°С соответственно.
| Найдено, %: | С 33.20; | Н 2.98; | N 4.15. |
| Для С9Н9Br2NO2 (HL1) | |||
| вычислено, %: | С 33.47; | Н 2.81; | N 4.34. |
| Найдено, %: | С 43.67; | Н 4.64; | N 4.93. |
| Для С9Н9Br2NO2 (HL2) | |||
| вычислено, %: | С 43.81; | Н 4.41; | N 5.11. |
ИК-спектры HL1 (ν, см–1): 3310 ш, 1652, 1610, 1512, 1350, 1270, 1215, 1158, 1090, 1070, 1030, 908, 870, 770, 748, 630, 580, 495, 447; HL2 (ν, см–1): 3400 ш, 1650, 1598, 1520, 1412, 1344, 1240, 1210, 1100, 1065, 1040, 1017, 965, 898, 870, 850, 780, 763, 722, 665, 570, 552, 415.
Синтез МК МоО2Сl2 · 2H2L1, 2 выполняли методом непосредственного взаимодействия МоО2Сl2 с салицилидениминами аналогично [14] в среде малополярных растворителей. В диэтиловом эфире комплексы образуются почти с количественным выходом. К раствору 0.199 г (0.001 моля) диоксодихлорида молибдена в 20 мл абсолютного диэтилового эфира прибавляли по каплям при перемешивании на магнитной мешалке раствор в бензоле 0.002 моля соответствующего азометина. Образовавшийся осадок комплекса отделяли на фильтре Шотта, снабженном осушительной системой, промывали эфиром и сушили в токе сухого аргона.
Синтез ВКС [MoO2L1, 2 · МеОН] осуществляли методом лигандного обмена между ацетилацетонатом молибденила и азометинами [1, 2]. К горячему раствору 0.326 г (0.001 моля) МоО2(Асас)2 в 20 мл метанола добавляли по каплям метанольный раствор 0.001 моля азометина, кипятили реакционную смесь в течение 10–15 мин и оставляли при комнатной температуре для кристаллизации. Выпавшие кристаллы комплексов отделяли фильтрованием, промывали холодным метанолом и сушили вначале в токе сухого воздуха, а затем в вакуум-эксикаторе над СаСl2. Выходы комплексов составляли ~90%. При перекристаллизации комплексов [MoO2L1, 2 · МеОН] из пиридина или α‑пиколина образовывались соответствующие кристаллические сольватокомплексы [MoO2L1, 2 · Рy] и [MoOL1, 2 · α-Рic]. Комплекс [MoO2L2 · ДМФА] получали в процессе синтеза в метаноле с добавлением в реакционную смесь 2 мл диметилформамида. Во всех случаях выход комплексов составлял не менее 90%.
Используемые в работе органические растворители подвергали тщательной очистке и обезвоживанию по общеизвестным методикам [15].
Элементный анализ комплексов на содержание молибдена и хлора проводили, как и в [14], по стандартным методикам. Анализ соединений на содержание углерода, водорода и азота проводили на анализаторе Carlo-Erba 1106 Elemental Analyzer CHN. Электропроводность растворов комплексов измеряли на кондуктометре с рабочей частотой переменного тока 1000 Гц по мостовой схеме в термостатируемой ячейке с платинированными электродами.
ИК-спектры азометинов и комплексных соединений регистрировали на спектрометре ИКС-29 в области 3600–400 cм–1 (суспензии в вазелиновом масле).
РСА I выполнен на автоматическом дифрактометре Enraf-Nonius CAD4 при комнатной температуре (MoKα-излучение, λ = 0.71073 Å, графитовый монохроматор). Кристаллы I (C10H11Br2MoNO5, M = 480.96) ромбические, пр. гр. Pbca, a = 6.711(1), b = 13.278(1), c = 31.534(1) Å, V = 2810.0(5) Å3, Z = 8, ρ(выч.) = 2.274 г/см3, μ(MoKα) = 6.635 мм–1, F(000) = 1840. Интенсивности 12 584 отражений (из них 2766 независимых, Rint = 0.0948) измерены методом ω-сканирования в интервале 2.58° < θ < < 26.00° (–1 ≤ h ≤ 8, –16 ≤ k ≤ 16, –38 ≤ l ≤ 38). Структура расшифрована прямым методом и уточнена полноматричным анизотропным МНК по F2 для всех неводородных атомов (SHELXL-97 [16]). Гидроксильный атом водорода Н(1) найден из разностного ряда Фурье и уточнен изотропно; остальные атомы H помещены в рассчитанные позиции и уточнены по схеме “наездника”. Окончательное значение факторов расходимости составило R1 = 0.0460, wR2 = 0.1036 для 1856 отражений с I > 2σ(I) и 178 параметров уточнения, GООF = 1.010, Δρmax, Δρmin = 1.261, –1.564 e/Å3. Основные длины связей и валентные углы приведены в табл. 1.
Таблица 1.
Длины связей (d) и валентные углы (ω) в структуре I
| Связь | d, Å | Связь | d, Å | Связь | d, Å | Связь | d, Å |
|---|---|---|---|---|---|---|---|
| Mo(1)–O(4) | 1.691(5) | Mo(1)–O(2) | 1.723(5) | Mo(1)–O(3) | 1.899(5) | Mo(1)–O(5) | 1.969(4) |
| Mo(1)–N(1) | 2.265(5) | Mo(1)–O(1) | 2.372(5) | Br(1)–C(5) | 1.894(6) | Br(2)–C(3) | 1.888(7) |
| O(1)–C(10) | 1.428(9) | O(3)–C(9) | 1.431(9) | N(1)–C(7) | 1.273(8) | C(1)–C(7) | 1.440(8) |
| O(1)–H(1) | 0.74(10) | O(5)–C(2) | 1.338(7) | N(1)–C(8) | 1.465(8) | C(8)–C(9) | 1.488(11) |
| Угол | ω, град | Угол | ω, град | Угол | ω, град | Угол | ω, град |
| O(4)Mo(1)O(2) | 107.2(3) | O(2)Mo(1)O(5) | 99.9(2) | O(3)Mo(1)N(1) | 75.5(2) | O(3)Mo(1)O(1) | 83.0(2) |
| O(4)Mo(1)O(3) | 100.0(2) | O(3)Mo(1)O(5) | 151.6(2) | O(5)Mo(1)N(1) | 79.9(2) | O(5)Mo(1)O(1) | 77.6(2) |
| O(2)Mo(1)O(3) | 98.1(2) | O(4)Mo(1)N(1) | 93.5(2) | O(4)Mo(1)O(1) | 168.5(2) | N(1)Mo(1)O(1) | 76.4(2) |
| O(4)Mo(1)O(5) | 95.5(2) | O(2)Mo(1)N(1) | 159.1(2) | O(2)Mo(1)O(1) | 83.2(2) | N(1)C(8)C(9) | 105.5(6) |
| C(10)O(1)Mo(1) | 130.4(4) | C(9)O(3)Mo(1) | 120.2(4) | C(7)N(1)Mo(1) | 126.9(5) | O(3)C(9)C(8) | 108.8(5) |
| C(10)O(1)H(1) | 130(9) | C(2)O(5)Mo(1) | 129.7(4) | C(8)N(1)Mo(1) | 111.2(4) | ||
| Mo(1)O(1)H(1) | 99(9) | C(7)N(1)C(8) | 121.8(6) | N(1)C(7)C(1) | 124.6(6) |
Координаты атомов и другие параметры структуры I депонированы в Кембриджском банке структурных данных (CCDC № 1898088; deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk/ data_request/cif).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Экспериментально установлено, что несмотря на наличие в молекулах салицилиденалкоголь-иминов двух гидроксогрупп, при взаимодействии МоО2Сl2 с H2L1, 2 в среде малополярных растворителей – диэтиловом эфире, диоксане, тетрагидрофуране – образуются МК (а не ВКС) состава МоО2Сl2 · 2H2L1, 2 (табл. 2), представляющие собой аморфные порошки, гидролизующиеся на воздухе, легко растворимые в метаноле с образованием токопроводящих растворов. Молярная электропроводность (λМ) растворов МК в метаноле для концентраций 1 × 10–3 моль/л составляет величину ~150 см см2 моль–1, что характеризует их как электролиты типа 1 : 2 [14].
Таблица 2.
Результаты физико-химического и ИК-спектроскопического исследования комплексов диоксомолибдена(VI) c двухзамещенными салицилиденмоноэтанолиминами
| Комплекс | Цвет | Tпл, °С | Брутто-формула | Содержание (найдено/вычислено), % | ИК-спектр, см–1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ν(C=N) | ν(C=N) | ν(C=O) | ν(Mo=O) | ||||||||
| Mo | С | Н | N | лиганда | комплекса | ||||||
| MoO2L1 · МеОН | Лимонный | 295–297 | C10H11Br2MoNO5 | 20.18/19.95 | 24.79/24.97 | 2.46/2.30 | 2.80/2.91 | 1652 | 1638 | 930, 915, 900 пл. | |
| MoO2L1 · Рy | Лимонный | 300–302 | C14H12Br2MoN2O4 | 18.10/18.17 | 31.78/31.85 | 2.40/2.29 | 5.42/5.31 | 1652 | 1642 | 920, 890 | |
| MoO2L1 · α-Рic | Лимонный | 296–298 | C15H14Br2MoN2O4 | 17.58/17.70 | 33.05/33.24 | 2.88/2.60 | 5.25/5.17 | 1652 | 1642 | 922, 905 | |
| MoO2L2 · MeOH | Лимонный | 278–280 | C11H14BrMoNO6 | 22.35/22.20 | 30.44/30.58 | 3.42/3.27 | 3.10/3.24 | 1650 | 1637 | 928, 902 | |
| MoO2L2 · ДМФА | Лимонный | 275–277 | C13H17BrMoN2O6 | 20.16/20.28 | 32.85/33.00 | 3.76/3.62 | 5.85/5.92 | 1650 | 1638 | 925, 900 | |
| MoO2L2 · Рy | Лимонный | 283–285 | C15H15BrMoN2O5 | 19.96/20.02 | 37.46/37.60 | 3.27/3.16 | 5.70/5.84 | 1650 | 1640 | 922, 897 | |
| MoO2L2 · α-Рic | Лимонный | 285–287 | C16H17BrMoN2O5 | 19.34/19.45 | 38.82/38.97 | 3.60/3.47 | 5.45/5.68 | 1650 | 1640 | 925, 903 | |
| MoO2Cl2 · 2HL1 * | Желтый | 224–226 | C18H18Br4Cl2MoN2O6 | 11.30/11.36 | 25.43/25.59 | 2.27/2.15 | 3.45/3.32 | 1652 | 1662 | 930, 905 | |
| MoO2Cl2 · 2HL2 ** | Оранжевый | 218–220 | C20H24Br2Cl2MoN2O8 | 12.87/12.84 | 32.06/32.15 | 3.42/3.24 | 3.90/3.75 | 1650 | 1658 | 932, 905 | |
ВКС [MoOL1,2 · Solv], в отличие от МК, образуют при медленной кристаллизации из органических растворителей хорошо сформированные кристаллы, плавящиеся при значительно более высокой температуре, чем соответствующие аддукты (табл. 2). Полученные ВКС устойчивы на воздухе, малорастворимы в холодных полярных растворителях (метаноле, диметилсульфоксиде, диметилформамиде, пиридине, пиколинах), но растворяются в них при нагревании, образуя растворы, не проводящие электрический ток. Выдерживание комплексов на воздухе при 140–150°С в течение 2 ч приводит к их полной десольватации.
В ИК-спектрах полученных ВКС (табл. 2) наблюдается характерное для внутрикомплексных соединений металлов с азометинами низкочастотное смещение полосы валентных колебаний связи С=N на 10–14 см–1 по сравнению с ее положением в спектрах свободных лигандов, а также исчезновение полос валентных колебаний гидроксогрупп в области 3100–3600 см–1. Сильная полоса 1655 см–1 в спектре [MoO2L2 · ДМФА] относится к пониженной частоте ν(С=О) координированной молекулы диметилформамида. В спектрах [MoO2L1,2 · Solv], где Solv – пиридин или α-пиколин, наблюдается смещение серии полос в область ~1000–1610 см–1, отвечающих колебаниям связей гетерокольца [17].
В спектрах МК в области 1600–1700 см–1 отмечается интенсивная полоса ~1660 см–1, которую с наибольшей вероятностью следует отнести к повышенной в результате комплексообразования полосе валентных колебаний связи С=О хиноидного (кето-аминного) таутомера. Стабилизацию хиноидной таутомерной формы о-окиазометинов при образовании комплексов молекулярного типа мы неоднократно обсуждали в [18–20]. Имеющиеся в литературе немногочисленные данные РСА молекулярных комплексов ряда металлов с о-оксиазометинами [21–24] также подтверждают координацию лигандов в хиноидной таутомерной форме.
Сильные полосы в области 890–930 см–1 относятся к симметричным и антисимметричным валентным колебаниям связи Мо=О цис-МоО2-группы [1, 2]. В области низких частот в ИК-спектрах комплексов появляются новые полосы поглощения при 540–580 cм–1 (в спектрах МК) и 520–540, 470–475 см–1 (в спектрах ВКС), которые следует отнести к валентным колебаниям связи Мо–ОHL или Мо–NL и Мо–ОL соответственно [1, 2, 14].
На основании полученных экспериментальных и с учетом литературных данных можно заключить, что исследуемые МК и ВКС диоксомолибдена(VI) c замещенными салицилиденмоноэтанолиминами имеют типичное для оксо- и диоксокатионов группы VIВ октаэдрическое строение с двумя кратносвязанными оксолигандами в цис-положении. В МК в вершинах октаэдра кроме концевых атомов кислорода расположены два карбонильных атома О хиноидной таутомерной формы оксоазометинов в транс-позициях к оксолигандам в соответствии с “правилом самосогласованности” [25] и два атома хлора в транс-позициях друг к другу. Вершины октаэдра в ВКС занимают два кратносвязанных концевых атома О – донорный атома N азометиновой группы (транс к оксолигандам) и два атома О депротонированных гидроксогрупп альдегидного и спиртового фрагментов в цис-позициях к оксолигандам. Шестое координационное место транс к оксолиганду занимают донорные атомы координированных молекул растворителей.
Строение ВКС I установлено методом РСА. В моноядерной молекуле (рис. 1) атом молибдена имеет октаэдрическую координацию двумя оксолигандами О(2), О(4), двумя атомами кислорода О(3), О(5) и атомом азота N(1) тридентатного бис(хелатного) двухзарядного лиганда (L1)2–, атомом O(1) молекулы метанола. В транс-позициях к лигандам О(оксо) расположены, в соответствии с “правилом самосогласованности” [25], нейтральные атомы N(1), O(1) лигандов L1 и МеОН соответственно. Связи Mo–N(1) 2.265 и Мо–О(1) 2.372 Å существенно удлинены вследствие структурного проявления транс-влияния кратносвязанных оксолигандов. Эти связи также заметно длиннее стандартных расстояний Мо–О(СТ) 2.04, Мо–N(СТ) 2.10 Å [25]. Связи Мо–О(оксо) (средн. 1.707 ± 0.016 Å) типичны по длине и соответствуют увеличенной кратности. Достаточно короткие связи Мо–О(3)цис и Мо–О(5)(L1)цис существенно различаются по длине (1.899 и 1.969 Å соответственно 1). Атом Мо смещен из центра октаэдра МоО5N к ребру О(2)(оксо)–О(4)(оксо): валентные углы О(оксо)Мо(О,N)цис, за одним исключением О(2)МоО(1) 83.2°, больше идеальной величины 90° (93.5°–100.0°), а противолежащие углы (О,N)Mo(O,N) – заметно меньше 90° (75.5°–83.0°). Самый большой валентный угол в структуре I О(2)МоО(4) 107.2° из-за отталкивания кратносвязанных оксолигандов. Углы между противолежащими донорными атомами лигандов О(3)МоО(5) 151.6°, О(2)МоN(1) 159.1°, O(4)MoO(1) 168.5° в разной степени отклоняются от идеального значения 180°. При координации тридентатного лиганда с атом металла замыкаются два сопряженных по связи Мо–N(1) хелатных цикла: шестичленный МоNC3O (А) и пятичленный MoNC2O (Б). Металлоцикл А имеет конформацию софы с отклонением от четырех копланарных атомов NC3 (±0.020–0.046 Å) в одну сторону атомов Мо на 0.430 Å, О(5) – на 0.266 Å. Хелатный цикл Б имеет асимметричную твист-конформацию с отклонением атомов С(8) на 0.129 Å, С(9) – на –0.469 Å от плоскости атомов Мо, N(1), O(3). Двугранный угол между плоскостями А и Б, в которых среднее отклонение атомов от средних плоскостей равно, соответственно, 0.151 и 0.153 Å, составляет 11.6°.
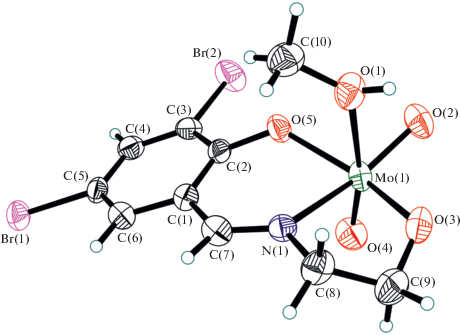
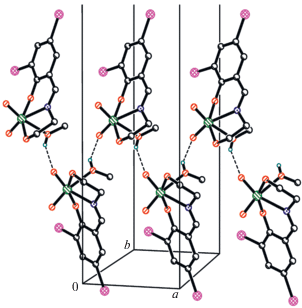
Молекулы I попарно объединены водородной связью (ВС) О(1)–Н(1)···О(2) (0.5 + х, у, 0.5 – z) между молекулой метанола и одним из оксолигандов (О(1)–Н(1) 0.74(10), Н(1)···О(2) 2.07(10), О(1)···О(2) 2.695(6) Å, угол О(1)Н(1)О(2) 143(13)°). За счет этих ВС формируются 1D-цепочки вдоль оси с кристалла (рис. 2). Участие оксолиганда О(2) в ВС, очевидно, является причиной несколько более длинной связи Мо–О(2) 1.723 Å, чем Мо–О(4) 1.691 Å.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Сергиенко В.С., Абраменко В.Л., Порай-Кошиц М.А., Гарновский А.Д. // Журн. структур. химии. 1990. Т. 31. № 5. С. 54.
Сергиенко В.С., Абраменко В.Л., Михайлов Ю.Н., Суражская М.Д. // Журн. неорган. химии. 2014. Т. 59. № 11. С. 1503 (Sergienko V.S., Abramenko V.L., Mikhailov Yu.N., Surazhskaya M.D. // Russ. J. Inorg. Chem. 2014. V. 59. № 11. P. 1219.).https://doi.org/10.1134/S0036023614110205
Torihara N., Mikuriya M., Okawa H., Kida S. // Bull. Chem. Soc. Jpn. 1980. V. 53. № 6. P. 1610.
Kessisoglou D.P., Li X., Butler W.M., Pecorara V.L. // Inorg. Chem. 1987. V. 26. № 15. P. 2487.
Коган В.А., Зеленцов В.В., Осипов О.А., Бурлов А.С. // Успехи химии. 1979. Т. 48. № 7. С. 1208.
Rajan G.C., Chakravorty A. // Inorg. Chem. 1981. V. 20. № 3. P. 665.
Topich J. // Inorg. Chem. 1981. V. 20. № 11. P. 3704.
Alyea E.C., Topich J. // Inorg. Chim. Acta. 1982. V. 65. № 3. P. L95.
Topich J., Lyon J.T. // Polyhedron. 1984. V. 3. № 1. P. 55.
Zhang C., Reinwald G., Lozan V. et al. // Z. Anorg. Allg. Chem. 2002. V. 628. № 6. P. 1259.
Nakajima K., Yokoyama K., Kano T., Kojima // Inorg. Chim. Acta. 1998. V. 282. № 2. P. 209.
Щукарев С.А., Василькова И.В., Шарупин Б.Н. // Вестн. ЛГУ. 1959. № 4. С. 73.
Fernelius W.C., Terada K., Bryant B.E. // J. Am. Chem. Soc. 1959. V. 81. P. 3188.
Абраменко В.Л., Гарновский А.Д., Сурпина Л.В. // Коорд. химия. 1983. Т. 9. № 2. С. 227.
Вайсбергер А., Проскауэр Э., Риддик Дж., Тупс Э. Органические растворители. М.–Л.: ИЛ, 1958. 520 с.
Sheldrick G.M. // Acta. Crystallogr. A. 2008. V. 64. № 1. P. 112.
Беллами Л. Инфракрасные спектры сложных молекул. М.: ИЛ, 1963. 592 с.
Абраменко В.Л. // Коорд. химия. 2001. Т. 27. № 11. С. 869.
Сергиенко В.С., Абраменко В.Л., Горбунова Ю.Е., Чураков А.В. // Журн. неорган. химии. 2017. Т. 62. № 12. С. 1569 (Sergienko V.S., Abramenko V.L., Gorbunoba Yu.E., Churakov A.V. // Russ. J. Inorg. Chem. 2017. V. 62. № 12. P. 1563.).https://doi.org/10.1134/S0036023617120019
Сергиенко В.С., Абраменко В.Л., Горбунова Ю.Е. // Журн. неорган. химии. 2018. Т. 63. № 8. С. 989 (Sergienko V.S., Abramenko V.L., Gorbunoba Yu.E. // Russ. J. Inorg. Chem. 2018. V. 63. № 8. P. 1026).https://doi.org/10.1134/S003602361808020X
Сергиенко В.С., Мистрюков А.Э., Литвинов В.В. и др. // Коорд. химия. 1990. Т. 16. № 2. С. 168.
Randaccio L. // J. Organomet. Chem. 1973. V. 55. № 2. P. 58.
Bullock J.J., Ladd M.F.C., Povey D.C., Tajmir-Riahi H.A. // Acta Crystallogr. B. 1979. V. 35. № 9. P. 2013.
Pavlović G., Katava R., Rajić M., Roje R. // Polyhedron. 2017. V. 123. P. 285.
Porai-Koshitz M.A. // Izv. Jugosl. Cent. Kristallogr (Zagreb). 1974. V. 9. P. 19.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия