

Координационная химия, 2019, T. 45, № 12, стр. 734-745
Циклометаллированные комплексы иридия(III) с норборнензамещенным пиколинатным лигандом и электролюминесцентные полимеры на их основе
Ю. Е. Беганцова 1, Л. Н. Бочкарев 1, *, Е. В. Баранов 1, В. А. Ильичев 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: lnb@iomc.ras.ru
Поступила в редакцию 16.04.2019
После доработки 03.07.2019
Принята к публикации 08.07.2019
Аннотация
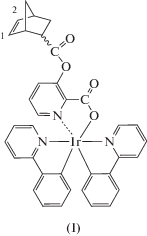
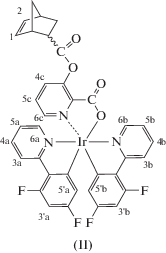
Синтезированы новые циклометаллированные комплексы иридия(III) – NBEpicIr(Ppy)2 (I) и NBEpicIr(Dfppy)2 (II) (NBEpicН = 3-(((1S,4S)-бицикло[2.2.1]гепт-5-ен-2-карбонил)окси)пиколиновая кислота, PpyH = 2-фенилпиридин, DfppyH = 2-(2,4-дифторфенил)пиридин). Комплекс I охарактеризован методом РСА (СIF file CCDC № 1878882). Реакцией метатезисной полимеризации с раскрытием цикла с участием соединений I и II и карбазолсодержащих норборненовых мономеров получены новые иридийсодержащие сополимеры. Изучены фотофизические свойства комплексов I, II и сополимеров на их основе.
Гомо- и гетеролигандные циклометаллированные комплексы иридия(III) привлекают большой интерес исследователей благодаря своим уникальным фотофизическим свойствам и широкому спектру применений [1–6]. Одно из основных направлений – их использование в качестве эмиттеров для органических светодиодов (OLEDs) [1, 3, 4]. Эффективность эмиттеров и цвет излучения в значительной степени зависят от природы лигандов, связанных с атомом иридия [1–3]. Кроме низкомолекулярных люминофорных комплексов иридия, к настоящему времени синтезировано также большое число иридийсодержащих полимерных эмиттеров [2, 7]. Среди электролюминесцентных иридийсодержащих полимеров наименее изучены и наименее эффективны эмиттеры, излучающие в синей области спектра [7]. В связи с этим получение новых эффективных “синих” иридийсодержащих полимерных электролюминофоров представляется важной и актуальной задачей.
Известно, что включение в циклометаллированные комплексы иридия пиколинатного лиганда приводит к сдвигу полос эмиссии в спектрах фотолюминесценции (ФЛ) и электролюминесценции (ЭЛ) иридиевых люминофоров в синюю область спектра [2, 8]. Аналогичный эффект наблюдается и в случае полимерных эмиттеров, содержащих комплексы иридия с пиколинатными лигандами [7]. Кроме применения в органических светодиодах, такие иридийсодержащие полимеры были использованы для создания устройств памяти и люминесцентных маркеров биологических объектов [9, 10].
В настоящей работе сообщается о синтезе двух новых бис-циклометаллированных комплексов иридия(III) – NBEpicIr(Ppy)2 (I) и NBEpicIr(Dfppy)2 (II) (NBEpicН = 3-(((1S,4S)-бицикло[2.2.1]гепт-5-ен-2-карбонил)окси)пиколиновая кислота, PpyH = 2-фенилпиридин, DfppyH = 2-(2,4-дифторфенил)пиридин) и получении на их основе методом метатезисной полимеризации с раскрытием цикла (ROMP – Ring-Opening Metathesis Polymerization) полимеров Р1–Р4, проявляющих интенсивную ФЛ и ЭЛ в зеленой и сине-зеленой областях спектра.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции с легко окисляющимися и гидролизующимися веществами проводили в вакууме или в аргоне с использованием стандартной техники Шленка. Используемые растворители тщательно очищали и дегазировали.
Соединения PpyH, DfppyH, 3-гидроксипиколиновая кислота (HOpicH), 4,7-дифенил-1,10-фенантролин (BATH) и трис(8-оксихинолинат) алюминия (Alq3) (Aldrich) использовали без дополнительной очистки. Циклометаллированные димерные хлориды [(L)2Ir(μ-Cl)]2, комплексы (L)2Ir(HOpic) (L = Ppy, Dfppy) синтезировали, как описано в [11, 12]. Карбазолсодержащие мономеры 9-(бицикло[2.2.1]гепт-5-ен-2-илметил)-9H-карбазол (L1) [13], бицикло[2.2.1]гепт-5-ен-2-ил(9Н-карбазол-9-ил)метанон (L2) [14] и (H2IMes) (3-Br-Рy)2(Cl)2Ru=CHPh (катализатор Граббса третьего поколения) [15] получали по известным методикам.
Спектры ЯМР 1H и 13C{1H} регистрировали на спектрометрах Bruker DPX-200 (ЯМР 1Н: 200 МГц, ЯМР 13С: 50 МГц) и Bruker Avance III-400 (ЯМР 1Н: 400 МГц, ЯМР 13С: 100 МГц). Химические сдвиги указаны в миллионных долях (м.д.) относительно тетраметилсилана в качестве внутреннего стандарта.
ИК-спектры снимали на ИК-Фурье спектрометре ФСМ 1201. Образцы соединений I и II готовили в виде суспензии в вазилиновом масле. Образцы полимеров P1–P4 готовили в виде тонких пленок между пластинами KBr.
Молекулярно-массовое распределение полимеров определяли методом гель-проникающей хроматографии (ГПХ) на хроматографе Knauer с дифференциальным рефрактометром Smartline RID 2300 в качестве детектора, с набором из двух колонок Phenomenex (сорбент Phenogel с размером пор 104 и 105 Å, элюент – THF, 2 мл/мин, 40°С). Калибровку колонок производили по 13 полистирольным стандартам.
ЭСП иридийсодержащих комплексов и полимеров в растворе CH2Cl2 снимали на спектрометре Perkin Elmer Lambda 25. Спектры фотолюминесценции регистрировали на флуоресцентном спектрометре Perkin Elmer LS 55. Относительные квантовые выходы определяли при комнатной температуре в дегазированных растворах CH2Cl2, длина волны возбуждения 370 нм. Значения квантовых выходов рассчитывали относительно Родамина 6G в этаноле (Φf = 0.95) [16] по методике, описанной в [17].
Дифференциально сканирующую калориметрию (ДСК) выполняли на приборе DSC 204 F1 Phoenix (Netzsch) в токе сухого аргона (скорость потока 20 см3/мин, скорость нагрева 5°C/мин).
Спектры электролюминесценции, вольт-амперные, вольт-яркостные характеристики и координаты цветности CIE получали на модельных OLED-устройствах без капсулирования с использованием автоматизированного, сопряженного с компьютером комплекса, включающего источник питания GW INSTEK PPE-3323, цифровой мультиметр GW INSTEK GDM-8246 и спектрофлуориметр Ocean Optics USB 2000.
Синтез NBEpicIr(Ppy)2 (I). К раствору (HOpic)Ir(Ppy)2 (0.10 г, 0.157 ммоль) и триэтиламина (0.16 г, 1.58 ммоль) в 10 мл CH2Cl2 добавляли раствор бицикло[2.2.1]гепт-5-ен-2-карбонил хлорида (0.12 г, 0.766 ммоль) в 5 мл CH2Cl2 и перемешивали при комнатной температуре в течение 2 ч. К реакционной смеси добавляли 20 мл воды и экстрагировали хлороформом. Экстракт сушили сульфатом магния, растворитель удаляли испарением в вакууме. Перекристаллизацией остатка из СНCl3 получили 0.12 г (87%) продукта I в виде желтого устойчивого на воздухе кристаллического вещества. Соединение содержит молекулу кристаллизационного хлороформа.
ИК-спектр (ν, см–1): 3054 сл, 3033 сл, 1101 ср, 1062 ср, 1030 ср, 1012 ср, 757 с, 731 с (CAr–H); 2927 о.с., 1379 с, 1334 ср, 1317 сл, 1163 ср (CAlk–H); 1607 ср (C⋅⋅⋅O); 1583 ср, 1462 о.с, 1265 ср (C=CAr); 494 о.сл (Ir–O).
Спектр ПМР (CDCl3; δ, м.д.): 8.79 д.д. (1H, Рyr, J = 6.4, 0.7 Гц), 7.82–7.89 м. (2H, Рic + Рyr), 7.70 т. (3H, J = 8.2 Гц, Рic + Рyr), 7.45–7.62 м. (4H, Рic + Рyr), 7.3 м. (1H, Рyr), 7.15 т. (1H, Рyr, J = 7.3 Гц), 6.72–6.98 м. (5Н, Ph), 6.37 д.д. (1H, Ph, J = 7.9, 0.9 Гц), 6.22 д.д. (1H, Н2, J = 5.4, 2.9 Гц), 6.14 д. (1H, pyr, J = 7.7 Гц), 6.09 м. (1H, Н1), 2.96 уш.с. (1.8 H, эндо-NBE), 2.62–2.73 м. (0.2 H, экзо-NBE), 1.95–2.28 м. (1 H, NBE), 1.48–.65 м. (4H, NBE), 0.85–0.92 м. (2H, NBE).
Спектр ЯМР 13С (CDCl3; δ, м.д.): 177.5, 169.3, 167.7, 160.9, 160.7, 148.9, 148.6, 148.1, 146.0, 139.7, 138.3, 138.1, 137.9, 137.3, 137.2, 137.1, 134.9, 132.5, 132.4, 130.0, 129.8, 129.6, 126.6, 124.3, 122.3, 122.8, 121.2, 119.0, 118.5, 49.9, 46.6, 46.5, 45.7, 42.6, 30.9, 29.7.
Синтез NBEpicIr(Dfppy)2 (II) проводили аналогично синтезу комплекса I из (HOpic)Ir(Dfppy)2 (0.0702 г, 0.098 ммоль) и бицикло[2.2.1]гепт-5-ен-2-карбонил хлорида (0.0784 г, 0.501 ммоль). Продукт выделяли перекристаллизацией из СН2Cl2 в виде желтых устойчивых на воздухе кристаллов массой 0.0790 г (87.6%).
ИК-спектр (KBr; ν, см–1): 3675 сл, 3371 сл, 3211 сл, 3081 ср, 1101 ср, 1012 ср, 763 ср, 722 ср (CAr–H); 2924 о.с, 1376 ср, 1340 ср, 1163 ср (CAlk–H); 1604 с (C⋅⋅⋅O); 1574 с, 1462 с, 1293 ср (C=CAr); 1249 ср, 1163 ср (C–F); 568 сл, 526 сл (Ir–O).
Спектр ПМР (CDCl3; δ, м.д.): 8.74 д. (1H, H6b Рyr, J = 5.2 Гц,), 8.21–8.31 м. (2H, Н3a и Н3b), 7.78 т. (2H, Н5c и Н6cJ = 7.76 Гц), 7.34–7.67 м. (2H, Н6a и Н4c), 7.21–7.23 м. (1H, Н5b), 6.99–7.01 м. (1H, Н5a), 6.33–6.53 м. (2H, Н3'a и Н3'b), 6.21–6.24 м. (1H, Н2), 6.08–6.12 м. (1H, Н1), 5.81 д.д. ( 1H, Н5'b, J = 8.5, 2.6 Гц), 5.52 д.д. (1H, Н5'a, J = 8.9, 2.3 Гц), 2.69 уш.с. (1.5 H, эндо-NBE), 2.61 – 2.70 м. (0.5 H, экзо-NBE), 2.06–2.14 м. (1 H, NBE), 1.26–1.51 м. (5H, NBE), 0.83–0.90 м. (1H, NBE).
Спектр ЯМР 13С (CDCl3; δ, м.д.): 177.3, 165.8, 164.4, 160.7, 152.0, 149.7, 148.6, 148.0, 145.9, 139.5, 138.4, 138.1, 135.8, 134.4, 132.5, 130.0, 128.8, 127.3, 123.4, 122.9, 122.6, 114.4, 98.1, 51.7, 49.7, 46.6. 45.6, 44.0, 42.6, 41.9, 29.9
Синтез сополимера P1. К смеси мономеров I (0.0312 г, 0.036 ммоль) и L1 (0.0965 г, 0.353 ммоль) в 5 мл СН2Cl2 добавляли катализатор Граббса третьего поколения (0.0031 г, 0.0034 ммоль). Смесь перемешивали при комнатной температуре. За ходом реакции следили методом тонкослойной хроматографии. По окончании сополимеризации (2 ч) к реакционной смеси добавляли несколько капель винилэтилового эфира и смесь перемешивали 30 мин. Образовавшийся сополимер осаждали гексаном, очищали переосаждением метанолом из СH2Cl2 и сушили в вакууме до постоянного веса. Выход сополимера Р1 0.11 г (89%).
ИК-спектр (ν, см−1): 3145 сл (C=C–H), 3051 ср, 1062 ср, 1030 ср, 1003 ср, 748 о.с, 722 о.с (CAr–H); 2938 с (C–H), 1381 сл, 1154 ср (CAlk–H); 1651 ср (C=O); 1598 с, 1482 о.с, 1461 о.с, 1228 ср (C=CAr); 970 ср (С–С); 565 сл, 529 сл (Ir–O).
Спектр ПМР (CDCl3; 200 МГц; δ, м.д.): 0.6–4.23 м. (97 H), 4.93–5.52 м. (20 Н), 7.05–7.33 м. (76 Н, Ar), 7.80–8.06 м. (23 Н, Ar).
Mn = 34 560, Mw/Mn = 1.7; Tg = 179.0°C.
Синтез сополимера P2 проводили аналогично синтезу сополимера P1. Время сополимеризации 4 ч. При использовании 0.0270 г (0.031 ммоль) мономера I и 0.0923 г (0.321 ммоль) мономера L2 получили 0.10 г (87%) сополимера P2.
ИК-спектр (ν, см−1): 3125 сл (C=C–H), 3060 ср, 1101 ср, 1062 сл, 1035 сл, 1006 о.сл, 725 о.с (CAr–H); 2947 ср (C–H), 1379 с, 1157 о.с (CAlk–H); 1690 о.с, 1651 ср (C=O); 1598 ср, 1476 ср, 1444 о.с, 1278 о.с (C=CAr); 970 сл (С–С); 568 сл, 524 сл (Ir–O).
Спектр ПМР (CDCl3; 200 МГц; δ, м.д.): 0.6–3.27 м. (77 H), 4.82–5.60 м. (22 Н), 7.08–8.18 м. (99 Н, Ar).
Mn = 22200, Mw/Mn = 1.4; Tg = 188°C.
Синтез сополимера P3 проводили аналогично синтезу сополимера P1. Время сополимеризации 2 ч. При использовании 0.0329 г (0.036 ммоль) мономера II и 0.0982 г (0.347 ммоль) мономера L1 получили 0.11 г (85%) сополимера P3.
ИК-спектр (ν, см−1): 3122 о.сл (C=C–H), 3053 ср, 1062 ср, 1027 ср, 1004 ср, 751 о.с (CAr–H); 2944 с (C–H), 1373 ср, 1154 с (CAlk–H); 1660 с (C=O); 1598 о.с, 1488 о.с, 1465 о.с, 1231 ср (C=CAr); 1243 ср, 1154 с (C–F); 973 сл (С–С); 565 сл, 526 сл (Ir–O).
Спектр ПМР (CDCl3; 200 МГц; δ, м.д.): 0.9–4.17 м. (97 H), 4.96–5.55 м. (22 Н), 7.15–7.33 м. (74 Н, Ar), 7.95–8.14 м. (21 Н, Ar).
Mn = 59410, Mw/Mn = 1.7; Tg = 188°C.
Синтез сополимера P4 проводили аналогично синтезу сополимера P1. Время сополимеризации 4 ч. При использовании 0.0251 г (0.026 ммоль) мономера II и 0.0770 г (0.268 ммоль) мономера L2 получено 0.90 г (93%) сополимера P4.
ИК-спектр (ν, см−1): 3128 сл (C=C–H), 3063 ср, 1101 сл, 1068 сл, 1038 ср, 754 о.с (CAr–H); 2950 ср (C–H), 1379 с, 1168 с (CAlk–H); 1690 о.с (C=O); 1559 ср, 1488 ср, 1479 с (C=CAr); 1240 сл, 1142 ср (C–F); 973 ср (С–С); 568 сл, 524 сл (Ir–O).
Спектр ПМР (CDCl3; 200 МГц; δ, м.д.): 0.95–3.54 м. (77 H), 4.95–5.52 м. (22 Н), 7.10–8.18 м. (95 Н, Ar).
Mn = 29 940, Mw/Mn = 1.2; Tg = 194°C.
РСА I проводили на дифрактометре Bruker D8 Quest (ω-сканирование, МоKα-излучение, λ = = 0.71073 Å). Экспериментальные наборы интенсивностей интегрированы с помощью программы SAINT [18]. Структура комплекса I расшифрована прямым методом с последующим уточнением полноматричным МНК по $F_{{hkl}}^{2}.$ Все неводородные атомы уточнены в анизотропном приближении. Атомы водорода помещены в геометрически рассчитанные положения и уточнены в модели “наездника”. Расчеты проведены с использованием программного пакета SHELXTL [19]. Учет поглощения проведен по программе SADABS [20]. В кристалле соединения I обнаружены сольватные молекулы хлороформа в общих положениях и в соотношении 1 : 1 к молекуле комплекса иридия. Норбоненовые фрагменты и одна из сольватных молекул CHCl3 разупорядочены по двум положениям. Кристаллографические данные и основные параметры уточнения представлены в табл. 1. Основные длины связей и валентные углы – в табл. 2.
Таблица 1.
Кристаллографические данные, параметры рентгеноструктурного эксперимента и уточнения для комплекса I
| Параметр | Значение |
|---|---|
| Формула | C37H29N3O4Cl3Ir |
| М | 878.18 |
| Сингония | Триклинная |
| Пр. гр | P1 |
| Температура, К | 100(2) |
| a, Å | 14.1292(10) |
| b, Å | 16.2045(11) |
| c, Å | 16.3443(11) |
| α, град | 76.056(2) |
| β, град | 65.665(2) |
| γ, град | 77.927(2) |
| V, Å3 | 3283.5(4) |
| Z | 4 |
| ρ(выч.), г/см3 | 1.776 |
| μ, мм–1 | 4.357 |
| F(000) | 1728 |
| Размер кристалла, мм | 0.35 × 0.35 × 0.22 |
| Область θ, град | 2.37–28.00 |
| Индексы отражений | –18 ≤ h ≤ 18, –21 ≤ k ≤ 21, –21 ≤ l ≤ 21 |
| Число собранных отражений | 38 962 |
| Число независимых отражений (Rint) | 15 738 (0.0382) |
| Число отражений с I > 2σ(I) | 12 649 |
| Поправка на поглощение (max/min) |
SADABS (0.3969/0.1915) |
| Данные/ограничения/параметры | 15 738/277/872 |
| Добротность (F 2) | 1.054 |
| R1/wR2 (I > 2σ(I)) | 0.0429/0.1045 |
| R1/wR2 (по всем отражениям) | 0.0584/0.1101 |
| Δρmax/Δρmin, e Å–3 | 2.807/–1.264 |
Таблица 2.
Некоторые длины связей (d) и валентные углы (ω) в комплексе I
| Связь | Молекула A | Молекула B | Угол | Молекула A | Молекула B |
|---|---|---|---|---|---|
| d, Å | ω, град | ||||
| Ir(1)–O(1) | 2.159(4) | 2.147(4) | O(1)Ir(1)C(36) | 172.16(17) | 172.30(19) |
| Ir(1)–N(1) | 2.129(4) | 2.134(5) | N(1)Ir(1)C(25) | 173.69(18) | 169.83(19) |
| Ir(1)–N(2) | 2.040(4) | 2.046(4) | N(2)Ir(1)N(3) | 174.54(16) | 173.93(18) |
| Ir(1)–N(3) | 2.026(4) | 2.026(4) | O(1)Ir(1)N(1) | 75.79(15) | 75.99(16) |
| Ir(1)–C(25) | 2.000(5) | 1.987(5) | N(2)Ir(1)C(25) | 80.4(2) | 80.2(2) |
| Ir(1)–C(36) | 1.976(5) | 1.983(6) | N(3)Ir(1C(36) | 80.63(19) | 81.1(2) |
| O(1)–С(1) | 1.276(7) | 1.254(7) | N(1)Ir(1)C(36) | 97.24(18) | 99.0(2) |
| O(2)–С(1) | 1.225(6) | 1.238(7) | O(1)Ir(1)C(25) | 99.38(17) | 95.52(18) |
| C(1)–C(2) | 1.524(8) | 1.506(8) | C(25)Ir(1)C(36) | 87.82(19) | 90.0(2) |
| C(2)–N(1) | 1.346(6) | 1.351(7) | N(1)Ir(1)N(3) | 87.73(16) | 91.92(17) |
| C(2)–C(3) | 1.389(8) | 1.390(8) | N(1)Ir(1)N(2) | 95.36(17) | 93.96(18) |
| C(3)–O(3) | 1.376(7) | 1.374(7) | N(3)Ir(1)C(25) | 96.84(19) | 94.18(19) |
| O(3)–C(7) | 1.367(8) | 1.379(8) | O(1)Ir(1)N(3) | 95.33(15) | 93.16(16) |
| C(7)–O(4) | 1.193(8) | 1.180(8) | O(1)Ir(1)N(2) | 89.79(15) | 89.75(16) |
| N(2)–C(19) | 1.357(7) | 1.354(7) | N(2)Ir(1)C(36) | 94.50(19) | 96.5(2) |
| C(19)–C(20) | 1.441(8) | 1.445(8) | |||
| C(20)–C(25) | 1.419(7) | 1.405(7) | |||
| N(3)–C(30) | 1.350(6) | 1.367(6) | |||
| C(30)–C(31) | 1.461(7) | 1.453(7) | |||
| C(31)–C(36) | 1.412(7) | 1.429(8) | |||
Кристаллографическая информация для соединения I депонирована в Кембриджском банке структурных данных (CCDC № 1878882; deposit@ccdc.cam.ac.uk или https://www.ccdc.cam.ac.uk/ structures/).
Изготовление OLED-устройств. В качестве несущей основы для OLED-устройств с конфигурацией ITO/Ir-полимер (40 нм)/BATH (30 нм)/ Alq3(30 нм)/Yb (150 нм) использовали стеклянную пластину с нанесенным слоем ITO (120 нм, 15 Oм/см2) (Lum Tec), играющим роль анода. Эмиссионный слой сополимера наносили из его раствора в CH2Cl2 (5 мг/мл) на центрифуге Spincoat G3-8 (3000 об./мин, 30 с) и высушивали в вакууме при 70°C в течение 3 ч. Толщину слоя определяли при помощи эллипсометра МЕТА-900. Дырочно-блокирующий слой BATH, электронопроводящий слой Alq3 и слой Yb, играющий роль катода, наносили испарением в вакууме (10–6 мм рт. ст.) с раздельных терморезистивных испарителей. Толщину слоев контролировали с помощью калиброванного кварцевого резонатора. Активная площадь устройств представляла собой круг диаметром 5 мм.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Норборненсодержащие комплексы иридия с пиколинатным “якорем” I и II получены в соответствии со схемой 1 :
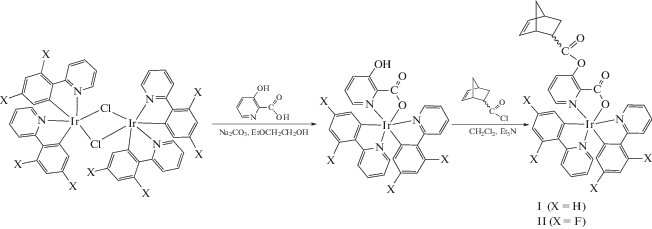
Схема 1 .
Соединения I и II выделены с высокими выходами в виде устойчивых на воздухе желтых кристаллических веществ, растворимых в хлороформе, CH2Cl2, DME. Исследования методом ЯМР показали, что в растворе продукты существуют в виде смеси эндо- и экзо-изомеров в соотношении 75 : 25.
Строение комплекса I определено методом РСА. В ассиметричной части элементарной ячейки расположены две молекулы А и В комплекса I, представляющие собой Δ- и Λ-изомеры соответственно (рис. 1). Центральный атом Ir(1) имеет октаэдрическое окружение и координирует три лиганда – два циклометаллированных (Ppy) и один дополнительный (NBEpic). Лиганды Ppy характеризуются цис-С–С и транс-N–N расположением относительно друг друга. Угол между апикальными атомами азота составляет 174.54(16)° (А) и 173.93(18)° (В). Выходы атомов Ir из плоскостей оснований октаэдров в молекулах А и В заметно различаются – 0.005 и 0.113 Å. Углы в лигандах Ppy NIr(1)C варьируют в узком диапазоне значений 80.2(2)°–81.1(2)°. Хелатные углы O(1)Ir(1)N(1) в норборненпиколинатных заместителях существенно меньшие – 75.79(15)° (А) и 75.99(16)° (В).
Рис. 1.
Молекулярные структуры Δ- (а) и Λ- (б) изомеров комплекса I. Тепловые эллипсоиды приведены c 30%-ной вероятностью. Атомы водорода и сольватные молекулы CHCl3 для наглядности не показаны.
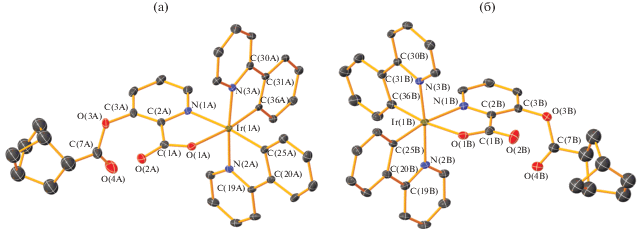
В каждой молекуле А и В комплекса I норборненовый фрагмент разупорядочен по двум положениям с равными заселенностями. В молекуле А норборнен в обоих разупорядоченных положениях находится в эндо-форме, в молекуле B – в эндо- и экзо-формах. Таким образом, в кристалле I обнаружены 75% комплексов иридия с норборненовым фрагментом в эндо-форме и 25% оставшихся комплексов с норборненом в экзо-форме, что соответствует данным спектров ЯМР.
Молекулы комплексов Ir(1A) и Ir(1B) упаковываются в центросимметричные пары с параллельной укладкой лигандов Ppy (рис. 2а, 2б). В паре молекул комплекса Ir(1A) параллельно ориентированные лиганды Ppy перекрываются пиридиновыми фрагментами N(2A)C(15A–21A) (рис. 2в). Центры пиридиновых колец смещены на 0.97 Å относительно друг друга. Расстояние между ними (3.54 Å) близко к расстоянию между параллельными плоскостями лигандов Ppy (3.38 Å). Данные геометрические характеристики могут указывать на наличие π–π-взаимодействия между пиридиновыми фрагментами N(2A)C(15A–21A) [21].
Рис. 2.
Фрагменты кристаллических упаковок молекул A (a, в) и B (б, г) комплекса I. Тепловые эллипсоиды приведены c 30%-ной вероятностью. Атомы водорода и сольватные молекулы CHCl3 для наглядности не показаны.
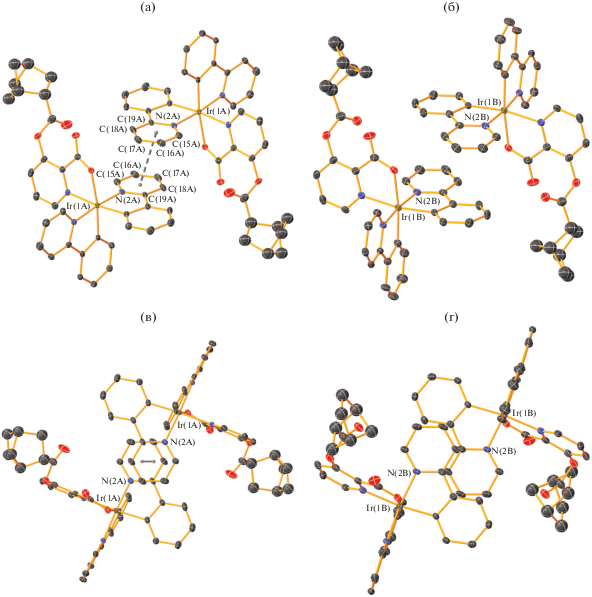
В отличие от комплекса Ir(1A), параллельно ориентированные лиганды Ppy двух соседних молекул второго комплекса Ir(1B) полностью смещены относительно друг друга – пиридиновый фрагмент одного Ppy-лиганда входит в “паз” между фенильным и пиридиновым фрагментами другого Ppy-заместителя (рис. 2г). Расстояния между центрами соседних фрагментов Ph···Py и Py···Py составляют 4.05 и 4.44 Å соответственно, что заметно больше межплоскостного расстояния между параллельными лигандами Ppy (3.44 Å). По-видимому, π–π-взаимодействие между двумя молекулами комплекса Ir(1B) отсутствует.
Известно, что полимеры на основе карбазола имеют оптимальные энергетические уровни для комплексов Ir с циклометаллированными Dfppy-лигандами [7]. Карбазольные группы в полимерных эмиттерах также улучшают их зарядо-транспортные свойства и электролюминесцентные характеристики [22, 23]. Поэтому для получения иридийсодержащих сополимеров методом ROMP были использованы норборненовые сомономеры с карбазольными фрагментами. Реакции сополимеризации протекают в присутствии катализатора Граббса III поколения при комнатной температуре и приводят к образованию иридийсодержащих сополимеров P1–P4 (схема 2 ).
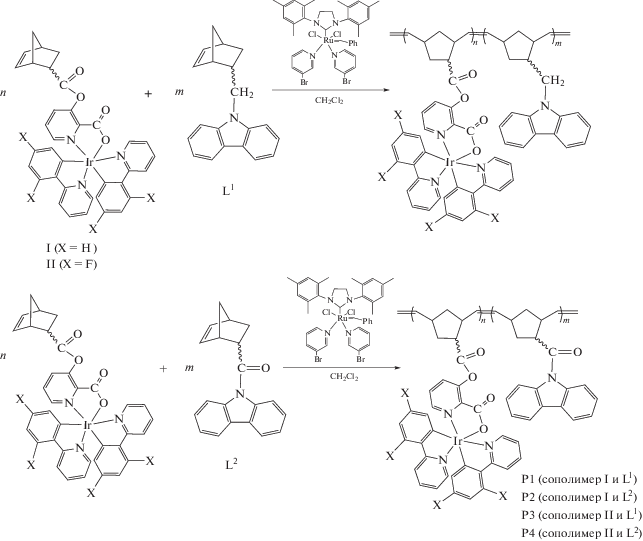
Исходное соотношение иридий- и карбазолсодержащих сомономеров n : m = 1 : 10 во всех случаях. Использовался 1 мол. % катализаторa Граббса по отношению к общему количеству введенных в реакцию сомономеров. Методом тонкослойной хроматографии определено, что реакции сополимеризации завершаются в течениe 2–4 ч. Сополимеры P1–P4 выделены в виде устойчивых на воздухе желтых порошков хорошо растворимых в THF, CH2Cl2 и CHCl3. Состав сополимеров подтвержден элементным анализом и спектроскопией ЯМР 1H.
ЭСП комплексов I и II (рис. 3, табл. 3) подобны спектрам известных пиколинатых комплексов иридия(III) (Pic)Ir(Ppy)2 и (Pic)Ir(Dfppy)2 (PicH = = пиколиновая кислота) и (HOPic)Ir(Ppy)2 и (HOPic)Ir(Dfppy)2 [24–26].
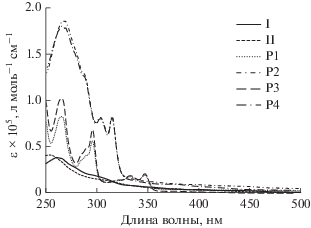
Таблица 3.
Фотофизические характеристики комплекса I, II и полимеров P1–P4
| Соединение | λпогл (нм)/lg ε | λэм (нм) | Квантовый выход, % | |
|---|---|---|---|---|
| пленка | CH2Cl2 | CH2Cl2 | ||
| I | 261 (4.57), 293 пл (4.29), 330 пл (3.90), 355 пл (3.76), 400 (3.51), 430 пл (3.35), 430 пл (3.24), 481 пл (2.71) | 538 (в кристаллах) | 503 | 2.3 |
| II | 253 (4.61), 284 пл (4.29), 330 пл (3.91), 354 пл (3.71), 395 (3.51), 435 пл (3.22), 481 пл (2.71) | 501 (в кристаллах) | 467, 497 | 65.8 |
| P1 | 264 (4.90), 285 пл (4.52), 290 пл (4.58), 295 (4.72), 322 пл (4.03), 332 (4.17), 347 (4.21), 367 пл (3.22), 383 пл (3.13), 395 пл (3.12), 440 пл (3.01), 480 пл (2.64) | 441 пл, 518 | 416, 440, 524, 550 пл | 12.5 |
| P2 | 266 (5.26), 278 пл (5.18), 303 (4.87), 315 (4.89), 333 пл (4.05), 360 пл (3.87), 380 пл (3.77), 410 пл (3.67) | 521 | 421, 433, 505 пл, 521 | 15.3 |
| P3 | 265 (5.01), 286 пл (4.63), 290 (4.67), 295 (4.83), 322 пл (4.14), 333 (4.27), 347 пл (4.31), 382 пл (3.21), 410 пл (3.06), 455 пл (2.86) | 482 пл, 498 | 468, 504 | 70.0 |
| P4 | 265 (5.24), 268 пл (5.23), 277 пл (5.17), 285 пл (5.09), 303 (4.88), 315 (4.89), 333 пл (4.01), 360 пл (3.80), 380 пл (3.78), 410 пл (3.52) | 480 пл, 499 | 428 пл, 468, 505 | 6.5 |
ЭСП комплексов I и II аналогичны и содержат интенсивные полосы с максимумами при 261 нм (I) и 253 нм (II), относящиеся к синглетным лиганд-центрированным переходам (LC) 1(π–π*) в ароматических циклометаллированных лигандах Ppy и Dfppy соответственно [27]. Слабые полосы поглощения в области 350–400 нм (ε < 6000) можно отнести к разрешенным по спину синглетным переходам с переносом заряда с металла на лиганд (1MLCT). Полосы в диапазоне 430–500 нм (ε < 2500) обусловлены запрещенными по спину 3LC/3MLCT переходами [25–27]. В спектрах поглощения сополимеров Р1–Р4 (рис. 3, табл. 3), кроме полос, характерных для комплексов I и II, наблюдаются дополнительные интенсивные полосы в области 250–340 нм, обусловленные 1(π–π*)-переходами в карбазольных фрагментах.
Спектры ФЛ комплексов I и II (рис. 4, табл. 3) в растворе CH2Cl2 содержат полосы с максимумами в области 460–500 нм, относящиеся к смешанным LC/MLCT переходам в циклометаллированных комплексах иридия [25–27]. В отличие от виб-ронно-структурированного спектра ФЛ комплекса II, спектр комплекса I широкий и неструктурированный, что, вероятно, связано с преобладанием MLCT-переходов [28]. Относительный квантовый выход фторированного комплекса II (65.8%) существенно выше, чем у нефторированного комплекса I (2.3%). Подобное поведение ранее отмечено для пар комплексов (НОPic)Ir(Dfppy)2/ (НОPic)Ir(Ppy)2 и (МеPic)Ir(Dfppy)2/(МеPic)Ir(Ppy)2, относительные квантовые выходы ФЛ 94/20% и 72/15% соответственно [26]. Авторы установили, что скорости безызлучательной дезактивации фторированных комплексов иридия значительно ниже, чем аналогичные процессы у их нефторированных аналогов. По-видимому, различие в относительных квантовых выходах комплексов I и II обусловлено теми же причинами.
Рис. 4.
Спектры ФЛ комплексов I и II при комнатной температуре: в растворе (I, II) и кристаллическом состоянии (Iкр, IIкр), λвозб = 370 нм.
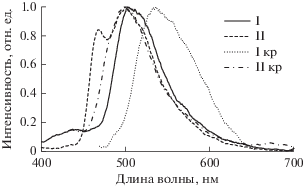
В спектрах ФЛ комплексов I и II в кристаллическом состоянии (рис. 4) наблюдается агрегационная эмиссия в виде широких неструктурированных полос [29]. Батохромный сдвиг (~35 нм) полосы испускания комплекса I обусловлен обнаруженными межмолекулярными π–π-взаимодействиями в кристалле.
Спектры ФЛ сополимеров Р1, Р2 и Р4 в растворе CH2Cl2 (рис. 5а, табл. 3), помимо широких полос в циклометаллированных комплексах иридия (480–521 нм), содержат полосы в диапазоне 416–433 нм, относящиеся к эксимерной эмиссии карбазольных групп [30, 31]. Отсутствие полос эмиссии полимерной матрицы в спектре сополимера Р3 свидетельствует о более эффективной передаче энергии возбуждения с карбазольных групп на иридийсодержащие фрагменты по механизму Форстера [32] и приводит к увеличению относительного квантового выхода Р3 по сравнению с другими сополимерами. В спектрах ФЛ сополимеров Р1–Р4 в тонких пленках (рис. 5б, табл. 3) присутствуют только полосы эмиссии металлсодержащих фрагментов, эмиссия полимерной матрицы практически не наблюдается.
Рис. 5.
Спектры ФЛ сополимеров Р1–Р4 в растворе CH2Cl2 при комнатной температуре (а) и в тонких пленках (б), λвозб = 370 нм.
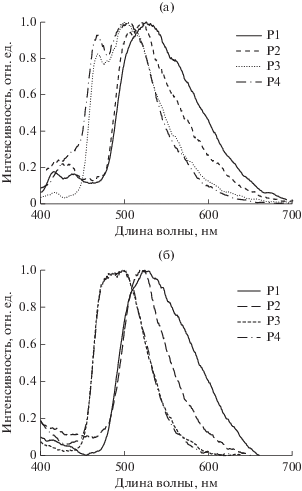
Электролюминесцентные свойства сополимеров P1–P4 исследованы на модельных OLED-устройствах конфигураций ITO/Ir–сополимер (40 нм)/BATH (30 нм)/Alq3 (30 нм)/Yb. Спектры ЭЛ полимеров P1–P4, а также рабочие характеристики OLED-устройств на их основе, представлены на рис. 6, 7 и в табл. 4.
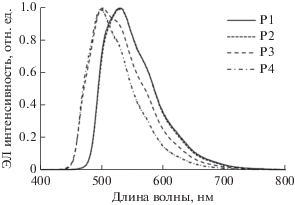
Рис. 7.
Вольт-амперные (а) и вольт-яркостные (б) характеристики светодиодов на основе полимеров P1–P4.
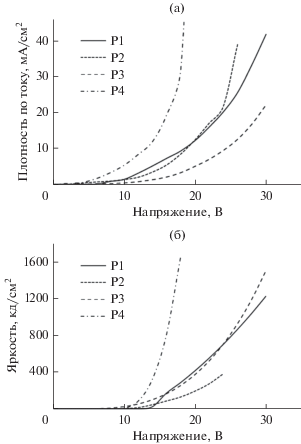
Таблица 4.
Рабочие характеристики светодиодов на основе полимеров P1–P4
| Ir-полимер | λмакс, нм | Напряжение включения, В* | Максимальная яркость, кд/м2 ** | Максимальная эффектив-ность по току, кд/A** | Максимальная эффективность по мощности, лм/Вт ** | Координаты цветности
в диаграмме CIE |
|---|---|---|---|---|---|---|
| P1 | 529, 555 пл | 12 | 1220 (30 В) | 3.42 (18 В) | 0.60 (18 В) | х = 0.32 y = 0.62 |
| P2 | 530, 556 пл | 8 | 380 (24 В) | 1.60 (16 В) | 0.31 (16 В) | х = 0.31 y = 0.62 |
| P3 | 503, 514 пл, 555 пл | 6 | 1493 (30 В) | 8.04 (16 В) | 1.88 (12 В) | х = 0.22 y = 0.45 |
| P4 | 501, 516 пл, 556 пл | 10 | 1653 (18 В) | 2.69 (14 В) | 0.60 (14 В) | х = 0.18 y = 0.42 |
Спектры ЭЛ сополимеров P1–P4 содержат полосы с максимумами в области 500–530 нм, относящиеся к 3LC/3MLCT переходам в циклометаллированных комплексах иридия, связанных с полимерной цепью. Отсутствие эмиссии карбазольных групп свидетельствует об эффективной передаче энергии возбуждения с полимерной матрицы на иридийсодержащие фрагменты по механизму Форстера [32]. Координаты цветности излучения светодиодов в диаграмме CIE (Commision Internationale del’Eclairage) (табл. 4) соответствуют желто-зеленому (Р1, Р2), зеленому (Р3), сине-зеленому (Р4) цветам и практически не изменяются во всем интервале рабочих напряжений. Наиболее высокие ЭЛ-характеристики показали светодиоды на основе сополимеров Р3 и P4 (табл. 4).
Таким образом, синтезированы новые циклометаллированные комплексы иридия(III) с норборнензамещенным пиколинатным лигандом и на их основе методом ROMP получены новые карбоцепные сополимеры P1–P4 с карбазольными и иридийсодержащими фрагментами в боковых цепях. Полученные полимерные эмиттеры проявляют интенсивную ФЛ и ЭЛ желто-зеленого, зеленого и сине-зеленого цвета. Максимальные значения яркости ЭЛ, эффективности по току и по мощности достигнуты при использовании эмиттеров Р3 и P4.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Highly Efficient OLEDs with Phosphorescent Materials / Ed. Yersin H. Weinheim: Wiley-VCH, 2008. P. 438.
Ulbricht C., Beyer B., Friebe C., Winter A. et al. // Adv. Mater. 2009. V. 21. P. 4418.
Iridium(III) in Optoelectronic and Photonics Applications / Ed. Zysman-Colman E. Chichester: Wiley-VCH, 2017. P. 736.
Yusoff A.R.B.M., Huckaba A.J., Nazeeruddin M.K. // Top Curr Chem (Z). 2017. V. 375. P. 39.
Lee S.Yu., Lee S.E., Oh Y.N. et al. // J. Nanosci. Nanotechnol. 2017. V. 17. P. 5673.
Nanomaterials, Polymers and Devices: Materials Functionalization and Device Fabrication / Ed. Kong E.S.W. New Jersey: Wiley-VCH, 2015. P. 195.
Xua F., Kima H.U., Kima J.-H. et al. // Progr. Polym. Sci. 2015. V. 47. P. 92.
Jayabharathi J., Jayamoorthy K., Thanikachalam V. // J. Organomet. Chem. 2014. V.761. P. 74.
Liu S.-J., Wang P., Zhao Q. et al. // Adv. Mater. 2012. V. 24. P. 2901.
Liu S., Qiao W., Cao G. et al. // Macromol. Rapid Commun. 2013. V. 34. P. 81.
Nonoyama M. // Bull. Chem. Soc. Jpn. 1974. V. 47. P. 767.
Kwon T.-H., Kim M.K., Kwon J. et al. // Chem. Mater. 2007. V. 19. P. 3673.
Liaw D.J., Tsai C.H. // Polymer. 2000. V. 41. P. 2773.
Рожков А.В., Бочкарев Л.Н., Басова Г.В. и др. // Журн. общ. химии. 2012. Т. 82. № 12. С. 1937.
Love J.A., Morgan J.P., Trnka T.M. et al. // Angew. Chem. Int. Ed. 2002. V. 41. P. 4035.
Magde D., Wong R., Seybold P.G. // Photochem. Photobiol. 2002. V. 75. P. 327.
Demas J.N., Crosby G.A. // J. Phys. Chem. 1971. V. 75. P. 991.
SAINT. Data Reduction and Correction Program. Version 8.34A. Madison (WI, USA): Bruker AXS, 2014.
Sheldrick G.M. SHELXTL. Version 6.14. Structure Determination Software Suite. Madison (WI, USA): Bruker AXS, 2003.
Sheldrick G.M. SADABS. Version 2014/5. Bruker/Siemens Area Detector Absorption Correction Program. Madison (WI, USA): Bruker AXS, 2014.
Janiak C. // Dalton Trans. 2000. P. 3885.
Grimsdale A.C., Chan K.L., Martin R.E. et al. // Chem. Rev. 2009. V. 109. P. 897.
Бочкарев М.Н., Витухновский А.Г., Каткова М.А. Органические светоизлучающие диоды (OLED). Н. Новгород: Деком, 2011. 364 с.
Baranoff E., Curchod B.F.E. // Dalton Trans. 2015. V. 44. P. 8318.
You Y., Kim K.S., Ahn T.K. et al. // J. Phys. Chem. C. 2007. V. 111. P. 4052.
Yi S., Kim J.-H., Cho Y.-J. et al. // Inorg. Chem. 2016. V. 55. P. 3324.
Lamansky S., Djurovich P., Murphy D. et al. // Inorg. Chem. 2001. V. 40. P. 1704.
Zhao Q., Liu S.J., Shi M. et al. // Inorg. Chem. 2006. V. 45. P. 6152.
Hao Z., Jiang H., Liu Y. et al. // Tetrahedron. 2016. V. 72. P. 8542.
Johnson G.E. // J. Chem. Phys. 1975. V. 62. P. 4697.
Vekikouas G.E., Powell R.C. // Chem. Phys. Lett. 1975. V. 34. P. 601.
Förster T. // Disc. Farad. Soc. 1959. V. 27. P. 7.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия